

Журнал аналитической химии, 2020, T. 75, № 8, стр. 702-713
Хромато-масс-спектрометрическое определение полициклических ароматических углеводородов в почвах и донных отложениях с применением техники дисперсионной жидкостно-жидкостной микроэкстракции
З. А. Темердашев a, *, Т. Н. Мусорина a, Т. А. Червонная a
a Кубанский государственный университет, факультет химии и высоких технологий
350040 Краснодар, ул. Ставропольская, 149, Россия
* E-mail: temza@kubsu.ru
Поступила в редакцию 11.12.2019
После доработки 28.01.2020
Принята к публикации 21.02.2020
Аннотация
Обсуждены проблемы и достоинства хроматографических методов определения полициклических ароматических углеводородов (ПАУ) в модельных и реальных образцах чернозема, песка и донных отложений Азовского моря и Курчанского лимана. Обоснована и реализована подготовка проб к анализу с использованием техники дисперсионной жидкостно-жидкостной микроэкстракции. Концентрации ПАУ в почвах различного типа и донных отложениях определяли методом газовой хромато-масс-спектрометрии. Исследованы особенности пробоподготовки почв и донных отложений для достижения максимального извлечения ПАУ в органическую фазу, оптимизированы состав экстракционной системы, условия извлечения аналитов, выбрана оптимальная масса навески. Предложена схема газохроматографического с масс-спектрометрическим детектированием (ГХ–МС) определения 20 ПАУ в почвах (донных отложениях). Пределы определения изученных ПАУ в почвах и донных отложениях составили 0.2–0.5 мкг/кг. Оптимизированная схема ГХ–МС-определения ПАУ апробирована на реальных образцах донных отложений Темрюкского залива Азовского моря.
Полициклические ароматические углеводороды (ПАУ) являются канцерогеноопасными и генотоксичными соединениями, поступающими в организм с продуктами питания, через пищевые цепи, а также непосредственно из окружающей среды. Контроль их содержания в этих объектах – актуальная, но сложная задача аналитической химии. Почвы и донные отложения являются устойчивой матрицей для аккумулирования и длительного хранения накопленных ПАУ и считаются объективными показателями состояния загрязнения окружающей среды. Данный факт объясняется гидрофобностью ПАУ и склонностью к агрегации на твердых частицах. Их накопление в почвах может привести к потенциальному загрязнению пищевой и сельскохозяйственной продукции [1, 2], и, как следствие, прямому или косвенному воздействию на человека. Сорбционная способность донных отложений и почв связана с их физико-химическими характеристиками, такими как гранулометрический состав и массовая доля в них органического вещества. Это подтверждается, в частности, результатами исследования [3] донных отложений Бискайского залива – наблюдается линейная корреляция между содержанием в них органического вещества и концентрацией ПАУ.
Важным этапом определения ПАУ в различных твердых объектах является их отделение от природной матрицы [4, 5]. Поскольку ПАУ относятся к суперэкотоксикантам, для некоторых из них установлены ориентировочно допустимые концентрации в почве: нафталин, антрацен, флуорантен, бензантрацен, хризен, бензперилен, бензфлуорантен [6]. Общепринятым маркером такого рода загрязнений признан бенз[а]пирен, значение ПДК которого 20 мкг/кг установлено нормативом [7]. Синергическое действие смеси ПАУ может привести к повышению канцерогенности этих веществ, поэтому для корректной оценки загрязненности природных объектов целесообразно определение индивидуальных генотоксичных ПАУ. Низкие фоновые содержания ПАУ в природных объектах, являющихся многокомпонентными матрицами, обусловили преимущественное использование газовой хромато-масс-спектрометрии (ГХ–МС) и ВЭЖХ с ультрафиолетовым [8], флуориметрическим [9] или диодно-матричным [10] детекторами для их анализа.
Применение ГХ–МС в режиме мониторинга заданных ионов (selected ion monitoring, SIM) позволяет разделять близкие по строению соединения, в том числе изомеры, а также минимизировать влияние матричных эффектов [11–13]. Метод исключает упаривание, последующее повторное растворение экстракта, совместим с бóльшим количеством растворителей, чем в случае ВЭЖХ. С другой стороны, ГХ–МС при определении ПАУ уступает ВЭЖХ по чувствительности, поэтому особое внимание необходимо уделять оптимизации условий пробоподготовки для концентрирования и снижения пределов определения аналитов.
Для выделения аналитов из анализируемой матрицы чаще всего используют экстракционные методы. Жидкостно-жидкостная экстракция получила развитие, благодаря экспрессности и сравнительно невысокой трудоемкости [14]. Одним из вариантов метода является гомогенная жидкостно-жидкостная экстракция (homogeneous liquid–liquid extraction, HLLE), предполагающая использование растворителя-экстрагента с плотностью легче воды и сорастворителя для повышения эффективности извлечения ПАУ в гидрофобную органическую фазу [15]. Тенденция к снижению экологической нагрузки на аналитические лаборатории способствовала появлению методов твердофазной микроэкстракции (ТФМЭ) [16], позволяющих предварительно извлекать аналиты из твердого образца в растворитель с последующей сорбцией их на устройство, содержащее волокна, покрытые пленкой, например полидиметилсилоксаном [17, 18]. Представляют интерес исследования с использованием микроэмульсий типа “масло в воде”, содержащих в своем составе поверхностно-активные вещества (ПАВ) [19, 20], главным образом анионные [21–26]. Достаточно перспективно применение для концентрирования аналитов ионных жидкостей (ИЖ), которые позволяют избежать применения токсичных растворителей [27].
Перечисленные выше методы пробоподготовки имеют определенные преимущества: не требуют применения большого количества токсичных органических растворителей, более экспрессны и эффективны. Однако существенные трудности в случае ТФМЭ вносит эффект “памяти” и хрупкость волокон, что делает практически невозможным их многократное использование, а плохая воспроизводимость толщины пленки может привести к искажению результатов. В варианте HLLE не гарантирована полнота извлечения аналитов при их следовых содержаниях ввиду малой площади поверхности контакта экстрагирующей системы и пробы. Применение микроэмульсий, содержащих ПАВ, может привести к необратимым процессам в хроматографической колонке, а использование ИЖ пока мало изучено.
Перспективным представляется метод дисперсионной жидкостно-жидкостной микроэкстракции (dispersive liquid−liquid microextraction, DLLME), высокая эффективность которой достигается за счет использования дополнительного растворителя в качестве диспергирующего агента. Образование мельчайших капель экстрагента позволяет значительно повысить площадь поверхности контакта фаз в экстракционной системе [28]. Описан [29] вариант применения ультразвуковой (УЗ) обработки вместо диспергирующего растворителя для техники DLLME, но в случае экстракции аналитов из твердых образцов данный подход реализовать трудно.
В работе [30] при определении ПАУ в природных объектах вместо диспергирующего агента предложили использовать вертикальный встряхиватель или микроэмульсию, содержащую неионогенные ПАВ. Для снижения токсичности в качестве экстракционной системы вместо хлорорганических растворителей применяли н-гептанол. Интересным является сочетание DLLME и техники затвердевающей капли растворителя (solidification of a floating organic droplet, SFOD). Применяемые легкие растворители затвердевают при небольших отрицательных температурах на поверхности раствора в виде отдельных капель, которые собирают механическим способом и расплавляют при комнатной температуре для последующего хроматографического анализа [31].
Несмотря на разнообразие предлагаемых решений, задача повышения чувствительности и воспроизводимости определения ПАУ в твердых образцах в широком концентрационном диапазоне остается актуальной. Сложность работы с почвами или донными отложениями заключается в необходимости проводить предварительное извлечение определяемых компонентов в жидкую фазу. Малая эффективность перехода аналитов из образца в растворитель на данном этапе может существенно сказаться на результатах анализа, поэтому оптимизация условий извлечения ПАУ является одной из ключевых. Для извлечения ПАУ оптимальным экстрагентом считается ацетонитрил [28, 32], а эффективность извлечения аналитов из образца часто достигается встряхиванием на вортексе [33, 34] либо УЗ-воздействием, что, по мнению ряда исследователей, более предпочтительно [35, 36]. Авторы работы [37] предложили интересный способ, состоящий в предварительном извлечении аналитов в ацетонитрил на вортексе и последующей микроэкстракции ПАУ из надосадочного раствора в дихлорметан. При этом не достигнута высокая степень извлечения аналита, а предлагаемый ВЭЖХ-анализ включает упаривание гидрофобного экстрагента с последующим растворением в ацетонитриле, в результате которого возможны потери “легких” ПАУ.
Анализ данных [38–41] показал, что суммарное содержание ПАУ в почвах и донных отложениях варьирует в диапазоне 20–80 мкг/кг вдали индустриальных центров и может составлять до 5000 мкг/кг в местах интенсивной антропогенной нагрузки. При разработке методики определения ПАУ необходимо учитывать высокую канцерогенную и токсикологическую опасность этих веществ, что требует их контроля ниже уровня ПДК.
Цель данного исследования – разработка схемы дисперсионной жидкостно-жидкостной микроэкстракции при хромато-масс-спектрометрическом определении ПАУ в почвах различного типа и донных отложениях.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объекты исследования. Исследования проводили на модельных и реальных пробах чернозема, песка, донных отложений. Для этого проанализировали значительное количество образцов донных отложений и почв (свыше 140), отобранных в восточной части Азовского моря и Курчанского лимана при проведении экологического мониторинга на содержание ПАУ. Содержание ПАУ в образцах определяли по стандартизированной методике [42]. Образцы, содержащие следовые количества исследуемых ПАУ, выбрали в качестве матриц реальных объектов для моделирования образцов при разработке методики. Образцы почв и донных отложений не сушили, дабы исключить возможные потери “легких” ПАУ. Содержание воды учитывали путем пересчета концентрации ПАУ на массу сухого образца при обработке данных анализа.
Реактивы и материалы. Использовали ацетонитрил для ВЭЖХ (Sigma-Aldrich, США); хлороформ ос. ч., дихлорметан ос. ч., тетрахлорметан ос. ч., бидистиллированную воду. Для идентификации и построения градуировочных кривых использовали индивидуальные стандартные образцы состава раствора ПАУ: нафталин СОП 0109-03 ЕR-РAH 6, бифенил СОП 0107-03 ЕR-РAH 4, 2‑метилнафталин СОП 0101-03 ЕR-РAH 5, флуорен СОП 0113-03 ЕR-РAH 9, аценафтен СОП 0103-03 ЕR-РAH 1, аценафтилен СОП 0104-03 ER-PAH 10, фенантрен СОП 0111-03 ЕR-РAH 7, антрацен СОП 0102-03 ЕR-РAH 2, флуорантен СОП 0112-03 ЕR-РAH 8, пирен СОП 0110-03 ЕR-РAH 12, бенз[а]антрацен СОП 0105-03 ЕR-РAH 15, хризен СОП 0114-03 ЕR-РAH 13, бенз[b]флуорантен СОП 0115-03 ЕR-РAH 14, бенз[k]флуорантен СОП 0116-03 ЕR-РAH 16, бенз[а]пирен СОП 0106-03 ЕR-РAH 3, дибенз[a,h]антрацен СОП 0108-03 ЕR-РAH 11, бенз[g,h,i]перилен СОП 0117-03 ЕR-РAH 17 в ацетонитриле (НПО “Экрос”, Санкт-Петербург, Россия); аналитические стандарты: бенз[е]пирен CAS 192-97-2 и индено[1,2,3-c,d]пирен CAS 193-39-5 в циклогексане, трифенилен CAS 217-59-4 – чистые вещества (Sigma-Aldrich, США).
Оборудование. Применяли газохроматографическую систему, состоящую из хроматографа и моноквадрупольного масс-спектрометрического детектора Shimadzu GCMS-QP2020, жидкостный хроматограф LC-30 NEXERA с RF-20Axs (Shimadzu, Япония). УЗ-воздействие осуществляли генератором ультразвуковых колебаний (Сапфир, Россия) с рабочей частотой 35 кГц. Для центрифугирования использовали настольные лабораторные центрифуги Universal 320 (Hettich, Германия) и MiniSpin (Eppendorf, Германия).
Условия хромато-масс-спектрометрического анализа. В основу хромато-масс-спектрометрического анализа образцов почв и донных отложений положена разработанная ранее методика хроматографического разделения и детектирования 16 ПАУ в различных типах вод [43]. Условия хроматографирования оптимизировали и уточняли с учетом расширения перечня определяемых компонентов: бенз[е]пирена, аценафтилена, инден[1,2,3-c,d]пирена, трифенилена. Использовали колонку ZB-5ms длиной 60 м с привитой фазой 5%-полисиарилен + 95%-диметилполисилоксан, позволяющую удовлетворительно разделять определяемые ПАУ (рис. 1). Оптимальные условия хроматографического анализа образцов представлены в табл. 1.
Рис. 1.
Хроматограмма модельной смеси 20 ПАУ (концентрация каждого индивидуального компонента составила 100 мкг/мл).
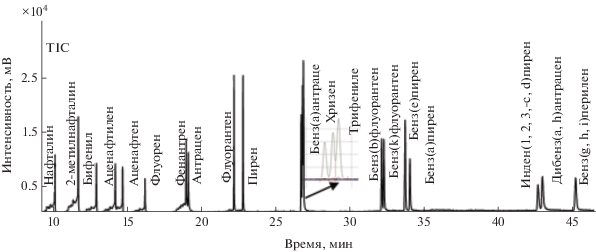
Таблица 1.
Условия проведения хромато-масс-спектрометрического анализа
| Параметр | Значение |
|---|---|
| Температура испарителя | 280°С |
| Температурная программа термостата | 60°С/1 мин – нагрев 15 град/мин – 170°С/3 мин – нагрев 10 град/мин – 280°С/8 мин – нагрев 10 град/мин – 290°С/25 мин |
| Температура ионного источника | 200°С |
| Температура интерфейса | 280°С |
| Скорость газа-носителя через колонку | 1.35 мл/мин |
| Коэффициент деления потока | 1 : 5 |
| Энергия ионизации | 70 эВ |
| Режим SIM | 128, 142, 152, 154, 166, 178, 202, 228, 252, 276, 278 m/z |
| Объем вводимой пробы | 2 мкл |
| Задержка работы детектора | 7 мин |
| Общее время анализа | 56 мин |
Ионизация осуществлялась электронным ударом, детектирование ПАУ выполняли в режиме мониторинга заданных ионов. Определяли времена удерживания каждого индивидуального вещества (табл. 2) и сопоставляли их масс-спектры с библиотечными данными баз NIST и WILLEY.
Таблица 2.
Хромато-масс-спектрометрические характеристики определяемых ПАУ
| ПАУ | Время удерживания, мин | Молекулярный ион, m/z |
|---|---|---|
| Нафталин | 10.17 | 128 |
| 2-Метилнафталин | 11.75 | 142 |
| Аценафтилен | 14.21 | 152 |
| Бифенил | 12.97 | 154 |
| Аценафтен | 14.73 | 154 |
| Флуорен | 16.23 | 166 |
| Фенантрен | 18.99 | 178 |
| Антрацен | 19.13 | 178 |
| Флуорантен | 22.21 | 202 |
| Пирен | 22.81 | 202 |
| Бенз[а]антрацен | 26.72 | 228 |
| Хризен | 26.88 | 228 |
| Трифенилен | 26.89 | 228 |
| Бенз[b]флуорантен | 32.14 | 252 |
| Бенз[k]флуорантен | 32.23 | 252 |
| Бенз[е]пирен | 33.72 | 252 |
| Бенз[а]пирен | 34.03 | 252 |
| Инден[1,2,3-c,d]пирен | 42.58 | 276 |
| Дибенз[a,h]антрацен | 42.92 | 278 |
| Бенз[g,h,i]перилен | 45.08 | 276 |
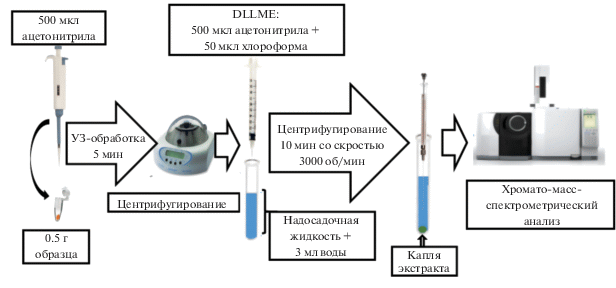
Пробоподготовка. Навеску исследуемого образца 0.5 г помещали в пробирку, добавляли 0.5 мл ацетонитрила и проводили ультразвуковую обработку в течение 5 мин. Затем образец в течение 10 мин центрифугировали со скоростью вращения ротора центрифуги 10 000 об/мин. Надосадочную жидкость декантировали в пробирку емк. 10 мл, добавляли 3 мл бидистиллированной воды и смесь для DLLME, содержащую хлорорганический экстрагент (дихлорметан, четыреххлористый углерод, хлороформ). Затем экстракт центрифугировали 10 мин при скорости 3000 об/мин. На дне пробирки образовывалась капля экстракта (рис. 2), из которой отбирали аликвоту для анализа.

РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
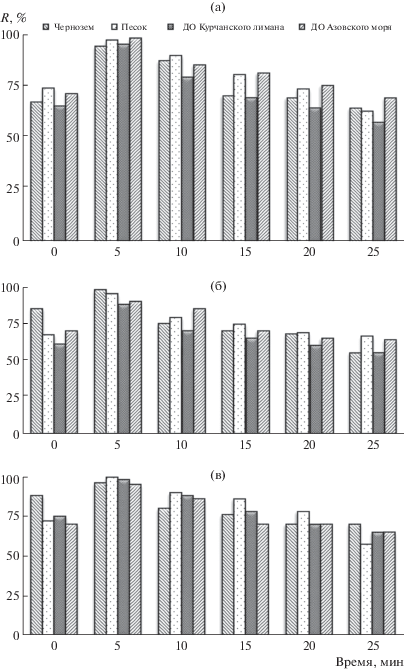
Оптимизация условий извлечения ПАУ в органическую среду. Основная проблема первого этапа подготовки пробы к анализу – достижение максимального перехода ПАУ в ацетонитрил. Поскольку ультразвуковая кавитация увеличивает площадь поверхности контакта твeрдой и жидкой фаз, для повышения эффективности процесса извлечения аналитов мы использовали УЗ-обработку исследуемых образцов. Для оптимизации УЗ-воздействия использовали образцы с добавками смесей ПАУ с концентрацией каждого 0.5 и 500 мкг/кг. Табл. 3 иллюстрирует зависимость степени извлечения ПАУ от продолжительности ультразвуковой обработки на примере проб чернозема с добавками. Зависимости степени извлечения бенз[а]пирена, флуорена и хризена из всех типов анализируемых образцов от продолжительности ультразвуковой обработки экстракта приведены на рис. 3, для всех остальных ПАУ они аналогичны. По результатам ВЭЖХ-анализа экстрактов установили, что максимальное извлечение аналитов из почв различного состава и донных отложений в ацетонитрил достигается в течение 5 мин УЗ-воздействия независимо от концентрации ПАУ.
Таблица 3.
Зависимость степени извлечения аналитов* из проб чернозема с добавками ПАУ от продолжительности ультразвуковой обработки
| ПАУ | Без УЗ | 3 мин | 5 мин | 10 мин | 15 мин | 20 мин |
|---|---|---|---|---|---|---|
| Нафталин | 70/71 | 84/83 | 98/97 | 94/95 | 87/84 | 64/65 |
| 2-Метилнафталин | 64/61 | 85/83 | 95/96 | 91/90 | 88/84 | 72/72 |
| Аценафтилен | 78/75 | 81/80 | 95/96 | 92/91 | 91/88 | 70/68 |
| Бифенил | 71/72 | 83/84 | 96/93 | 94/93 | 85/85 | 79/79 |
| Аценафтен | 71/71 | 80/81 | 97/95 | 94/93 | 89/86 | 84/82 |
| Флуорен | 85/84 | 90/92 | 98/95 | 75/75 | 70/72 | 68/64 |
| Фенантрен | 84/85 | 92/88 | 98/96 | 98/94 | 94/91 | 77/79 |
| Антрацен | 64/65 | 79/80 | 92/94 | 92/93 | 84/83 | 78/80 |
| Флуорантен | 75/77 | 88/84 | 94/95 | 92/94 | 87/84 | 74/75 |
| Пирен | 64/63 | 89/90 | 94/96 | 92/91 | 83/85 | 75/75 |
| Бенз[а]антрацен | 68/70 | 84/86 | 92/93 | 90/91 | 89/86 | 75/75 |
| Хризен | 88/69 | 90/92 | 96/95 | 80/92 | 76/75 | 70/68 |
| Трифенилен | 68/69 | 83/84 | 94/96 | 90/92 | 80/82 | 77/78 |
| Бенз[b]флуорантен | 70/69 | 89/87 | 95/92 | 87/91 | 86/84 | 78/78 |
| Бенз[k]флуорантен | 64/67 | 90/91 | 97/94 | 88/87 | 82/84 | 78/76 |
| Бенз[е]пирен | 68/70 | 90/93 | 95/95 | 93/89 | 91/90 | 82/80 |
| Бенз[а]пирен | 68/69 | 88/87 | 95/96 | 88/91 | 71/89 | 70/72 |
| Инден[1,2,3-c,d]пирен | 68/67 | 93/94 | 98/96 | 95/94 | 95/84 | 74/75 |
| Дибенз[a,h]антрацен | 64/65 | 95/94 | 99/96 | 91/90 | 87/89 | 76/75 |
| Бенз[g,h,i]перилен | 64/65 | 93/94 | 97/97 | 91/90 | 91/93 | 72/74 |
Рис. 3.
Зависимость степени извлечения бенз[а]пирена (а), флуорена (б) и хризена (в) из анализируемых образцов от продолжительности ультразвуковой обработки экстракта (ДО – донные отложения).
Оптимизация состава экстракционной системы для концентрирования ПАУ. Для концентрирования аналитов и исключения возможного гидролиза неподвижной жидкой фазы капиллярной колонки при анализе влажных образцов в схему пробоподготовки включали стадию микроэкстракции ПАУ с использованием техники DLLME. Для этого опробовали различные составы трехкомпонентных систем, обеспечивающих образование устойчивой, воспроизводимой капли экстракта и количественный переход аналитов из ацетонитрила. Наиболее эффективное отделение хлорорганического экстракта от диспергирующего агента по данным [28, 29, 34, 37] и нашим экспериментальным данным достигается введением в систему бидистиллированной воды.
Установлено, что при использовании в качестве экстрагента дихлорметана или четыреххлористого углерода образование капли нестабильно или не происходит вовсе. В случае хлороформа достигается образование устойчивой и воспроизводимой капли экстракта, поэтому в дальнейших исследованиях использовали систему ацетонитрил–хлороформ–вода. При варьировании объема добавляемого в экстракционную систему хлороформа (100, 80 и 50 мкл) устойчивость и воспроизводимость капли экстракта сопоставимы. При меньших объемах хлороформа не удавалось получить воспроизводимую трехкомпонентную систему. Для снижения предела определения аналитов выбрали объем хлороформа 50 мкл.
Для обеспечения максимальной эффективности концентрирования на стадии микроэкстракции подбирали оптимальные объемы компонентов экстрагирующей системы. Объем ацетонитрила варьировали от 0.6 до 1 мл ацетонитрила, воды – от 2 до 5 мл воды. Установлено, что количественное извлечение аналитов достигается при соотношении (мл) ацетонитрил–вода (1 : 3). При меньших содержаниях воды в системе увеличивается растворимость хлороформа в ацетонитриле и экстракт невозможно выделить количественно. Когда объем хлороформа, добавляемого в экстракционную систему, составляет 50 мкл, оптимально использование 1 мл ацетонитрила и 3 мл воды, причем 0.5 мл ацетонитрила добавляют к навеске анализируемого образца естественной влажности на первом этапе извлечения, а остальные 0.5 мл ацетонитрила вводят в экстракционную систему в качестве диспергирующего агента в составе смеси для микроэкстракции.
Количественное отделение надосадочной жидкости от твердых частиц пробы обеспечивали высокой степенью седиментации при центрифугировании в течение 10 мин со скоростью вращения ротора центрифуги 10 000 об./мин. Установили оптимальный состав экстракционной системы для DLLME, включающий смесь из 50 мкл хлороформа и 0.5 мл ацетонитрила.
Выбор оптимальной массы навески осуществляли в оптимизированных условиях экстракционного извлечения ПАУ. Изучали модельные системы с ПАУ, содержащие от 0.5 до 1.5 г исследуемого образца. Использование массы навески менее 0.5 г при хромато-масс-спектрометрическом детектировании аналита нежелательно из-за повышения предела определения ПАУ (до 1.3 мкг/кг), кроме того, возникает проблема обеспечения представительности пробы. При массе навески более 1.5 г необходимо использовать бóльшее количество ацетонитрила для смачивания, что нарушает стабильность экстрагирующей системы. С учетом полученных результатов и концентрационного диапазона изучаемых ПАУ в качестве оптимальной выбрали массу навески 0.5 г.
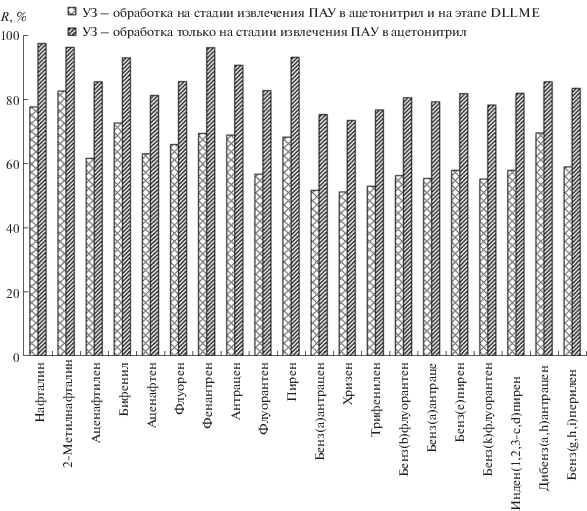
Выбор оптимальных условий микроэкстракции ПАУ. Для определения условий максимальной степени извлечения определяемых аналитов исследовали влияние УЗ-обработки на этапе микроэкстракции. Из рис. 4 видно, что эффективность микроэкстракции ПАУ при обработке смеси ультразвуком заметно снижается по сравнению с пробами, не подвергавшимися такому воздействию. Объяснить этот факт сложно. Возможно, происходит обратный переход ПАУ из хлороформа в ацетонитрил, кроме того, нельзя исключить разрушение аналитов в условиях ультразвукового воздействия.
Рис. 4.
Оценка эффективности извлечения ПАУ из ацетонитрила в хлороформ на стадии микроэкстракции под ультразвуковым воздействием.
Для минимизации потерь аналитов на стадии микроэкстракции пробы встряхивали, а полноту осаждения капли экстракта достигали центрифугированием в течение 10 мин при скорости вращения ротора центрифуги 3000 об/мин.
Оптимизированная аналитическая схема газохроматографического с масс-спектрометрическим детектированием определения ПАУ в почвах (донных отложениях). С учетом оптимизированных условий для этапов подготовки образцов (предварительное извлечение в ацетонитрил и концентрирование аналитов с использованием техники DLLME) предложена схема определения ПАУ в почвах (донных отложениях) (рис. 5).
Пределы определения (cн) ПАУ в почвах (донных отложениях) и линейные диапазоны концентраций (n = 5, Р = 0.95) рассчитывали в соответствии с рекомендациями ИЮПАК [44]. Для нафталина, 2‑метилнафталина, аценафтилена, бифенила, аценафтена, флуорена линейный диапазон составил 0.5–50 мкг/кг; для фенантрена, антрацена, флуорантена, пирена, инден[1,2,3-c,d]пирена, бенз[а]антрацена, хризена, дибенз[a,h]антрацена, трифенилена, бенз[g,h,i]перилена, бенз[b]флуорантена, бенз[k]флуорантена, бенз[е]пирена, бенз[а]пирена – 0.2–50 мкг/кг. Концентрации ПАУ по данным хроматографического анализа рассчитывали по методу внешнего стандарта с учетом влажности и потерь при пробоподготовке образцов (мкг/кг сухой массы).
Правильность результатов определения ПАУ по разработанной аналитической схеме проверяли сопоставлением данных, полученных по стандартизированной методике (табл. 4) и методом введено–найдено при анализе донных отложений Темрюкского залива Азовского моря (табл. 5). Видно, что разработанную схему можно применять для ГХ–МС-определения ПАУ в почвах разных типов и донных отложениях.
Таблица 4.
Результаты (мкг/кг) анализа образца донных отложений, отобранного в восточной части Азовского моря (n = 2, P = 0.95)
| ПАУ | [42] | Разработанная методика |
|---|---|---|
| Бенз[а]пирен | 0.8 ± 0.2 | 1.0 ± 0.3 |
| Фенантрен | 0.7 ± 0.2 | 0.9 ± 0.3 |
| Хризен | 1.2 ± 0.3 | 1.0 ± 0.3 |
| Трифенилен | 0.9 ± 0.3 | 0.6 ± 0.2 |
| Бенз[g,h,i]перилен | 0.8 ± 0.3 | 0.7 ± 0.2 |
| Нафталин | Ниже пределов определения аналитов по методике [42] | Ниже пределов определения аналитов по разработанной методике |
| Флуорен | ||
| Флуорантен | ||
| Пирен | ||
| Бенз[b]флуорантен | ||
| Дибенз[a,h]антрацен | ||
| Бенз[k]флуорантен | ||
| Антрацен |
Таблица 5.
Результаты (мкг/кг) определения ПАУ в образцах донных отложений Азовского моря по методу введено–найдено (n = 5, P = 0.95)
| ПАУ | Концентрация в образце донных отложений № 1 |
Концентрация в образце донных отложений № 2 |
||||
|---|---|---|---|---|---|---|
| исходная | введено | найдено | исходная | введено | найдено | |
| Нафталин | <0.5 | 2.0 | 2.1 ± 0.6 | 12 ± 4 | 10 | 25 ± 8 |
| 2-Метилнафталин | <0.5 | 2.0 | 2.3 ± 0.7 | 5.6 ± 1.7 | 10 | 17 ± 5 |
| Аценафтилен | <0.5 | 2.0 | 2.0 ± 0.6 | 2.6 ± 0.8 | 10 | 12 ± 4 |
| Бифенил | <0.5 | 2.0 | 1.9 ± 0.6 | 8.1 ± 2.4 | 10 | 19 ± 6 |
| Аценафтен | <0.5 | 2.0 | 2.5 ± 0.8 | 5.6 ± 1.7 | 10 | 17 ± 5 |
| Флуорен | <0.5 | 2.0 | 2.1 ± 0.6 | 4.7 ± 1.4 | 10 | 14 ± 4 |
| Фенантрен | 0.9 ± 0.3 | 2.0 | 2.7 ± 0.8 | 7.8 ± 2.4 | 10 | 18 ± 5 |
| Антрацен | <0.2 | 2.0 | 1.7 ± 0.5 | 2.8 ± 0.8 | 10 | 14 ± 4 |
| Флуорантен | <0.2 | 2.0 | 1.8 ± 0.5 | 8.4 ± 2.5 | 10 | 18 ± 5 |
| Пирен | <0.2 | 2.0 | 2.1 ± 0.6 | 14 ± 4 | 10 | 22 ± 7 |
| Бенз[а]антрацен | <0.2 | 2.0 | 2.2 ± 0.7 | <0.2 | 10 | 8.8 ± 2.6 |
| Хризен | 1.0 ± 0.3 | 2.0 | 2.7 ± 0.8 | <0.2 | 10 | 11 ± 3 |
| Трифенилен | 0.6 ± 0.2 | 2.0 | 2.4 ± 0.7 | <0.2 | 10 | 10 ± 3 |
| Бенз[b]флуорантен | <0.2 | 2.0 | 2.3 ± 0.7 | <0.2 | 10 | 9.0 ± 3.0 |
| Бенз[k]флуорантен | <0.2 | 2.0 | 1.9 ± 0.6 | <0.2 | 10 | 9.7 ± 2.9 |
| Бенз[е]пирен | <0.2 | 2.0 | 1.9 ± 0.6 | 1.6 ± 0.5 | 10 | 11 ± 3 |
| Бенз[а]пирен | 1.0 ± 0.3 | 2.0 | 3.0 ± 0.9 | <0.2 | 10 | 10 ± 3 |
| Инден[1,2,3-c,d]пирен | <0.2 | 2.0 | 2.1 ± 0.6 | <0.2 | 10 | 9.5 ± 2.9 |
| Дибенз[a,h]антрацен | <0.2 | 2.0 | 2.0 ± 0.6 | 4.2 ± 1.3 | 10 | 15 ± 5 |
| Бенз[g,h,i]перилен | 0.7 ± 0.2 | 2.0 | 2.5 ± 0.8 | 2.8 ± 0.9 | 10 | 12 ± 4 |
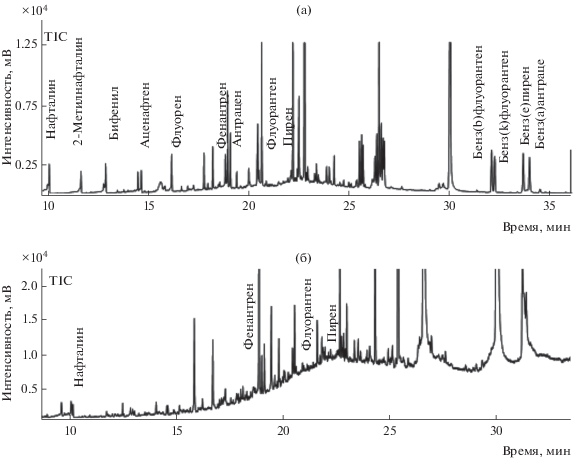
Предложенную схему анализа апробировали при определении ПАУ в образцах песка, отобранных в пойме реки Кубань (Краснодар) и в пустыне Сахара (Тунис) (рис. 6). В образце песка, отобранного в пустыне Сахара (Тунис), обнаружены бифенил, фенантрен, флуорен, флуорантен и пирен в концентрациях, не превышающих 1–2 мкг/кг, что соответствует фоновому уровню содержания ПАУ на территориях, удаленных от индустриальных центров. Факт их наличия можно объяснить возможностью длительной миграции ПАУ с воздушными массами и их адсорбцией на твердых частицах [45, 46]. В образце песка, отобранном в пойме реки Кубань близ г. Краснодара, идентифицированы нафталин, 2-метилнафталин, аценафтилен, бифенил, флуорен, фенантрен, флуорантен, пирен, бенз(а)антрацен, хризен, трифенилен, суммарная концентрация которых превышает 100 мкг/кг, что указывает на антропогенный характер нагрузки.
Рис. 6.
Хроматограммы экстрактов образцов песка, отобранных в пойме р. Кубань (а) и пустыне Сахара (б).
Разработанная схема анализа является экспрессной и позволяет определять широкий спектр ПАУ в почвах и донных отложениях в широком диапазоне их концентраций.
Исследования проводились в рамках выполнения гранта РФФИ (№ 19-43-230003 р_а) с использованием научного оборудования ЦКП “Эколого-аналитический центр” Кубанского госуниверситета.
Список литературы
Kipopoulou A.M., Manoli E., Samara C. Bioconcentration of polycyclic aromatic hydrocarbons in vegetables grown in an industrial area // Environ. Pollut. 1999. V. 106. P. 369.
Cai C., Zhang Y., Reid B.J., Nunes L.M. Carcinogenic potential of soils contaminated with polycyclic aromatic hydrocarbons (PAHs) in Xiamen metropolis, China // J. Environ. Monit. 2012. V. 14. P. 3111.
Viguri J., Verde J., Irabien A. Environmental assessment of polycyclic aromatic hydrocarbons (PAHs) in surface sediments of the Santander Bay, Northern Spain // Chemosphere. 2002. V. 48. P. 157.
Ravelo-Pérez L.M., Hernández-Borges J., Herrera-Herrera A.V., Rodríguez-Delgado M.Á. Pesticide extraction from table grapes and plums using ionic liquid based dispersive liquid–liquid microextraction // Anal. Bioanal. Chem. 2009. V. 395. P. 2387.
Daneshfar A., Khezeli T., Lotfi H.J. Determination of cholesterol in food samples using dispersive liquid–liquid microextraction followed by HPLC–UV // J. Chromatogr. B. 2009. V. 877. P. 456.
ГН 2.1.7.12-1-2004. Перечень предельно допустимых концентраций (ПДК) и ориентировочно допустимых концентраций (ОДК) химических веществ в почве. Гигиенические нормативы. М.: Министерство здравоохранения Республики Беларусь. 2004. 59 с.
ГН 2.1.7.2041-06. Предельно допустимые концентрации (ПДК) химических веществ в почве. Гигиенические нормативы. М.: Федеральная служба по надзору в сфере защиты прав потребителей и благополучия человека. 2006. С. 5.
Poster D.L., Schantz M.M., Sander L.C., Wise S.A. Analysis of polycyclic aromatic hydrocarbons (PAHs) in environmental samples: a critical review of gas chromatographic (GC) methods // Anal. Bioanal. Chem. 2006. V. 386. P. 859.
Gimeno R.A., Altelaar A.F.M., Marcé R.M., Borrull F. Determination of polycyclic aromatic hydrocarbons and polycylic aromatic sulfur heterocycles by high-performance liquid chromatography with fluorescence and atmospheric pressure chemical ionization mass spectrometry detection in seawater and sediment samples // J. Chromatogr. A. 2002. V. 958. P. 141.
Wise S.A., Sander L.C., Chang H.-C. K., Markides K.E., Lee M.L. Shape selectivity in liquid and gas chromatography: Polymeric octadecylsilane (C18) and liquid crystalline stationary phases // Chromatographia. 1988. V. 25. P. 473.
Santos F.J., Galceran M.T. The application of gas chromatography to environmental analysis // Trends Anal. Chem. 2002. V. 21. P. 672.
Baumard P., Budzinski H. Internal standard quantification method and Gas Chromatograph-Mass Spectrometer (GC-MS): A reliable tool for polycyclic aromatic hydrocarbon (PAH) quantification in natural matrices // Analysis. 1997. V. 25. P. 246.
Santos F.J., Galceran M.T., Santos F.J., Galceran M.T Modern developments in gas chromatography–mass spectrometrybased environmental analysis // J. Chromatogr. A. 2003. V. 1000. P. 125.
Hassan J., Izadib M., Homayonnejad S. Application of low density homogeneous liquid-liquid extraction combined with GC for TPH and PAH determination in semi-micro solid samples // J. Braz. Chem. Soc. 2013. V. 24. P. 639.
Shamsipur M., Hassan J. A novel miniaturized homogenous liquid–liquid solvent extraction-high performance liquid chromatographic-fluorescence method for determination of ultra traces of polycyclic aromatic hydrocarbons in sediment samples // J. Chromatogr. A. 2010. V. 1217. P. 4877.
Eriksson M., Eriksson M., Fäldt J., Dalhammar G., Borg-Karlson A.-K. Determination of hydrocarbons in old creosote contaminated soil using headspace solid phase microextraction and GC–MS // Chemosphere. 2001. V. 44. P. 1641.
Rocha M.J., Ferreira P.C., Reis P.A., Cruzeiro C., Rocha E. Determination of polycyclic aromatic hydrocarbons in coastal sediments from the Porto Region (Portugal) by microwave-assisted extraction. Followed by SPME and GC–MS // J. Chromatogr. Sci. 2011. V. 49. P. 695.
Doong R., Chang. S., Sun Y. Solid-phase microextraction and headspace solid-phase microextraction for the determination of high molecular-weight polycyclic aromatic hydrocarbons in water and soil samples // J. Chromatogr. Sci. 2000. V. 38. P. 528.
Zhu L., Synergistic S.F. Solubilization of polycyclic aromatic hydrocarbons by mixed anionic–nonionic surfactants // Chemosphere. 2002. V. 53. P. 459.
Delgado B., Pino V., Ayala J.H., González V., Afonso A.M.M. Nonionic surfactant mixtures: A new cloud-point extraction approach for the determination of PAHs in seawater using HPLC with fluorimetric detection // Anal. Chim. Acta. 2004. V. 518. P. 165.
Zhao Q., Weise L., Li P., Yang K., Zhang Y., Dong D., Li P., Li X. Ageing behavior of phenanthrene and pyrene in soils: A study using sodium dodecylbenzenesulfonate extraction // J. Hazard. Mater. 2010. V. 183. P. 881.
Casero I., Sicilia D., Rubio S., Pe’rez-Bendito D. An acid-induced phase cloud point separation approach using anionic surfactants for the extraction and preconcentration of organic compounds // Anal. Chem. 1999. V 71. P. 4519.
Goryacheva I.Y., Shtykov S.N., Loginov A.S., Panteleeva I.V. Preconcentration and fluorimetric determination of polycyclic aromatic hydrocarbons based on the acidinduced cloud-point extraction with sodium dodecylsulfate // Anal. Bioanal. Chem. 2005. V. 382. P. 1413.
Merino F., Rubio S., Perez-Bendito D. Acid-induced cloud point extraction and preconcentration of polycyclic aromatic hydrocarbons from environmental solid samples // J. Chromatogr. A. 2002. V. 962. P. 1.
Song G., Lu C., Lin J.-M. Application of surfactants and microemulsions to the extraction of pyrene and phenanthrene from soil with three different extraction methods // Anal. Chim. Acta. 2007. V. 596. P. 312.
Толмачева Н.Г., Чжан М., Пирогов А.В., Шпигун О.А. Применение микроэмульсии для извлечения. концентрирования и определения десяти ПАУ из различных типов почв // Журн. аналит. химии. 2017. Т. 72. № 6. С. 515. (Tolmacheva N.G., Zhang M., Pirogov A.V., Popik M.V., Shpigun O.A. Application of microemulsions to the recovery, preconcentration, and determination of ten surfactants from various soils // J. Analyt. Chem. 2017. V. 72. P. 602.)
Pena T., Casais C., Mejuto C., Cela R. Development of an ionic liquid based dispersive liquid–liquid microextraction method for the analysis of polycyclic aromatic hydrocarbons in water samples // J. Chromatogr. A. 2009. V. 1216. P. 6356.
Rezaee M., Assadi Y., Hosseini M.R.M., Aghaee E., Ahmadi F., Berijani S. Determination of organic compounds in water using dispersive liquid–liquid microextraction // J. Chromatogr. A. 2006. V. 1116. P. 1.
Avino P., Notardonato I., Peruginib L., Russo M.V. New protocol based on high-volume sampling followed by DLLME-GC-IT/MS for determining PAHs at ultra-trace levels in surface water samples // Microchem. J. 2017. V. 133. P. 251.
Tseng W.-C., Chen P.-S., Huang S.-D. Optimization of two different dispersive liquid–liquid microextraction methods followed by gas chromatography–mass spectrometry determination for polycyclic aromatic hydrocarbons (PAHs) analysis in water // Talanta. 2014. V. 120. P. 425.
Song X., Zhang S., Li T. A novel method for PAHs in aqueous samples based on ultrasound-assisted solidified floating organic drop microextraction // IOP Conference Series: Earth and Environmental Science. 2018. V. 121. P. 1.
Kamankesha M., Mohammadia A., Hosseinia H., Modarres Z. Rapid determination of polycyclic aromatic hydrocarbons in grilled meat using microwave assisted extraction and dispersive liquid−liquid microextraction coupled to gas chromatography−mass spectrometry // Europ. Food Res. Tech. 2015. V. 240. P. 441.
Gomez-Eyles J.L., Collins C.D., Hodson M.E. Using deuterated PAH amendments to validate chemical extraction methods to predict PAH bioavailability in soils // Environ. Pollut. 2011. V. 159. P. 918.
Zheng J., Liu B., Ping J., Chen B., Wu H., Zhang B. Vortex- and shaker-assisted liquid–liquid microextraction (VSA-LLME) coupled with gas chromatography and mass spectrometry(GC−MS) for analysis of 16 polycyclic aromatic hydrocarbons (PAHs) in offshore produced water // Water Air Soil Pollut. 2015. V. 226. P. 318.
Priego-Lopez E., Luque de Castro M.D. Ultrasound-assisted derivatization of phenolic compounds in spiked water samples before pervaporation. Gas chromatographic separation and flame ionization detection // Chromatographia. 2003. V. 57. P. 513.
Tor A., Aydin M.E., Ozcan S. ultrasonic solvent extraction of organochlorine pesticides from soil // Anal. Chim. Acta. 2006. V. 559. P. 173.
Leng G., Lui G., Chen Y., Yin H., Dan D. Vortex-assisted extraction combined with dispersive liquid–liquid microextraction for the determination of polycyclic aromatic hydrocarbons in sediment by high performance liquid chromatography // J. Sep. Sci. 2012. V. 35. P. 2796.
Morillo E., Romero A.S., Maqueda C., Madrid L., Ajmone-Marsan F., Grcman H. Soil pollution by PAHs in urban soils: a comparison of three European cities // J. Environ. Monit. 2007. V. 9. P. 1001.
Motelay-Massei A., Ollivon D., Garban B., Teil M.J., Blanchard M., Chevreuil M. Distribution and spatial trends of PAHs and PCBs in soils in the Seine River basin, France // Chemosphere. 2004. V. 55. P. 555.
Chen L. Contents and sources of polycyclic aromatic hydrocarbons and organochlorine pesticides in vegetable soils of Guangzhou, China // Chemosphere. 2005. V. 60. P. 879.
Maliszewska-Kordybach B., Smreczak B., Klimkowicz-Pawlas A., Terelak H. Monitoring of the total content of polycyclic aromatic hydrocarbons (PAHs) in arable soils in Poland // Chemosphere. 2008. V. 73. P. 1284.
ФР.1.31.2007.03548. Методика выполнения измерений массовой доли полициклических ароматических углеводородов в пробах почв и донных отложений пресных и морских водных объектов. Ростов-на-Дону: ФГУП “АзНИИРХ”, 2007 . С. 9.
Темердашев З.А., Мусорина Т.Н., Киселева Н.В., Елецкий Б.Д., Червонная Т.А. Хромато-масс-спектрометрическое определение полициклических ароматических углеводородов в поверхностных водах // Журн. аналит. химии. 2018. Т. 73. № 12. С. 897. (Temerdashev Z.A., Musorina T.N., Kiseleva N.V., Eletskii B.D., Chervonnaya T.A. Gas Chromatography–Mass Spectrometry Determination of Polycyclic Aromatic Hydrocarbons in Surface Water // J. Analyt. Chem. 2018. V. 73. №. 12. P. 1154.)
Представление результатов химического анализа (рекомендации IUPAC 1994 г.) // Журнал. аналит. химии. 1998. Т. 53. № 9. С. 999.
Demircioglu E., Sofuoglu A., Odabasi M. Particle-phase dry deposition and air-soil gas exchange of polycyclic aromatic hydrocarbons (PAHs) in Izmir, Turkey // J. Hazard. Mater. 2011. V. 186. P. 328.
Masih A., Masih J., Taneja A. Study of air-soil exchange of polycyclic aromatic hydrocarbons (PAHs) in the North-Central part of India—A Semi Arid region // J. Environ. Monit. 2012. V. 14. P. 172.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии