

Журнал аналитической химии, 2020, T. 75, № 9, стр. 806-819
Быстрый скрининг и определение остаточных количеств β-лактамных антибиотиков в пищевых продуктах методом ультравысокоэффективной жидкостной хроматографии–квадруполь-времяпролетной масс-спектрометрии высокого разрешения
В. Г. Амелин a, b, *, Д. С. Большаков b, И. В. Подколзин b
a Владимирский государственный университет имени Александра Григорьевича и Николая Григорьевича Столетовых
600000 Владимир, ул. Горького, 87, Россия
b Федеральный центр охраны здоровья животных
600901 Владимир, мкрн. Юрьевец, Россия
* E-mail: amelinvg@mail.ru
Поступила в редакцию 17.07.2019
После доработки 23.08.2019
Принята к публикации 02.02.2020
Аннотация
Разработаны методики быстрого скрининга и чувствительного определения остаточных количеств 19 β-лактамных антибиотиков в матрицах различных пищевых продуктов (мясо, почки, печень, шпик, молоко, яйца, мёд) методом ультравысокоэффективной жидкостной хроматографии–квадруполь-времяпролетной масс-спектрометрии высокого разрешения. Осаждение белков и экстрагирование аналитов проводили ацетонитрилом, экстракт использовали для анализа без дополнительной очистки. Идентификацию выполняли по точным массам ионов аналитов, времени удерживания и картине изотопного распределения ионов (mSigma). Установлено, что пенициллины образуют положительно заряженные протонированные аддукты с метанолом, цефалоспорины – протонированные ионы или аддукты с натрием. Оценен матричный эффект при анализе различных проб. Установлено, что матричный эффект незначительно различается для молока, мяса, печени, почек, яиц и шпика (sr ≤ 0.07) и существенно отличается для мёда. Количественное определение обнаруженных аналитов проводили как методом стандартных добавок, так и с использованием матричной градуировки (sr ≤ 0.05). Степень извлечения β-лактамных антибиотиков для разных матриц и двух вариантов пробоподготовки составляла от 75 до 110%, sr ≤ 0.11 в условиях матричной градуировки. Пределы обнаружения составили 0.05–5 нг/г, диапазоны определяемых содержаний аналитов (1–10)–200 нг/г (R2 ≥ 0.99), продолжительность скрининга проб 15–20 мин, продолжительность анализа проб 20–30 мин.
β-Лактамные антибиотики широко используют в ветеринарной практике, они ингибируют процессы синтеза клеточной стенки бактерий, особенно грамм-положительных. В класс β-лактамных антибиотиков включают соединения, имеющие в своей структуре β-лактамное кольцо – пенициллины и цефалоспорины (рис. 1). Нерациональное применение β-лактамов для лечения и профилактики заболеваний приводит к появлению остаточных количеств данных препаратов в молоке, мясе, яйцах, печени и почках сельскохозяйственных животных, а также в мёде. Употребление в пищу таких продуктов негативно сказывается на организме человека. В связи с этим большое внимание уделяется безопасности пищевых продуктов как в отдельных государствах, так и в их союзах. Так, в Европейском (ЕС) и Таможенном Союзах (ТС) для пищевых продуктов установлены максимально допустимые уровни (МДУ) β-лактамных антибиотиков. В ЕС и ТС МДУ β-лактамных антибиотиков в мясе и мясной продукции установлены для каждого отдельного соединения, их значения практически не различаются. Для молочной продукции в ТС регламентируются пенициллины, в ЕС установлены нормы конкретно для каждого антибиотика. В соответствии с регламентами ЕС и ТС присутствие β-лактамных антибиотиков в мёде и яйцах недопустимо (табл. 1).
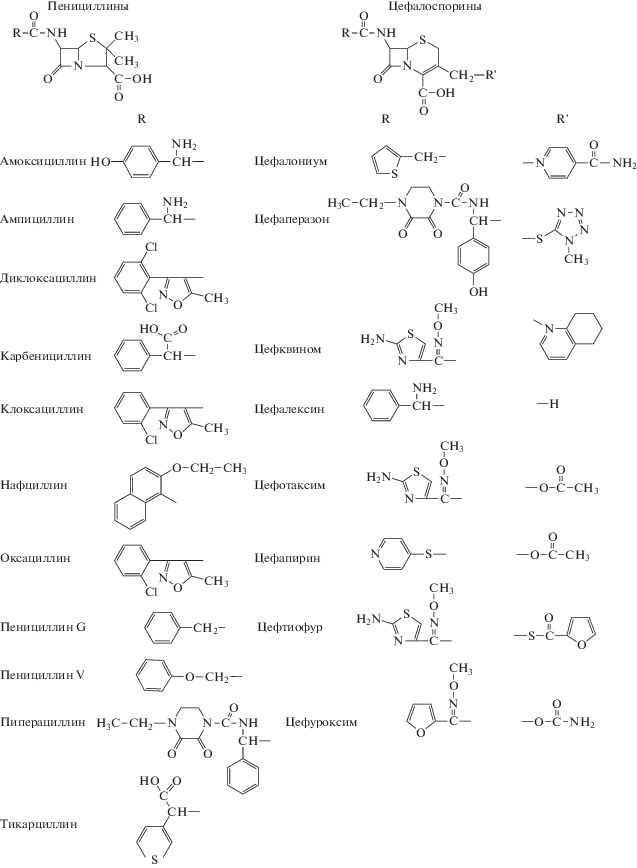
Таблица 1.
Основные характеристики β-лактамных антибиотиков, определяемых методом масс-спектрометрии высокого разрешения, и их максимально допустимые уровни в пищевых продуктах
| Аналит | Брутто-формула | Ион | tR, мин | m/z | Δ, млн–1 |
МДУ (ЕС [26]/ТС [27–29]), нг/г | |||
|---|---|---|---|---|---|---|---|---|---|
| молоко | мясо | печень | почки | ||||||
| Амоксициллин | C16H19N3O5S | [M + CH3OH + H]+ [M + H]+ |
1.8 0.5 |
398.1380 366.1118 |
0.9 | 4/4 | 50/50 | 50/50 | 50/50 |
| Ампициллин | C16H19N3O4S | [M + CH3OH + H]+ [M + H]+ |
3.2 1.9 |
382.1431 350.1169 |
1.2 | 4/4 | 50/50 | 50/50 | 50/50 |
| Диклоксациллин | C19H17Сl2N3O5S | [M + CH3OH + H]+ [M + H]+ |
4.6 4.6 |
502.0601 470.0339 |
1.6 | 30/4 | 300/300 | 300/300 | 300/300 |
| Карбенициллин | C17H18N2O6S | [M + CH3OH + H]+ [M + H]+ |
3.9 3.8 |
411.1220 379.0958 |
1.9 | –*/4 | – | – | – |
| Клоксациллин | C19H18Сl N3O5S | [M + CH3OH + H]+ [M + H]+ |
4.4 4.4 |
468.0990 436.0728 |
2.0 | 30/4 | 300/300 | 300/ 300 | 300/300 |
| Нафциллин | C21H22N2O5S | [M + CH3OH + H]+ [М+Н]+ |
4.4 4.5 |
447.1584 415.1322 |
1.4 | 30/4 | 300/300 | 300/ 300 | 300/ 300 |
| Оксациллин | C19H19N3O5S | [M + CH3OH + H]+ [M + H]+ |
4.3 4.3 |
434.1380 402.1118 |
2.1 | 30/ 4 | 300/300 | 300/ 300 | 300/300 |
| Пенициллин G | C16H18N2O4S | [M + CH3OH + H]+ [M + H]+ |
4.0 4.0 |
367.1322 335.1060 |
0.7 | 4/4 | 50/50 | 50/ 50 | 50/50 |
| Пенициллин V | C16H18N2O5S | [M + CH3OH + H]+ [M + H]+ |
4.1 4.1 |
383.1271 351.1009 |
0.5 | –/4 | 25/250 | 25/250 | 25/250 |
| Пиперациллин | C23H27N5O7S | [M + CH3OH + H]+ [M + H]+ |
4.0 4.0 |
550.1966 518.1707 |
1.3 | –/4 | –/– | –/– | –/– |
| Тикарциллин | C15H16N2O6S2 | [M + CH3OH + H]+ [M + H]+ |
3.8 3.7 |
417.0784 385.0523 |
2.0 | –/4 | –/– | –/– | –/– |
| Цефалексин | C16H17N3O4S | [M + H]+ [M + Na]+ |
1.9 1.9 |
348.1013 370.0832 |
1.8 | 100/– | 200/200 | 200/200 | 1000/1000 |
| Цефалониум | C20H18N4O5S2 | [M + H]+ | 1.6 | 459.0791 | 1.9 | 20/– | –/– | –/– | –/– |
| Цефаперазон | C25H27N9O8S2 | [М + Н]+ [M + Na]+ |
3.6 3.6 |
646.1497 668.1316 |
2.0 | 50/– | –/– | –/– | –/– |
| Цефапирин | C17H17N3O6S2 | [M + H]+ [M + Na]+ |
0.9 0.9 |
424.0632 446.0450 |
2.0 | 60/– | 50/50 | –/– | 100/100 |
| Цефквином | C23H24N6O5S2 | [M + H]+ [M + Na]+ |
2.2 2.2 |
529.1322 551.1142 |
1.7 | 20/– | 50/50 | 100/100 | 200/200 |
| Цефотаксим | C16H17N5O7S2 | [М + Н]+ [M + Na]+ |
3.2 3.2 |
456.0642 478.0463 |
1.9 | –/– | –/– | –/– | –/– |
| Цефтиофур | C19H17N5O7S3 | [М + Н]+ [M + Na]+ |
3.8 3.8 |
524.0362 546.0181 |
1.3 | 100/– | 1000/1000 | 2000/2000 | 6000/6000 |
| Цефуроксим | C16H16N4O8S | [M + NH4]+ [M + Na]+ |
3.4 3.4 |
442.1027 447.0581 |
1.6 | –/– | –/– | –/– | –/– |
Одновременное определение остаточных количеств большинства классов ветеринарных препаратов (в том числе и β-лактамов) в пищевых продуктах в настоящее время, как правило, проводят методом высоко(ультравысоко)эффективной жидкостной хроматографии–тандемной масс-спектрометрии (ВЭЖХ–МС/МС, УВЭЖХ–МС/МС) при ионизации электрораспылением подготовленных экстрактов [1–11]. В обзоре [12] представлены характеристики различных методик определения β-лактамов в объектах окружающей среды, пищевых продуктах, клинических и фармацевтических объектах анализа, опубликованных до 2012 г. Показано, что для чувствительного определения β-лактамов в пищевых продуктах главным образом используют метод ВЭЖХ–МС/МС с экстракцией ацетонитрилом или буферными растворами, очисткой полученного экстракта методом твердофазной экстракции, дальнейшим концентрированием и последующей идентификацией аналитов как по основным, так и по дочерним ионам (m/z). Нами проанализированы литературные данные по определению β-лактамов в пищевых продуктах, опубликованные с 2008 по 2018 гг. Методики включают экстракцию аналитов и одновременное осаждение белков, чаще всего ацетонитрилом. Используют пробоподготовку QuEChERS [3, 13] и более простой способ, включающий экстракцию ацетонитрилом и удаление жиров обработкой гексаном [1, 4, 8, 14]. Для нивелирования матричного эффекта применяют матричную градуировку путем введения стандартов в экстракты или анализируемые продукты, не содержащие β-лактамы. В работах [7, 15, 16] экстракты не очищают: уменьшить матричный эффект (МЭ) позволяет разбавление экстракта деионированной водой. Разделение осуществляют в основном на колонках для УВЭЖХ Acquity BEH C18 (50–100 × 2.1 мм, 1.7 мкм). Применение УВЭЖХ позволило снизить продолжительность хроматографического разделения до 5–15 мин по сравнению с ВЭЖХ, где разделение проходит за 20–30 мин [12]. Используют режим градиентного элюирования, чаще всего 0.1%-ным водным раствором муравьиной кислоты (А) и 0.1%-ным ацетонитрильным раствором муравьиной кислоты (Б). Пределы обнаружения составили 0.1–10 нг/г для различных матриц и конкретных соединений. Использование масс-спектрометрии высокого разрешения [17–19] до 15 000 FWHM (времяпролетная [15, 20–22]), до 50 000 FWHM (квадруполь-времяпролетная [13, 14]) и до 10 0000 FWHM (орбитальная ионная ловушка [16, 18]) позволяет определять β-лактамные антибиотики в различных матрицах по точным массам ионов, образующихся в условиях электрораспылительной ионизации экстракта. Показано, что применение масс-спектрометрии высокого разрешения в большинстве случаях позволяет устранить стадию очистки экстракта, что значительно ускоряет и упрощает анализ [13, 14, 23].
Во всех упомянутых выше работах наиболее часто измеряют массы протонированных или депротонированных форм β-лактамов при их определении (или аддуктов пенициллинов с натрием [7]). Однако установлены и другие аддукты β-лактамных антибиотиков для их идентификации и последующего определения. Так, в работе [24] описаны протонированные аддукты амоксициллина с метанолом для его чувствительного определения. Показано, что образование и стабильность аддуктов с метанолом зависит от состава подвижной фазы (метанол–вода–10%-ная уксусная кислота (60 : 39 : 1, по объему) и добавки метанола в стандартный раствор амоксициллина. Описано [25] применение протонированных аддуктов амоксициллина, ампициллина и цефалексина при анализе природных вод. Протонированные аддукты пенициллинов с метанолом указаны также в работе [23] для клоксациллина, диклоксациллина, оксациллина и пенициллина V. Однако не изучено примененние данных аддуктов в условиях анализа реальных проб.
В данной статье предложен быстрый скрининг и одновременное чувствительное определение остаточных количеств 19 β-лактамных антибиотиков в мясе (говядина, свинина, птица), почках, печени, шпике, молоке, яйцах и мёде методом УВЭЖХ–квадруполь-времяпролетной масс-спектрометрии высокого разрешения по точным массам протонированных аддуктов пенициллинов с метанолом, протонированных форм цефалоспоринов или их аддуктов с ионами натрия.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Аппаратура. Использовали ультравысокоэффективный жидкостной хроматограф UltiMate 3000 (Thermo Scientific, США) в сочетании с квадруполь-времяпролетным масс-спектрометрическим детектором maXis 4G и ионизацию электрораспылением в устройстве ionBooster (Bruker Daltonics, Германия). Разделение проводили на колонках ACQUITY UPLC® BEN C18 (30 × 2.1 мм, 50 × 2.1 мм, 100 × 2.1 мм), ACQUITY UPLC® Shield RP18 (50 × 2.1 мм), ACQUITY UPLC® Phenyl (50 × 2.1 мм) c диаметром зерна сорбента 1.7 мкм (Waters, США), Acclaim™ 120 C18 (50 × × 2.1 мм) c диаметром зерна сорбента 2.2 и 3.0 мкм (Thermo Scientific, США) в режиме градиентного элюирования.
Реактивы. Использовали стандартные образцы пенициллинов и цефалоспоринов (Dr. Ehrenstor-fer, Fluka, Sigma-Aldrich) с содержанием основного вещества не менее 99.1%. Исходные растворы с концентрацией 1 мг/мл готовили растворением соответствующей навески (с учетом содержания основного вещества) в метаноле. Хранили растворы при –20°С не более шести месяцев. Рабочие растворы готовили разбавлением исходных метанолом в день использования.
Использовали ацетонитрил (99.9%, Scharlab S.L., Испания), изопропиловый спирт (для ВЭЖХ, Scharlab S.L., Испания), метанол (PA-ACS-ISO, Panreac, ЕС), янтарную кислоту (ч., МСД КЕМИКАЛ, Россия), ЭДТА (этилендиаминтетраацетат натрия) (99%, ХИММЕД, Россия), сульфат аммония (х. ч., ЗАО “ХИМРЕАКТИВ”, Россия), гексан (96%, Scharlab S.L., Испания), хлорид натрия (х. ч., ХИММЕД, Россия).
Пробы. Анализируемые пробы приобретали в местных супермаркетах, получали с птицефабрик и животноводческих ферм. Мясо, печень, почки, шпик хранили при –20°С в холодильнике в течение месяца. Перед анализом замороженные продукты измельчали с использованием бытового миксера; молоко, мёд, яйца хранили при 4°С в течение месяца. Перед анализом яйца освобождали от скорлупы, объединяли 6 штук в одну пробу и тщательно перемешивали с использованием бытового миксера.
Пробоподготовка. Использовали два варианта пробоподготовки:
Вариант I. В центрифужную пробирку емк. 15 мл вносили 1.00 г тщательно измельченной пробы (навеску мёда массой 1.00 г растворяли в 1 мл воды), добавляли 2.0 мл ацетонитрила, 0.5 г NaCl, 40.0 мг ЭДТА и перемешивали в течение 5 мин, затем центрифугировали в течение 5 мин при 2700 об/мин. Отбирали верхний ацетонитрильный слой во флакон и упаривали досуха при 40°С в токе азота с использованием устройства RapidVap (LABCONCO, США). К сухому остатку добавляли 50 мкл метанола и 950 мкл воды, перемешивали 5 мин и фильтровали через мембранный фильтр (0.20 мкм, GHP ACRODISC 13, PALL, США) в микрофлакон для хроматографирования.
Вариант II. В центрифужную пробирку емк. 15 мл вносили навеску тщательно измельченной пробы массой 1.00 г (навеску мёда массой 1.00 г растворяли в 1 мл воды), добавляли 12.0 мг янтарной кислоты, 40.0 мг ЭДТА, 2.0 мл воды, перемешивали вручную в течение 5 мин. Затем добавляли 2.0 мл ацетонитрила, 2.0 г сульфата аммония, перемешивали 5 мин и центрифугировали в течение 5 мин при 2700 об/мин. Отбирали верхний ацетонитрильный слой во флакон и упаривали досуха при 40°С в токе азота с использованием устройства RapidVap (LABCONCO, США). К сухому остатку добавляли 50 мкл метанола и 950 мкл воды, перемешивали 5 мин и фильтровали через мембранный фильтр (0.20 мкм, GHP ACRODISC 13, PALL, США) в микрофлакон для хроматографирования.
Идентификация и определение. β-Лактамные антибиотики идентифицировали по полученным хроматограммам с использованием программных продуктов DataAnalysis-4.1, TargetAnalysis (Bruker Daltonics, Германия). Для составления картины изотопного распределения аналитов использовали программу IsotopePattern (Bruker Daltonics, Германия). Неизвестную концентрацию аналита в пробе определяли методом стандартной добавки [14] и градуировочного графика (матричная градуировка).
Матричный эффект оценивали по формуле:
где C, D – значения наклонов градуировочных графиков, полученные для экстрактов из матрицы и деионированной воды соответственно в диапазоне 0.1–200 нг/мл (нг/г).Условия хроматографического разделения и детектирования. Подвижная фаза состояла из 0.1%-ного раствора муравьиной кислоты в воде (А) и 0.1%-ного раствора муравьиной кислоты в ацетонитриле (Б). Осуществляли градиентное элюирование в соответствии со схемой: 0 мин – 5% Б, 0.5 мин – 5% Б, 2 мин – 50% Б, 5 мин – 100% Б, 6 мин – 5% Б, 10 мин – 5% Б. Расход подвижной фазы 0.4 мл/мин. Оптимальная температура хроматографической колонки 50°С, объем вводимой пробы 50 мкл. Температура термостата автоматического дозатора 10°С.
Установлены следующие оптимальные значения параметров ионизации электрораспылением: напряжение на щите капилляра 400 В, на капилляре 1000 В, давление газа-распылителя азота 4.76 атм., поток газа-осушителя азота 6 л/мин, температура газа-осушителя азота 200°С, поток газа-испарителя азота 250 л/ч, температура газа-испарителя азота 250°C.
Диапазон регистрируемых масс ионов составил от 200 до 600 Да. Для калибровки использовали 10 мМ раствор формиата натрия в смеси вода–изопропанол (1 : 1, по объему) в интервале хроматографирования 9.5–10 мин.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Оптимизация условий анализа. Следует отметить, что использование любых из перечисленных в “Экспериментальной части” колонок не приводит к существенному изменению основных аналитических характеристик методики. Однако эффективность колонки ACQUITY UPLC® BEN C18 (30 × 2.1 мм, 1.7 мкм) оказалась несколько выше (число теоретических тарелок N = 5843 для пенициллина G), чем других (N = 3329–4997), селективность для всех изученных колонок была одинаковой. Для дальнейших исследований использовали колонку ACQUITY UPLC® BEN C18 (30 × 2.1 мм).
При выборе подвижной фазы использовали воду, ацетонитрил и метанол без добавок и с добавками муравьиной кислоты и формиата аммония. В итоге установлен оптимальный вариант: А – 0.1%-ный водный раствор муравьиной кислоты, Б – 0.1%-ный ацетонитрильный раствор муравьиной кислоты. Добавки формиата аммония и использование метанола в качестве подвижной фазы не привели к существенному увеличению хроматографических пиков исследуемых аналитов. Лучшие результаты получили при расходе подвижной фазы 0.4 мл/мин и следующем градиенте: 0 мин – 5% Б, 0.5 мин – 5% Б, 2 мин – 50% Б, 5 мин – 100% Б, 6 мин – 5% Б, 10 мин – 5% Б.
Температуру термостата хроматографической колонки выбирали из 30, 40, 50 и 60°С, вводимый объем пробы – из 5, 10, 20, 50, 60 и 100 мкл. Установлено, что для достижения высокой чувствительности определения следует использовать температуру хроматографической колонки 50°С (при этом давление подвижной фазы в колонке составило 160–170 бар) и оптимальный объем вводимой в дозатор пробы – 50 мкл (при вводе более 50 мкл нарушается симметричность хроматографического пика).
Использование УВЭЖХ привело к значительному сокращению времени хроматографирования по сравнению с ранее предложенным [14]. В данной работе продолжительность разделения β‑лактамных антибиотиков на колонке длиной 30 мм составила 5 мин, на колонках длиной 50 мм – 7–9 мин. Общая продолжительность анализа составила 10–12 мин с учетом кондиционирования колонки.
Условия образования аддуктов. Ранее мы рассматривали определение пенициллинов в пищевых продуктах с использованием стандартных растворов, приготовленных на ацетонитриле и воде [14]. При ионизации электрораспылением при этом проявлялись либо протонированные формы пенициллинов (амоксициллин, ампициллин, карбенициллин, нафциллин, пенициллин G, тикарциллин), либо депронированные (диклоксациллин, клоксациллин, нафциллин, оксациллин, пенициллин V, пиперациллин). Чувствительность определения пеницилинов была недостаточна для определения, например, амоксициллина, ампициллина, пеницилина G и V, для которых установлены низкие значения МДУ (табл. 1), и составляла 1–10 нг/г. Кроме того, необходимо было дважды получать хроматограммы для положительных и отрицательных ионов, что приводило к увеличению продолжительности анализа. Установлено, что приготовление стандартных растворов аналитов на метаноле и добавление метанола в экстракт пробы приводит к появлению положительно заряженных протонированных аддуктов пенициллинов с метанолом [M + CH3OH + H]+, использование которых в анализе позволяет значительно увеличить чувствительность определения аналитов (табл. 1, 2). На рис. 2 представлены масс-хроматограммы экстракта молока с добавлением 50 нг/г β-лактамов. Установлено, что введение 50 мкл пробы при температуре термостата колонки 50°С приводит к получению интенсивных и симметричных хроматографических пиков.
Таблица 2.
Аналитические характеристики определения β-лактамных антибиотиков в различных матрицах при разных условиях пробоподготовки (варианты I и II) (n = 3, P = 0.95)
| Аналит | Деионированная вода | Молоко | Мясо, свинина, печень, почки, яйца | Мёд | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| уравнение градуировочного графика в диапазоне (0.1–1)–100 нг/мл |
R2 | смин, нг/мл | вариант пробо-подгото-вки | уравнение градуировочного графика в диапазоне (1–10)–200 нг/г |
R2 | МЭ, % (sr) | смин, нг/г | сн, нг/г | МЭ, % | вариант пробо-подгото-вки | уравнение градуировочного графика в диапазоне (1–10)–200 нг/г |
R2 | МЭ, % (sr) | смин, нг/г | сн, нг/г | |
| Амоксициллин | y = 140 449х + 4527 | 1.000 | 0.05 | I II |
y = 4917х + 5727 y = 10 435х + 31 222 |
1.000 0.9999 |
96 (0.03) 90 (0.01) |
1 0.8 |
3 2 |
93 ± 4 90 ± 1 |
I II |
y = 21 344х + 82 807 y = 5253х + 65 562 |
0.9976 0.9976 |
84 (0.04) 96 (0.03) |
0.8 1 |
2 4 |
| Ампициллин | y = 609 149x + 44341 | 1.000 | 0.03 | I II |
y = 22 807х + 58 923 y = 48 588х + 156 680 |
0.9988 0.9948 |
96 (0.03) 90 (0.01) |
0.07 0.05 |
0.2 0.1 |
93 ± 3 90 ± 1 |
I II |
y = 66 086х + 35 479 y = 39 735х + 228 445 |
0.9999 0.9963 |
89 (0.05) 93 (0.02) |
0.08 0.1 |
0.2 0.3 |
| Диклоксациллин | y =307 718x + 34856 | 0.9995 | 0.03 | I II |
y = 88 381х + 166 081 y = 59 142х + 273 633 |
0.9993 0.9908 |
71 (0.03) 78 (0.02) |
0.05 0.05 |
0.1 0.1 |
73 ± 4 77 ± 3 |
I II |
y = 66 574х + 80 810 y = 64 512х + 382 646 |
0.9981 0.9987 |
78 (0.03) 79 (0.03) |
0.06 0.05 |
0.3 0.2 |
| Карбенициллин | y =36 196x + 228 676 | 0.9944 | 0.5 | I II |
y = 3236х + 4604 y = 28 142х + 88 905 |
0.9999 0.9910 |
91 (0.04) 0.1 (0.04) |
1 0.5 |
4 2 |
89 ± 6 0.1 ± 0.2 |
I II |
y = 28 877х + 106 120 y = 32 393х + 579 706 |
0.9988 0.9908 |
20 (0.04) 10 (0.04) |
2 0.6 |
6 2 |
| Клоксациллин | y = 403 158x + 62 851 | 0.9998 | 0.03 | I II |
y = 88 128х + 391 026 y = 74 777х + 346 535 |
0.9994 0.9909 |
78 (0.05) 81 (0.03) |
0.05 0.05 |
0.1 0.1 |
76 ± 4 79 ± 7 |
I II |
y = 88 753х + 244 502 y = 59 105х + 551 346 |
0.9955 0.9903 |
77 (0.05) 85 (0.03) |
0.05 0.06 |
0.2 0.3 |
| Нафциллин | y = 502 103x + 61 398 | 0.9994 | 0.03 | I II |
y = 90 333х + 353 296 y = 67 331х + 367 508 |
0.9987 0.9908 |
82 (0.04) 85 (0.03) |
0.05 0.05 |
0.1 0.1 |
83 ± 4 83 ± 4 |
I II |
y = 70 306х + 156 330 y = 57 034х + 518 302 |
0.9976 0.9912 |
85 (0.02) 88 (0.03) |
0.1 0.1 |
0.5 0.5 |
| Оксациллин | y = 597 087x + 60 989 | 0.9997 | 0.03 | I II |
y = 142 648х + 132 150 y = 85 993х + 220 141 |
0.9999 0.9963 |
76 (0.04) 80 (0.02) |
0.04 0.05 |
0.1 0.1 |
75 ± 3 81 ± 5 |
I II |
y = 147 214х – 22 060 y = 93 504х + 647 584 |
0.9991 0.9901 |
75 (0.02) 84 (0.03) |
0.05 0.05 |
0.2 0.2 |
| Пенициллин G | y = 904 189x + 112 509 | 0.9999 | 0.02 | I II |
y = 227 705х – 38 650 y = 249 339х + 12 418 |
0.9936 0.9971 |
74 (0.03) 69 (0.04) |
0.04 0.03 |
0.1 0.1 |
73 ± 3 67 ± 6 |
I II |
y = 246 703х – 25 727 y = 238 481х + 749 164 |
0.9989 0.9993 |
72 (0.03) 74 (0.04) |
0.05 0.05 |
0.2 0.2 |
| Пенициллин V | y = 688 073x + 115 313 | 0.9994 | 0.02 | I II |
y = 222 819х – 35 038 y = 254 658х – 139 677 |
0.9989 0.9998 |
67 (0.04) 60 (0.03) |
0.04 0.03 |
0.1 0.1 |
66 ± 4 58 ± 7 |
I II |
y = 238 655х – 100 000 y = 239 208х – 60 714 |
0.9901 0.9992 |
65 (0.05) 65 (0.05) |
0.04 0.05 |
0.1 0.1 |
| Пиперациллин | y = 224 264x + 23 836 | 0.9999 | 0.03 | I II |
y = 44 291х + 242 216 y = 38 486х + 315 037 |
0.9989 0.9903 |
80 (0.03) 80 (0.02) |
0.05 0.05 |
0.1 0.1 |
82 ± 4 80 ± 3 |
I II |
y = 35 134х + 248 446 y = 41 889х + 434 709 |
0.9907 0.9908 |
85 (0.05) 81 (0.04) |
0.1 0.1 |
0.4 0.4 |
| Тикарциллин | y = 140 097x + 11 876 | 0.9994 | 0.04 | I II |
y = 42 617х + 13 565 y = 58 209х + 220 794 |
0.9987 0.9908 |
69 (0.04) 50 (0.03) |
0.08 0.05 |
0.2 0.1 |
70 ± 4 50 ± 7 |
I II |
y = 6978х + 18 207 y = 63 077х + 290 906 |
0.9945 0.9914 |
95 (0.05) 54 (0.03) |
0.2 0.08 |
0.7 0.2 |
| Цефалексин | y = 179 897x + 13 837 | 1.000 | 0.04 | I II |
y = 5267х – 4567 y = 3307х + 31 774 |
0.9987 0.9994 |
97 (0.03) 97 (0.02) |
5 5 |
12 12 |
96 ± 3 96 ± 6 |
I II |
y = 8408х + 143 298 y = 4062х – 1862 |
0.9967 0.9993 |
95 (0.05) 97 (0.02) |
5 5 |
10 10 |
| Цефалониум | y = 18 704х + 1105 | 1.000 | 0.1 | I II |
y = 1020х + 5655 y = 2285х + 21 531 |
0.9980 0.9950 |
94 (0.05) 92 (0.02) |
1 1 |
4 4 |
93 ± 4 93 ± 4 |
I II |
y = 203х + 61 352 y = 1471х + 29 802 |
0.9987 0.9985 |
98 (0.02) 92 (0.04) |
2 1 |
7 4 |
| Цефаперазон | y = 45 003х + 4897 | 0.9999 | 0.1 | I II |
y = 4048х + 1314 y = 12 244х + 44 241 |
0.9997 0.9955 |
91 (0.04) 72 (0.03) |
0.5 0.3 |
2 1 |
92 ± 4 72 ± 6 |
I II |
y = 3694х + 7024 y = 5565х + 47 643 |
0.9955 0.9947 |
91 (0.03) 87 (0.05) |
0.5 0.5 |
2 3 |
| Цефапирин | y = 228 884х + 11 992 | 0.9999 | 0.05 | I II |
y = 4835х + 21 000 y = 33 574х + 132 651 |
0.9983 0.9916 |
97 (0.04) 83 (0.03) |
0.5 0.3 |
2 1 |
95 ± 4 85 ± 6 |
I II |
y = 3523х + 16 602 y = 20 303х + 41 796 |
0.9993 0.9998 |
98 (0.05) 91 (0.04) |
0.5 0.5 |
3 4 |
| Цефквином | y = 53 521х + 3334 | 0.9997 | 0.1 | I II |
y = 4506х – 2832 y = 8899х + 7067 |
0.9990 0.9990 |
91 (0.03) 87 (0.03) |
2 1 |
6 4 |
93 ± 4 89 ± 5 |
I II |
y = 988х – 102 y = 11 136х + 91 898 |
0.9938 0.9980 |
99 (0.04) 79 (0.05) |
5 1 |
15 3 |
| Цефотаксим | y = 300 175х + 15 837 | 0.9996 | 0.04 | I II |
y = 16 870х + 77 496 y = 112 313х + 316 375 |
0.9996 0.9966 |
94 (0.05) 59 (0.05) |
0.5 0.08 |
2 1 |
93 ± 5 60 ± 2 |
I II |
y = 69 281х + 124 149 y = 59 823х + 113 789 |
0.9972 0.9989 |
77 (0.04) 80 (0.02) |
0.2 0.2 |
0.7 0.7 |
| Цефтиофур | y = 232 684х + 18 668 | 0.9999 | 0.04 | I II |
y = 38 367х + 198 896 y = 54 899х + 297 674 |
0.9997 0.9900 |
83 (0.05) 76 (0.04) |
0.1 0.08 |
0.5 0.3 |
83 ± 4 75 ± 4 |
I II |
y = 38 183х + 57 247 y = 43 695х + 220 914 |
0.9965 0.9947 |
84 (0.03) 81 (0.03) |
0.1 0.1 |
0.4 0.3 |
| Цефуроксим | y = 34 427х + 7495 | 0.9999 | 0.1 | I II |
y = 2727х + 55 670 y = 10 671х + 68 227 |
0.9925 0.9975 |
92 (0.05) 66 (0.04) |
1 0.5 |
4 2 |
93 ± 4 66 ± 6 |
I II |
y = 6189х + 39 340 y = 6426х + 45 818 |
0.9958 0.9948 |
82 (0.05) 81 (0.02) |
0.5 1 |
3 4 |
Рис. 2.
Масс-хроматограммы экстракта молока с добавлением 50 нг/г 19 β-лактамных антибиотиков: 1 – цефапирин, 2 – цефалониум, 3 – амоксициллин, 4 – цефалексин, 5 – цефквином, 6 – цефотаксим, 7 – ампициллин, 8 – цефуроксим, 9 – цефаперазон, 10 – тикарциллин, 11 – цефтиофур, 12 – карбенициллин, 13 – пенициллин G, 14 – пиперациллин, 15 – пенициллин V, 16 – оксациллин, 17 – нафциллин (и протонированная форма), 18 – клоксациллин, 19 – диклоксациллин.

Пенициллины способны образовывать в данных условия следующие аддукты: [M + CH3OH + H]+, [M + H]+, [M + Na]+, [M + NН4]+. На рис. 3 показаны масс-хроматограммы данных аддуктов для пиперациллина и нафциллина. Видно, что в предлагаемых условиях пробоподготовки (добавление метанола после упаривания экстракта) образование других аддуктов, отличных от протонированных с метанолом, в значительной степени подавлено. Конкурировать с протонированными аддуктами с метанолом могут только протонированные формы амоксициллина, нафциллина и пиперациллина (рис. 4). Однако в условиях пробоподготовки и анализа конкретных проб проявляется преимущественно протонированный метанольный аддукт нафциллина и в равной степени протонированная форма нафциллина, протонированные формы амоксициллина и пиперациллина значительно подавляются. Установлено, что времена удерживания для некоторых аддуктов различаются (рис. 5, табл. 1). Так, выход из колонки аддукта [M + H]+ по сравнению с [M + CH3OH + H]+ для амоксициллина и ампициллина запаздывает на –1.3 мин, для нафциллина опережает на +0.1 мин и запаздывает для карбенициллина и тикарцеллина (–0.1 мин) (рис. 5). Данный эффект, вероятно, свидетельствует об образовании аддуктов вне хроматографической колонки и тем более вне распылительного устройства, а, скорее всего, в процессе приготовления стандартных растворов и проб к анализу.
Рис. 3.
Масс-хроматограммы аддуктов пиперациллина (1 – [M + Na]+, 2 – [M + NH4]+, 3 – [M + H]+, 4 – [M + CH3OH + H]+) и нафциллина (5 – [M + NH4]+, 6 – [M + H]+, 7 – [M + CH3OH + H]+).
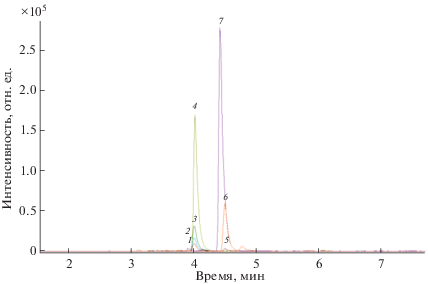
Рис. 4.
Соотношение аддуктов β-лактамов для стандартных растворов: 1 – амоксициллин, 2 – нафциллин, 3 – ампициллин, 4 – диклоксациллин, 5 – карбенициллин, 6 – клоксациллин, 7 – оксациллин, 8 – пенициллин G, 9 – пенициллин V, 10 – пиперациллин, 11 – тикарциллин, 12 – цефалексин, 13 – цефалониум, 14 – цефаперазон, 15 – цефапирин, 16 – цефквином, 17 – цефотаксим, 18 – цефтиофур, 19 – цефуроксим.
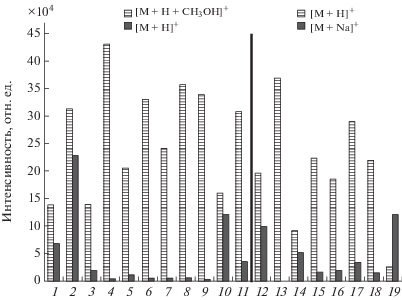
Рис. 5.
Масс-хроматограммы: 1 – амоксициллин, 2 – ампициллин, 3 – нафциллин, 4 – карбенициллин, 5 – тикарциллин; А – [M + CH3OH + H]+, В – [M + H]+.

Все цефалоспорины образуют протонированные формы и в незначительной степени аддукты с натрием (рис. 4). Отличаются цефуроксим, цефалексин и цефаперазон, образующие аддукты как с натрием, так и с ионами водорода (однако в условиях пробоподготовки аддукты с натрием подавляются). Цефуроксим образует аддукты как с аммонием, так и с натрием в равной степени, но в условиях пробоподготовки (добавление натриевых солей) преобладает аддукт [M + Na]+ (табл. 1, рис. 4).
Оптимизация пробоподготовки. С учетом проведенных нами ранее исследований [14] для пробоподготовки использовали экстракцию ацетонитрилом в нейтральной и кислой средах (добавление янтарной кислоты). В первом и во втором случаях добавляли ЭДТА для связывания ионов металлов с целью предотвращения образования комплексов с аналитами. Варьирование различных параметров (массы навески пробы, количества высаливателя и экстрагента, природы высаливателя, количества ЭДТА), а также учет матричного эффекта привели к оптимальным условиям пробоподготовки (вариантам I и II) для различных матриц. Кроме того, добавление метанола в экстракт после его упаривания способствовало образованию протонированных аддуктов пенициллинов с метанолом.
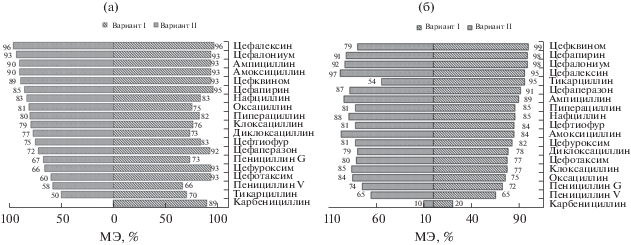
Матричный эффект обусловлен влиянием на ионизацию целевых аналитов присутствующих в экстракте соэкстрагируемых компонентов. В условиях ионизации электрораспылением они могут как усиливать (+), так и понижать (–) интенсивность сигнала аналита. В табл. 3 представлены значения МЭ при исследовании различных матриц. Во всех случаях наблюдали понижение интенсивности сигнала аналита (–), и значительный МЭ: от –50 до –99%. Матричный эффект отсутствовал лишь для карбенициллина в условиях пробоподготовки по варианту II. Установлено, что МЭ для проб молока, мяса, печени, почек, шпика, яиц различается незначительно (sr ≤ 0.07, табл. 3), в отличие от проб мёда. На рис. 6 показано влияние условий пробоподготовки на МЭ для различных объектов анализа. Видно, что для молока, мяса, печени, почек, яиц и шпика предпочтительна пробоподготовка по варианту II, а для мёда – пробоподготовка по варианту I, так как в этих условиях большее число аналитов имеет МЭ < 80%. Для нивелирования МЭ, как правило, используют матричную градуировку [1, 7, 8, 13] и метод стандартной добавки [14]. Эти приемы использовали и в данной работе.
Таблица 3.
Степени извлечения (%) β-лактамных антибиотиков на разных уровнях концентраций при разных условиях пробоподготовки (варианты I и II) (n = 3, P = 0.95)
| Аналит | Молоко, мясо, свинина, печень, почки и яйца (вариант пробоподготовки I) |
Мёд (вариант пробоподготовки II) |
||||
|---|---|---|---|---|---|---|
| 1 нг/г | 10 нг/г | 50 нг/г | 1 нг/г | 10 нг/г | 50 нг/г | |
| Амоксициллин | –* | 92–98 (0.07–0.06) | 99–102 (0.04–0.03) | – | 88 (0.06) | 92 (0.03) |
| Ампициллин | 89–92 (0.05–0.04)** | 88–97 (0.06–0.05) | 95–97 (0.06–0.03) | 85 (0.05) | 98 (0.09) | 102 (0.06) |
| Диклоксациллин | 95–97 (0.05–0.03) | 98–99 (0.07–0.04) | 92–98 (0.07–0.04) | 95 (0.07) | 103 (0.07) | 106 (0.09) |
| Карбенициллин | – | 88–110 (0.10–0.04) | 92–106 (0.07–0.03) | – | 99 (0.06) | 110 (0.03) |
| Клоксациллин | 85–95 (0.07–0.05) | 95–105 (0.07–0.06) | 94–97 (0.06–0.05) | 97 (0.08) | 103 (0.04) | 94 (0.07) |
| Нафциллин | 95–99 (0.08–0.06) | 99–99 (0.08–0.07) | 92–105 (0.07–0.06) | 75 (0.05) | 106 (0.07) | 104 (0.07) |
| Оксациллин | 98–99 (0.06–0.05) | 105–106 (0.07–0.05) | 95–108 (0.09–0.03) | 95 (0.04) | 110 (0.08) | 96 (0.06) |
| Пенициллин G | 95–103 (0.08–0.04) | 99–110 (0.06–0.05) | 98–109 (0.06–0.04) | 105 (0.06) | 99 (0.03) | 99 (0.04) |
| Пенициллин V | 85–103 (0.09–0.05) | 93–98 (0.07–0.03) | 98–105 (0.07–0.03) | 98 (0.06) | 89 (0.05) | 92 (0.05) |
| Пиперациллин | 93–96 (0.05–0.04) | 95–104 (0.09–0.04) | 96–100 (0.07–0.07) | 93 (0.05) | 97 (0.09) | 103 (0.08) |
| Тикарциллин | 85–90 (0.10–0.05) | 98–103 (0.07–0.06) | 95–105 (0.08–0.07) | 94 (0.07) | 87 (0.06) | 110 (0.05) |
| Цефалексин | – | – | 92–105 (0.06–0.05) | – | 88 (0.06) | 90 (0.06) |
| Цефалониум | – | 88–89 (0.06–0.06) | 92–99 (0.08–0.03) | – | 85 (0.07) | 91 (0.08) |
| Цефаперазон | 86–89 (0.10–0.09) | 80–90 (0.07–0.06) | 90–93 (0.05–0.05) | – | 80 (0.06) | 82 (0.07) |
| Цефапирин | 75–89 (0.08–0.07) | 82–86 (0.09–0.06) | 92–93 (0.07–0.06) | – | 82 (0.06) | 89 (0.04) |
| Цефквином | – | 83–88 (0.10–0.06) | 96–97 (0.08–0.03) | – | 98 (0.06) | 96 (0.05) |
| Цефотаксим | 80–90 (0.09–0.05) | 91–97 (0.06–0.05) | 92–95 (0.06–0.05) | 80 (0.08) | 90 (0.05) | 91 (0.08) |
| Цефтиофур | 82–96 (0.08–0.07) | 78–89 (0.10–0.07) | 90–100 (0.08–0.08) | 79 (0.09) | 101 (0.07) | 103 (0.07) |
| Цефуроксим | – | 87–89 (0.08–0.07) | 91–110 (0.09–0.08) | – | 89 (0.06) | 90 (0.06) |
Рис. 6.
Матричный эффект при различных вариантах пробоподготовки для молока, мяса, печени, почек, шпика, яиц (а) и мёда (б).
Валидация. Линейность градуировочных графиков оценивали на уровнях концентраций β-лактамных антибиотиков 0.1, 0.5, 1.0, 5.0, 10, 50, 100 и 200 нг/г для образцов молока и мёда, не содержащих остаточных количеств определяемых аналитов. Коэффициенты линейной зависимости площадей хроматографических пиков β-лактамов от их концентрации в анализируемом образце составили не менее 0.99 (табл. 2).
Пределы обнаружения (смин) и определения (сн) рассчитывали по отношению аналитического сигнала (интенсивности пика) к шуму, равному 3 и 10 соответственно. Пределы обнаружения и определения составили 0.05–5 и 1–10 нг/г, соответственно (табл. 2). Диапазоны определяемых содержаний β-лактамов составили (1–10)–200 нг/г.
Средние степени извлечения определяемых антибиотиков и относительные стандартные отклонения оценивали для трех уровней концентраций 1.0, 10 и 50 нг/г (табл. 3). Как видно, степень извлечения аналитов для разных матриц и различной пробоподготовки составляет от 75 до 110% с sr ≤ 0.10 в условиях матричной градуировки. Степени извлечения β-лактамов для мяса, печени, почек, шпика и яиц определяли с использованием матричной градуировки для молока, поэтому в табл. 3 указаны минимальные и максимальные значения.
Идентификацию β-лактамных антибиотиков проводили по полученным масс-хроматограммам с использованием программного продукта Target-Analysis-1.3. Идентификационными параметрами служили времена удерживания (±0.1 мин), точность массы моноизотопа m/z (±5 млн–1) и совпадение картины изотопного распределения (mSi-gma < 20). Погрешность в определении масс ионов не превышала 2 млн–1 (табл. 1).
Определение β-лактамов. Количественный анализ проводили как методом добавок, так и с использованием матричной градуировки для молока и мёда. Проанализировали более 100 различных проб на содержание остаточных количеств β-лактамных антибиотиков. Результаты определения идентифицированных в пробах β-лактамов приведены в табл. 4, из которой следует, что применение двух способов определения концентрации β-лактамов равнозначно (sr ≤ 0.13). Для анализа молока, мяса птицы, говядины, свинины, печени, почек, шпика и яиц использовали матричную градуировку для молока, а для анализа мёда – матричную градуировку для мёда. Следует отметить, что для рутинного анализа предпочтительнее использовать метод добавок [14]. В качестве примера на рис. 7, 8 приведены масс-хроматограммы экстрактов из молока и говядины без добавки и с добавкой идентифицированных β-лактамов.
Таблица 4.
Результаты (нг/г) определения β-лактамных антибиотиков в пищевых продуктах методом добавок и матричной калибровки на молоке (n = 3, P = 0.95)
| Матрица | Аналит | Найдено методом добавок | sr | Найдено методом матричной градуировки по молоку | sr |
|---|---|---|---|---|---|
| Молоко № 1 | Цефтиофур | 5.4 ± 0.1 | 0.08 | 4.9 ± 0.2 | 0.09 |
| Нафциллин | 9.7 ± 0.2 | 0.05 | 9.9 ± 0.1 | 0.04 | |
| Молоко № 2 | Цефтиофур | 3.2 ± 0.1 | 0.10 | 2.9 ± 0.2 | 0.13 |
| Нафциллин | 2.4 ± 0.1 | 0.11 | 2.9 ± 0.2 | 0.12 | |
| Молоко № 3 | Цефтиофур | 9.6 ± 0.2 | 0.08 | 9.9 ± 0.3 | 0.09 |
| Нафциллин | 0.30 ± 0.08 | 0.11 | 0.35 ± 0.06 | 0.12 | |
| Говядина | Цефтиофур | 0.22 ± 0.06 | 0.13 | 0.21 ± 0.07 | 0.13 |
| Нафциллин | 0.43 ± 0.05 | 0.12 | 0.38 ± 0.07 | 0.13 | |
| Цефуроксим | 5.64 ± 0.08 | 0.10 | 5.39 ± 0.06 | 0.09 | |
| Творог | Цефтиофур | 5.0 ± 0.1 | 0.09 | 4.3 ± 0.2 | 0.09 |
| Нафциллин | 2.2 ± 0.1 | 0.09 | 2.3 ± 0.1 | 0.09 | |
| Яйца куриные | Пиперациллин | 7.6 ± 0.1 | 0.08 | 7.3 ± 0.1 | 0.09 |
| Цефтиофур | 5.8 ± 0.1 | 0.09 | 5.6 ± 0.1 | 0.09 | |
| Мясо курицы | Цефотаксим | 40 ± 2 | 0.08 | 43 ± 3 | 0.08 |

Список литературы
Schneider M.J., Lehotay S.J., Lightfield A.R. Evaluation of a multi-class, multi-residue liquid chromatography – tandem mass spectrometry method for analysis of 120 veterinary drugs in bovine kidney // Drug Test. Analysis. 2012. V. 4. P. 91.
Zhan J., Yu X., Zhong Y., Zang Z., Cui X., Peng J., Feng R., Liu X., Zhu Y. Generic and rapid determination of veterinary drugs residues and other contaminants in row milk by performance chromatography-tandem mass spectrometry // J. Chromatogr. B. 2012. V. 906. P. 48.
Lopes R.P., Reyes R.C., Romero-Gonzalez R., Vidal J.L.M., Frenich A.G. Multiresidue determination of veterinary drugs in aquaculture fish samples by ultra high performance liquid chromatography coupled to tandem mass spectrometry // J. Chromatogr. B. 2012. V. 895–896. P. 39. https://doi.org/10.1016/j.jchromb.2012.03.011
Freitas A., Barbosa J., Ramos F. Multi-residue and multi-class method for the determination of antibiotics in bovine muscle by ultra-high-performance liquid chromatography tandem mass spectrometry // Meat Sci. 2014. V. 98. P. 58.
Li N., Feng F., Yang B., Jiang P., Chu X. Simultaneous determination of β-lactam antibiotics and β-lactamase inhibitors in bovine milk by ultra performance liquid chromatography-tandem mass spectrometry // J. Chromatogr. B. 2014. V. 945–946. P. 110.
Han R.W., Zheng N., Yu Z.N., Wang J., Xu X.M., Qu X.Y., Li S.L., Zhang Y.D., Wang J.Q. Simultaneous determination of 38 veterinary antibiotic residues in raw milk by UPLC-MS/MS // Food Chem. 2015. V. 181. P. 119.
Moretti S., Dusi G., Giusepponi D., Pellicciotti S., Rossi R., Saluti G., Cruciani G., Galarini R. Screening and confirmatory method for multiclass determination of 62 antibiotics in meat // J. Chromatogr. A. 2016. V. 1429. P. 175.
Yin Z., Chai T., Mu P., Xu N., Song Y., Wang X., Jia Q., Qiu J. Multi-residue determination of 210 drugs in pork by ultra-high-performance liquid chromatography–tandem mass spectrometry // J. Chromatogr. A. 2016. V. 1463. P. 49.
Sun L., Jia L., Xie X., Xie K., Wang J., Liu J., Cui L., Zhang G., Dai G., Wang J. Quantitative analysis of amoxicillin, its major metabolites and ampicillin in eggs by liquid chromatography combined with electrospray ionization tandem mass spectrometry // Food Chem. 2016. V. 192. P. 313.
Huang Z., Pan X.-D., Huang B., Xu J.-J., Wang M.-L., Ren Y.-P. Determination of 15 β-lactam antibiotics in pork muscle by matrix solid-phase dispersion extraction (MSPD) and ultra-high pressure liquid chromatography tandem mass spectrometry // Food Control. 2016. V. 66. P. 145.
Dorival-García N., Junza A., Zafra-Gomez A., Barron D., Navalon A. Simultaneous determination of quinolone and β-lactam residues in raw cow milk samples using ultrasound-assisted extraction and dispersive-SPE prior to UHPLC-MS/MS analysis // Food Control. 2016. V. 60. P. 382.
Lara F.J., Olmo-Iruela M., Cruces-Blanco C., Quesada-Molina C., Garcia-Campana A.M. Advances in the determination of β-lactam antibiotics by liquid chromatography // Trends Anal. Chem. 2012. V. 38. P. 52.
Zhang Y., Li X., Liu X., Zhang J., Cao Y., Shi Z., Sun H. Multi-class, multi-residue analysis of trace veterinary drugs in milk by rapid screening and quantification using ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry // J. Dairy. Sci. 2015. V. 98. P. 8433.
Amelin V., Korotkov A., Andoralov A. Identification and determination of 492 contaminants of different classes in food and feed by high-resolution mass spectrometry using the standard addition method // J. AOAC Int. 2016. V. 99. № 6. P. 1600.
Ortelli D., Cognard E., Jan Ph., Edder P. Comprehensive fast multiresidue screening of 150 veterinary drugs in milk by ultra-performance liquid chromatography coupled to time of flight mass spectrometry // J. Chromatogr. B. 2009. V. 877. P. 2363.
Gomez-Perez M. L., Plaza-Bolanos P., Romero-Gonzaleza R., Martinez-Vidal J. L., Garrido-Frenich A. Comprehensive qualitative and quantitative determination of pesticides and veterinary drugs in honey using liquid chromatography–orbitrap high resolution mass spectrometry // J. Chromatogr. A. 2012. V. 1248. P. 130.
Peters R.J.B., Stolker A.A.M., Mol J.G.J., Lommen A., Lyris E., Angelis Y., Vonaparti A., Stamou M., Georgakopoulos C., Nielen M.W.F. Screening in veterinary drug analysis and sports doping controlbased on full-scan, accurate-mass spectrometry // Trends Anal. Chem. 2010. V. 29. № 11. P 1250.
Kaufmann A., Butcher P., Maden K., Walker S., Widmer M. Development of an improved high resolution mass spectrometry based multi-residue method for veterinary drugs in varios food matrices // Anal. Chim. Acta. 2011. V. 700. P. 86.
Kaufmann A. The current role of high-resolution mass spectrometry in food analysis // Anal. Bioanal. Chem. 2012. V. 403. P. 1233.
Kaufmann A., Butcher P., Maden K., Widmer M. Quantitative multiresidue method for about 100 veterinary drugs in different meat matrices by sub 2-µm particulate high-performance liquid chromatography coupled to time of flight mass spectrometry // J. Chromatogr. A. 2008. V. 1194. P. 66.
Stolker A.A.M., Rutgers P., Oosterink E., Lasaroms J.J.P., Petrs P.J.R., Rhijn J.A., Nielen M.W.F. Comprehensive screening and quantification of veterinary drugs in milk using UPLC-ToF-MS // Anal. Bioanal. Chem. 2008. V. 391. P. 2309.
Peters R.J.B., Bolck Y.J.C., Rutgers P., Stolker A.A.M., Nielen M.W.F. Multi-residue screening of veterinary drugs in egg, fish and meat using high-resolution liquid chromatography accurate mass time-of-flight mass spectrometry // J. Chromatogr. A. 2009. V. 1216. P. 8206.
Pérez-Ortega P., Lara-Ortega F. J., Gilbert-López B., Moreno-González1 D., García-Reyes J. F., Molina-Díaz A. Screening of over 600 pesticides, veterinary drugs, food-packaging contaminants, mycotoxins, and other chemicals in food by ultra-high performance liquid chromatography quadrupole time-of-flight mass spectrometry (UHPLC-QTOFMS) // Food Anal. Methods. 2017. V. 10. P. 1216. https://doi.org/10.1007/s12161-016-0678-0
Grujic S., Vasiljevic T., Lausevic M., Ast T. Study on the formation of an amoxicillin adduct with methanol using electrospray ion trap tandem mass spectrometry // Rapid Commun. Mass Spectrom. 2008. V. 22. P. 67.
Grujic S., Vasiljevic T., Lausevic M. Determination of multiple pharmaceutical classes in surface and ground waters by liquid chromatography–ion trap–tandem mass spectrometry // J. Chromatogr. A. 2009. V. 1216. P. 4989.
Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin.
Технический регламент Таможенного союза (ТР ТС 033/2013) “О безопасности молока и молочной продукции” (Принят Решением Совета Евразийской экономической комиссии от 9 октября 2013 г. N 67).
Технический регламент Таможенного союза (ТР ТС 034/2013) “О безопасности мяса и мясной продукции” (Принят Решением Совета Евразийской экономической комиссии от 9 октября 2013 г. N 68).
Технический регламент Таможенного союза (ТР ТС 021/2011) “О безопасности пищевой продукции” (Утвержден Решением Комиссии Таможенного союза от 9 декабря 2011 года N 880).
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии