

Журнал аналитической химии, 2021, T. 76, № 11, стр. 1028-1037
Определение индикаторных конгенеров полихлорированных бифенилов в воде на ультраследовом уровне концентраций методом газовой хроматографии–тандемной масс-спектрометрии
О. В. Кустова a, *, А. С. Степанов b, А. Г. Горшков a
a Лимнологический институт Сибирского отделения Российской академии наук
664033 Иркутск, ул. Улан-Баторская, 3, Россия
b Уральский научно-исследовательский институт метрологии – филиал Всероссийского научно-исследовательского института метрологии им. Д.И. Менделеева
620075 Екатеринбург, ул. Красноармейская, 4, Россия
* E-mail: kustova_ov@lin.irk.ru
Поступила в редакцию 31.03.2021
После доработки 07.04.2021
Принята к публикации 08.04.2021
Аннотация
Валидирована методика определения индикаторных конгенеров (ИК) полихлорированных бифенилов (ПХБ) (№№ по ИЮПАК: 28, 52, 101, 118, 138, 153 и 180) в воде на ультраследовом уровне концентраций в диапазоне от 0.02 до 0.90 нг/л. Методика включает отбор проб объемом 1 л, экстракцию ПХБ в н-гексан, концентрирование экстрактов (kконц = 104) и прямой анализ концентратов методом газовой хроматографии−тандемной масс-спектрометрии. Хроматография концентратов на колонках с карборановой фазой (НТ-8, 30 м), детектирование аналитических пиков в режиме мониторинга заданных реакций и применение ИК, меченных углеродом 13С, в качестве суррогатных внутренних стандартов обеспечили высокую надежность идентификации пиков и точность количественного определения с суммарной погрешностью (±δ, Р = 0.95), равной 35%. Методика апробирована в рамках мониторинга ПХБ в водной экосистеме озера Байкал, определены концентрации ИК в верхнем водном слое пелагиали и в притоках озера.
Полихлорированные бифенилы (ПХБ) обладают рядом уникальных физических и химических свойств, которые определили их широкое применение в прошлом в качестве диэлектриков в трансформаторах, конденсаторах, гидравлических жидкостях, теплоносителях, хладагентах и смазочных маслах, как компонентов пластификаторов красок, лаков и клеев. Суммарный объем мирового производства ПХБ составил 1.5 млн тонн, из которых приблизительно 10% остались в окружающей среде [1]. Вследствие исключительной химической и биологической устойчивости, а также глобального распространения ПХБ обнаружены в воздухе, воде, почве, и биоте. Установлено, что ПХБ оказывают токсическое воздействие на живые организмы в крайне малых дозах и способны накапливаться в биологических объектах, поэтому Стокгольмской конвенцией (в РФ конвенция ратифицирована 2011 г. [2]) их производство и использование было запрещено; эти вещества включены в число стойких органических загрязняющих веществ и конвенцией ОСПАР в список приоритетных загрязнителей, требующих первоочередных мер по их контролю [3, 4].
Основная часть исследований токсичности ПХБ выполнена с копланарными диоксиноподобными конгенерами, которые Международным агентством по изучению рака отнесены к группе веществ, оказывающих канцерогенное действие на человека [5]. В работах последних лет представлены убедительные доказательства ответственности и непланарных конгенеров за экологические риски для живой природы, в частности их нейротоксичности [6, 7].
На современном этапе к источникам ПХБ отнесены демонтаж и утилизация оборудования, содержащего эти вещества в качестве конструктивных материалов, а также различные химические и термические процессы, в результате которых ПХБ образуются как побочные продукты. После прекращения промышленного производства ПХБ отмечено снижение их содержания в рыбе и донных отложениях [8–12]. По мнению авторов работ [13, 14], количество ПХБ в окружающей среде в настоящее время не уменьшается и даже увеличивается в некоторых районах мира. В частности, в 2014 и 2015 гг. в водах озера Байкал, которое содержит до 20% мировых запасов поверхностных пресных вод (23.6 тыс. км3), отмечено повышение уровня концентраций ПХБ [15–17] в 4.5 раза относительно максимальных и в 20 раз относительно минимальных значений, зафиксированных в 1992–1993 гг. [18, 19].
Для определения содержания ПХБ в поверхностных водах в чистых фоновых районах при их содержании на уровне следов требуются соответствующие методики. Например, при содержании ПХБ в воде ниже 2.0 нг/л их определение затруднено вследствие низкой растворимости, высокой гидрофобности и тенденции к накоплению в липидах, возможного поступления из лабораторного фона, которое может быть сопоставимым с содержанием аналитов в образце. Очевидно, поэтому в методиках, рекомендованных для определения ПХБ в природных водах, нижние границы диапазона определяемых концентраций (сн) соответствуют интервалу от 2.0 до 10 нг/л [20–22].
Разработанные методики анализа воды на первом этапе включают жидкостно-жидкостную экстракцию (ЖЖЭ) или твердофазную экстракцию. Экстракты очищают (в случае применения ЖЖЭ), концентрируют и анализируют методом жидкостной или газовой хроматографии с селективным детектированием [23]. Число конгенеров, которое может быть обнаружено в пробе, зависит как от уровня содержания ПХБ и ее матрицы, так и от используемого аналитического метода. Вследствие близких и совпадающих параметров удерживания отдельных конгенеров идентификация их пиков на хроматограммах относится к наиболее сложному этапу анализа, приобретающему критическое значение при содержании ПХБ на уровне следов. Для определения ПХБ в подобных пробах требуются методы, характеризующиеся не только низкими пределами обнаружения, но и высокой надежностью идентификации аналитических пиков.
Определение фиксированного числа конгенеров – “индикаторных конгенеров” ПХБ (ИК) – дает возможность унифицировать аналитичеcкую процедуру в рамках серийного анализа. Как правило, присутствие ПХБ в поверхностных водах фоновых районов связано с одним доминирующим источником – глобальным атмосферным переносом. Оценка загрязнения поверхностных вод ПХБ по концентрации ИК в таких районах оправдана и представляет адекватные результаты [24].
В настоящей работе с целью контроля ПХБ в поверхностных водах на ультраследовом уровне концентраций предложена методика определения ИК (№№ по ИЮПАК: 28, 52, 101, 118, 153, 138 и 180). Поскольку мониторинг проводят в условиях серийного анализа, в методику включены стандартизованные этапы отбора проб, ЖЖЭ ПХБ в н-гексан и концентрирование экстрактов. Для обеспечения высокой надежности идентификации пиков ИК и точности результатов для определения выделенных аналитов применяли метод газовой хроматографии−тандемной масс-спектрометрии (ГХ−МС/МС) с детектированием пиков в режиме мониторинга заданных реакций (МЗР). Валидация методики включала оценку предела обнаружения и сн, линейности градуировочных зависимостей в диапазоне концентраций ИК от 0.02 до 0.90 нг/л, и точности определения концентрации ИК. Методику апробировали при мониторинге ПХБ в водах озера Байкал, отличающихся минимальным содержанием взвешенного органического вещества (не более 0.01–0.05% [25]) и низкой степенью минерализации (суммарная концентрация главных ионов в воде озера не превышает уровня 15–150 мг/л [26]).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и реагенты. Пробы воды в пелагиали Байкала (верхний водный слой с глубины 5 м) собирали в ходе кругобайкальских экспедиций с помощью кассетного пробоотборника SBE-32 (CarouselWaterSampler, Sea-BirdElectronics, США). На каждой станции пелагиали озера, в устьях притоков, на очистных сооружениях пробы отбирали в две стеклянные бутыли емк. 1 л, к которым добавляли 0.5 мл 1 М водного раствора азида натрия (MERCK, ос. ч.) в качестве консерванта. Бутыли с водой закрывали крышкой с прокладкой из алюминиевой фольги и хранили при +5°С до анализа в лаборатории. В качестве экстрагента использовали н-гексан сорт 1 (КРИОХРОМ®, Россия), который перегоняли перед анализом над гидроксидом калия ч. д. а. (ВЕКТОН, Россия). Сульфат натрия х. ч. (НЕВАРЕАКТИВ®, Россия) прокаливали при 600°С в течение 6 ч.
Подготовка проб и хромато-масс-спектрометрический анализ. Пробы байкальской, речной воды, очищенных сточных вод анализировали без фильтрования. К пробам воды (900–1000 мл, точный объем) добавляли суррогатный стандарт в 0.5 мл ацетона (смесь изотопномеченых ИК ПХБ “Marker-7 PCB Mixture (W/PCB-118) (13C12 99%)”, Cambridge Isotope Laboratories, Inc., США, 10–30 мкл, 0.1 нг/мкл каждого). Пробу с суррогатным стандартом встряхивали, выдерживали не менее часа при комнатной температуре и переносили в делительную воронку емк. 1.5 л. Затем к пробе добавляли 25 мл н-гексана, смесь энергично встряхивали в течение 3–5 мин и оставляли до полного расслоения фаз. Верхний слой н-гексана отбирали в колбу емк. 250 мл, экстракцию повторяли. Экстракты объединяли, сушили над безводным сульфатом натрия, концентрировали на роторном испарителе до объема ~1 мл, затем в токе аргона до объема ~0.1 мл.
Подготовленные образцы анализировали на хромато-масс-спектрометре Agilent Technologies 7890B GC System 7000C GC-MS Triple Quad с капиллярной колонкой HT-8, SGE Analytical Science (30 м × 0.25 мм × 0.25 мкм) в режиме программирования температуры колонки от 80 до 310°С со скоростью 15 град/мин с последующим выдерживанием при 310°С в течение 7 мин; температуре инжектора 280°С; температуре источника 230°С; температуре квадруполей 150°С; энергии ионизации 70 эВ. В колонку хроматографа образец вводили в режиме без деления потока, объем образца 2–4 мкл. Пики ИК и стандартов регистрировали в режиме МЗР (табл. 1), идентифицировали по относительным временам удерживания (tR), гомогенность аналитических пиков оценивали по соотношению площадей пиков двух переходов Rик = SМЗР-2/SМЗР-1 (R).
Таблица 1.
Условия детектирования в режиме мониторинга заданных реакций
| № ИК | МЗР-переходы | m/z1 первичного иона |
m/z1 вторичного иона | Время выдержки, мс | Энергия столкновения2 “collusion energy”, В | Относительная интенсивность переходов3Rtarget (справочно) |
|---|---|---|---|---|---|---|
| ИК – аналиты | ||||||
| 28 | МЗР-1 | 256 | 186 | 10 | 25 | 0.64 |
| МЗР-2 | 258 | 186 | 10 | 25 | ||
| 52 | МЗР-1 | 290 | 220 | 10 | 25 | 0.41 |
| МЗР-2 | 255 | 220 | 10 | 10 | ||
| 101, 118 | МЗР-1 | 326 | 256 | 10 | 30 | 0.83, 0.63 |
| МЗР-2 | 254 | 184 | 10 | 35 | ||
| 153, 138 | МЗР-1 | 360 | 290 | 10 | 30 | 0.72, 0.69 |
| МЗР-2 | 288 | 218 | 10 | 40 | ||
| 180 | МЗР-1 | 394 | 324 | 10 | 30 | 0.45 |
| МЗР-2 | 394 | 359 | 10 | 15 | ||
| ИК 13С12 – суррогатные стандарты | ||||||
| 28 | МЗР-1 | 268 | 198 | 10 | 25 | 0.64 |
| МЗР-2 | 270 | 198 | 10 | 25 | ||
| 52 | МЗР-1 | 302 | 232 | 10 | 25 | 0.42 |
| МЗР-2 | 267 | 232 | 10 | 10 | ||
| 101, 118 | МЗР-1 | 338 | 268 | 10 | 30 | 0.89, 0.63 |
| МЗР-2 | 266 | 196 | 10 | 35 | ||
| 153, 138 | МЗР-1 | 372 | 302 | 10 | 30 | 0.75, 0.72 |
| МЗР-2 | 300 | 230 | 10 | 40 | ||
| 180 | МЗР-1 | 406 | 336 | 10 | 30 | 0.49 |
| МЗР-2 | 406 | 371 | 10 | 15 | ||
| 4,4’-Дибромбифенил – стандарт выхода | ||||||
| 4,4'-ДББФ | МЗР-1 | 312 | 152 | 10 | 20 | 0.49 |
| МЗР-2 | 314 | 152 | 10 | 20 | ||
1 Значения m/z для МЗР-переходов получены на основе масс-спектров молекулярного и дочерних ионов, полученных для растворов аналитов и стандартов с известной концентрацией; 2для оптимизации интенсивности аналитического сигнала энергия столкновения варьировалась от 10 до 60 В с шагом 5 В; 3относительную интенсивность переходов оценивали как среднее арифметическое соотношений площадей пиков масс-фрагментограмм МЗР-переходов, полученных для n градуировочных растворов.
Количественное определение индикаторных конгенеров. Содержание ИК в пробах воды рассчитывали по методу внутреннего стандарта как среднее значение для двух результатов единичных измерений в двух емкостях, содержащих пробу, с использованием изотопномеченых ИК ПХБ (13С12) в качестве суррогатных стандартов. Хромато-масс-спектрометр градуировали в интервале ожидаемых концентраций ИК в воде от 0.005 до 25 нг/л. Учитывая несложную матрицу проб (поверхностные воды в фоновых районах) и отсутствие матричного эффекта, градуировку проводили по градуировочным растворам, приготовленным в растворителе, используемом в качестве экстрагента. Растворы готовили дважды смешиванием соответствующих объемов аттестованной смеси ИК (“CEN PCB Congener Mix 1”, Supelco, США) и суррогатных стандартов. Каждый раствор хроматографировали, измеряли площади аналитических пиков, строили градуировочные зависимости Sа/Sst = k(ma/mst), где Sа/Sst – отношение площадей МЗР-1 переходов; ma/mst – отношение масс ИК и суррогатных стандартов; k – градуировочный коэффициент (коэффициент чувствительности). Значение достоверности аппроксимации соответствовало условию R2 ≥ 0.99.
Показатели повторяемости и внутрилабораторной прецизионности для всех ИК, кроме конгенера № 180, оценивали по рабочим пробам согласно рекомендациям РМГ 61-2010 и РМГ 76-2014 [27, 28]. Показатель внутрилабораторной прецизионности оценивали с использованием алгоритма оценки среднего квадратического отклонения (СКО) через относительные размахи результатов измерений, полученных в соответствующих условиях по рекомендациям [28]. Результат оценки показателя точности включает как вклад от оценки показателя прецизионности измерений, так и вклад от оценки показателя правильности измерений, проведенной с применением метода добавок. В связи с ограниченным количеством реальных проб, содержащих ИК № 180 в концентрации выше сн (25 параллельных проб в диапазоне концентраций 0.10–0.34 нг/л), данные для оценки показателя прецизионности получали в рамках оценивания показателя правильности измерений по рекомендациям [27]. Ниже приведены полученные нами значения перечисленных показателей:
| Диапазон концентра-ций опре-деляемого ИК, нг/л | Показатель повторяе-мости, σr, отн, % | Показатель внутрила-бораторной прецизион-ности σRлаб, отн, % (n= 2) | Показатель точности, ±δ, % (n=2, Р = 0.95) |
| 0.02–0.90 | 15 | 17 | 35 |
Точность определения суммарных концентраций обнаруженных ИК (ΣИК) оценивали с учетом внутрилабораторной прецизионности измерений как основного вклада в точность определения индивидуальных концентраций по уравнению:
где сi – концентрации индивидуальных ИК, δ – интервальная оценка погрешности определения сi – 35%, cΣ – концентрация ΣИК.Исходя из полученных результатов при генерации в MS Excel 1 × 104 случайных значений сi для индивидуальных ИК в диапазоне от 0.02 до 0.90 нг/л статистические оценки δ∑ составили: δ∑(min) = 13%, δ∑(max) = 23%, δ∑(med) = 15%, P(δ∑ ≤ 17%) = 0.959, P(δ∑ ≤ 18%) = 0.990.
Показатели правильности оценивали методом добавок с применением реальных проб и образцов сравнения для следующих диапазонов измерений, нг/л: I – (0.02–0.10) нг/л; II – (0.10–2.2) нг/л (табл. 2). В качестве добавки использовали растворы ИК с концентрациями 0.005 нг/мкл и 0.050 нг/мкл, приготовленные разбавлением аттестованной смеси ИК ацетоном. Введение добавок не приводило к увеличению объема пробы по отношению к начальному более чем на 1%. Оценка правильности результатов определения ИК (табл. 2) свидетельствует об отсутствии значимого вклада систематической погрешности в результат определения в регламентированном диапазоне измерений, а также в суммарную погрешность.
Таблица 2.
Оценка правильности результатов определения индикаторных конгенеров в байкальской воде методом введено–найдено (n = 3, Р = 0.95)
| № ИК | Содержание ИК в пробе воды, нг/л | Введено, нг | Найдено, нг/л | Добавка, нг | ||||
|---|---|---|---|---|---|---|---|---|
| I | II | I | II | I | II | I | II | |
| 28 | 0.09 ± 0.03 | 1.1 ± 0.25 | 0.07 | 1.0 | 0.16 ± 0.06 | 2.2 ± 0.51 | 0.07 ± 0.02 | 1.1 ± 0.25 |
| 52 | 0.07 ± 0.02 | 1.1 ± 0.25 | 0.07 | 1.0 | 0.14 ± 0.05 | 2.1 ± 0.48 | 0.07 ± 0.02 | 1.0 ± 0.23 |
| 101 | 0.05 ± 0.02 | 0.40 ± 0.14 | 0.05 | 0.35 | 0.10 ± 0.04 | 0.70 ± 0.25 | 0.04 ± 0.01 | 0.32 ± 0.11 |
| 118 | 0.04 ± 0.01 | 0.38 ± 0.13 | 0.05 | 0.35 | 0.09 ± 0.03 | 0.73 ± 0.26 | 0.04 ± 0.01 | 0.32 ± 0.11 |
| 153 | 0.03 ± 0.01 | 0.12 ± 0.04 | 0.03 | 0.10 | 0.06 ± 0.02 | 0.22 ± 0.08 | 0.03 ± 0.01 | 0.10 ± 0.04 |
| 138 | 0.03 ± 0.01 | 0.12 ± 0.04 | 0.03 | 0.10 | 0.06 ± 0.02 | 0.21 ± 0.07 | 0.03 ± 0.01 | 0.10 ± 0.04 |
| 180 | 0.03 ± 0.01 | 0.11 ± 0.04 | 0.02 | 0.10 | 0.06 ± 0.02 | 0.21 ± 0.07 | 0.03 ± 0.01 | 0.10 ± 0.04 |
Полноту извлечения ИК н-гексаном оценивали по количеству суррогатного стандарта (табл. 3), добавленного к пробам дистиллированной воды (V = 1000 мл) в количестве 10–30 мкл (0.1 нг/мкл, ацетон). Экстракцию ИК и концентрирование экстрактов проводили в условиях, используемых в разработанной методике. Содержание ИК в экстрактах определяли методом внешнего стандарта (4,4'-дибромобифенил (ДББФ), 10 мкл, 0.5 нг/мкл), который добавляли к экстрактам непосредственно перед ГХ−МС/МС-анализом.
Таблица 3.
Аналитические характеристики методики определения индикаторных конгенеров в воде на ультраследовом уровне концентраций
| Характеристика | № ИК | ||||||
|---|---|---|---|---|---|---|---|
| 28 | 52 | 101 | 118 | 138 | 153 | 180 | |
| Извлечение (среднее значение), % СКО (n = 20) |
73 0.08 | 81 0.11 | 84 0.12 | 85 0.06 | 87 0.08 | 89 0.08 | 92 0.08 |
| Градуировочные зависимости: | |||||||
| k R2 | 0.92 0.999 | 1.00 0.999 | 1.12 0.999 | 1.11 0.999 | 0.99 0.999 | 1.08 0.999 | 1.09 0.999 |
| Средняя величина реагент-бланков, нг СКО (n = 68) | 0.021 0.001 | 0.021 0.001 | 0.019 0.002 | 0.015 0.002 | 0.008 0.001 | 0.008 0.002 | 0.001 0.0002 |
| сн, нг/л | 0.02 | 0.02 | 0.02 | 0.02 | 0.01 | 0.01 | 0.01 |
| Предел обнаружения при отношении S/N ≥ 3, пг/пик | 0.4 | 0.4 | 0.4 | 0.4 | 0.2 | 0.2 | 0.2 |
Лабораторный фон ПХБ, суррогатные стандарты, содержащие в качестве примеси ИК без изотопной метки в структуре, могут вносить систематическую погрешность в результат определения, причем ее величина может быть значимой при определении ИК на ультраследовом уровне концентраций. Систематическую погрешность определения ИК оценивали по величине реагент-бланков (табл. 3), значения которых (содержание ИК в экстракте, полученном по процедуре подготовки без пробы воды) определяли до и после экстракции каждых 10 проб в серии образцов байкальской воды с использованием всего набора стеклянной посуды, реактивов, суррогатного стандарта и хромато-масс-спектрометрического оборудования. С учетом систематической погрешности граница сн в методике установлена на уровне 0.01–0.02 нг/л, которая на два порядка величины ниже, чем в ГОСТ Р 54503-2011 [20].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Определение индикаторных конгенеров в воде на ультраследовом уровне концентраций. Методика определения ИК в воде включает отбор проб объемом 1 л, который признан оптимальным. Увеличение объема пробы и соответственно объема получаемого экстракта и степени концентрирования не решает проблему снижения сн вследствие роста интенсивности пиков сопутствующих компонентов и увеличивает затраты времени и ресурсов. При уменьшении объема пробы и содержании ИК в воде на уровне концентрации 0.02 нг/л количество ИК, извлекаемых из пробы, недостаточно для регистрации аналитических пиков с соотношением сигнал/шум (S/N) ≥ 3, вклад ПХБ из лабораторного фона может быть сравнимым с содержанием аналитов в экстракте.
Эффективность извлечения ИК н-гексаном из воды с низким уровнем минерализации лежит в диапазоне от 73 до 92%. Учитывая область применения методики – поверхностные воды в чистых фоновых районах, отличающихся минимальным уровнем загрязнения, – стадия очистки экстрактов исключена, и определение ИК основано на прямом вводе концентратов в хроматографическую колонку. При разделении концентратов на капиллярных колонках с карборановой фазой (HT-8) [29] и детектировании в режиме МЗР на хроматограммах регистрируются разрешенные пики ИК с соотношением S/N ≥ 3, число и интенсивность пиков сопутствующих компонентов в экстракте минимальны (рис. 1а). Пара конгенеров (№№ 28 и 31) разделяется до базовой линии, но при “старении” колонки после выполнения ~1000 анализов разрешение этой пары снижается. Конгенеры №№ 52 и 69 имеют равные времена удерживания, но конгенер № 69 отсутствует как в смесях ПХБ, так и во фракциях ПХБ, выделенных из природных объектов. Анализ тех же самых экстрактов с применением метода ГХ и детектирования в режиме мониторинга выбранных ионов (МВИ), регламентированного национальным стандартом [20], отличается падением селективности и регистрацией неразрешенных пиков ИК (рис. 1б).
Рис. 1.
Масс-фрагментограммы экстракта верхнего водного слоя байкальской воды (5 м), отобранной в сентябре 2020 г. в Южном Байкале. Детектирование пиков в режимах МЗР (а) и МВИ (б). Пики индикаторных конгенеров (а), № (пг/пик): 28 (0.80); 52 (1.0); 101 (1.2); 118 (0.60); 153 (0.40); 138 (0.20); 180 (<сн). Пики индикаторных конгенеров (б), № (пг/пик): 28 (0.80); 52 (1.2); 101 (1.0); 153 (0.40). Пики конгенеров № 118 и 138 негомогенны, пик конгенера № 180 характеризуется значением S/N < 3.
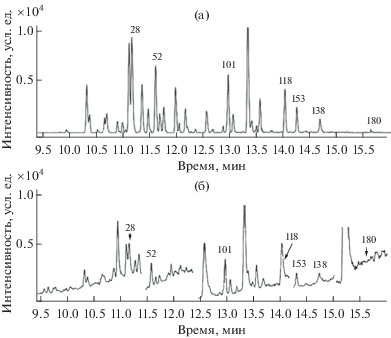
Режим детектирования МЗР резко повышает как чувствительность анализа за счет увеличения специфичности, так и его селективность в связи с уменьшением фоновых сигналов от матрицы и дает возможность проводить прямой анализ сконцентрированных экстрактов. Надежность идентификации аналитических пиков обеспечена двумя критериями: tR и относительной интенсивностью переходов R. Возможное отклонение значений tR идентифицируемого пика ИК от tR суррогатного стандарта регламентировано интервалом ±0.15%; отклонение R пиков ИК от Rtarget пиков ИК, полученных для градуировочных растворов ±30% [30].
Градуировочные зависимости в диапазоне масс ИК от 0.02 до 25 пг/пик характеризуются коэффициентами корреляции R2 = 0.999 (табл. 4). Определение ИК на следовом уровне концентраций предполагает взвешенный выбор внутреннего стандарта. Включение в методику структурных аналогов ПХБ или конгенеров, отсутствующих в анализируемых пробах [31, 32], в качестве суррогатных внутренних стандартов приводит к разбросу чувствительности определения ИК (вееру градуировок). Например, коэффициенты чувствительности ИК при использовании ДББФ в качестве внутреннего стандарта лежат в диапазоне от 0.50 до 2.18 (рис. 2а), в случае пентахлорированных бифенилов при использовании в качестве стандарта конгенера № 118 13С12 – в диапазоне от 0.57 до 1.42 (рис. 2б). Градуировка ИК с изотопномечеными стандартами ПХБ (13С12) нивелируют разброс чувствительности определения ИК (табл. 3).
Таблица 4.
Содержание ПХБ и ИК, обнаруженных в верхнем водном слое пелагиали (5 м) и притоках озера Байкал
| Район, сезон отбора проб | Число конгенеров ПХБ | ΣПХБ, нг/л | Число ИК | ΣИК, нг/л | Вклад ΣИК в ΣПХБ, % | |
|---|---|---|---|---|---|---|
| Пелагиаль озера Байкал | ||||||
| 2015 г. | Май | 24–34 | 1.4–6.6 | 7 | 0.43–2.0 | 28–35 |
| 2016 г. | Май | 15–27 | 0.45–3.1 | 6 | 0.23–1.2 | 38–58 |
| Сентябрь | 6–20 | ≥0.11–0.60 | 3–6 | ≥0.11–0.33 | 55–70 | |
| 2018 г. | Май | 7–16 | ≥0.11–0.73 | 4–6 | ≥0.11–0.42 | 58–70 |
| 2019 г. | Май | 7–21 | 0.13–0.82 | 4–6 | 0.10–0.47 | 57–70 |
| Сентябрь | 9–27 | 0.24–2.8 | 6–7 | 0.17–1.3 | 46–71 | |
| 2020 г. | Май | 10–16 | 0.20–0.75 | 5–6 | 0.14–0.37 | 49–72 |
| Сентябрь | 7–25 | 0.25–0.47 | 4–6 | 0.16–0.24 | 50–65 | |
| Притоки Южного Байкала | ||||||
| 2017 г., май | р. Хара-Мурин | 10 | 0.13 | 6 | 0.11 | 61 |
| р. Солзан | 9 | 0.11 | 5 | 0.11 | 60 | |
| р. Снежная | 3 | ≥0.11 | 2 | ≥0.11 | – | |
| 2019 г., сентябрь | р. Хара-Мурин | 16 | 1.8 | 7 | 0.85 | 46 |
| р. Солзан | 20 | 3.2 | 7 | 1.3 | 41 | |
| р. Снежная | 21 | 3.6 | 7 | 1.5 | 41 | |
Рис. 2.
Градуировочные зависимости для определения индикаторных конгенеров (внутренний стандарт 4,4'-дибромобифенил) (а): № конгенера (k) – 28 (2.18), 52 (1.24), 118 (0.74), 101 (0.70), 153 (0.69), 138 (0.59), 180 (0.50); градуиовочные зависимости для определения пентахлорбифенилов (внутренний стандарт конгенер № 118 13С12) (б): № конгенера (k) – 121 (1.42), 118 (1.11), 112 (1.06), 101 (1.08), 114 (0.90), 105 (0.81), 123 (0.78), 126 (0.57).

Валидация методики определения полихлорированных бифенилов в воде на ультраследовом уровне концентрациий. Валидацию методики проводили в рамках мониторинга ПХБ в водной экосистеме Байкала в разные сезоны (весна, осень) в течение 2015–2020 гг. Показано, что в составе обнаруженных ПХБ доминируют три-, тетра- и пентахлорированные гомологи, число идентифицированных конгенеров составило 6–34, и их суммарные концентрации (ΣПХБ) лежат в диапазоне от ≤0.12 до 6.6 нг/л (табл. 4). За исключением конгенера № 180, ИК присутствовали во всех исследованных пробах, содержание конгенеров №№ 153 и 138 в единичных образцах было ниже границы сн. При минимальном содержании ПХБ в воде (ΣПХБ ≥ 0.11 нг/л) состав фракции ПХБ представлен доминирующими ИК №№ 28, 52, 101, 118. Суммарная концентрация ИК в водах Байкала составила от ≥0.11 до 2.0 нг/л, а их доля в ΣПХБ – 28–72%. За период мониторинга ПХБ в водах озера (с 2015 по 2020 гг.) отмечено снижение концентрации ΣПХБ в водах Байкала и достижение ее минимального уровня, зафиксированного в 1992 г. (0.02–0.59 нг/л [18]) и в 1993 г. (0.13–1.9 нг/л [19]).
Разработанная методика апробирована при анализе проб с относительно высоким содержанием ИК (до 10 нг/л) и характеризуется достаточно высокой точностью определения содержания ИК (δ = 10–23%, Р = 0.95) в образцах воды, собранных в южной котловине озера (май 2015 г.), в притоках Южного Байкала (сентябрь 2019 г.) (табл. 4), и в сточных водах после прохождения очистных сооружений. В образцах сточных вод г. Северобайкальска содержание ПХБ оценили величиной ΣПХБ, равной 31 нг/л (32 конгенера), и величиной ΣИК, равной 9.8 нг/л (7 конгенеров). На расстоянии до 2.5 км от точки сброса стоков в р. Тыя концентрации ΣПХБ и ΣИК в речной воде снизились в 30 раз до уровней 0.95 и 0.31 нг/л соответственно [33].
Применение в предложенной методике традиционного способа ЖЖЭ как наиболее простого, не требующего специального оборудования и реагентов, наиболее перспективно для целей мониторинга ПХБ. Прямой анализ экстрактов проб воды, собранных в чистых фоновых районах и отличающихся несложной матрицей, дает возможность сократить суммарное время определения ПХБ. Хроматография экстрактов на колонках с карборановой фазой и применение метода ГХ−МС/МС обеспечивают высокую надежность идентификации пиков и точность количественного определения ИК на ультраследовом уровне концентрации.
Исследование выполнено в рамках государственного задания Министерства науки и высшего образования РФ, проекты № 0279-2019-0003 (АААА-А19-119070190033-0) и № 0279-2021-0005 (№ гос. регистрации 121032300224-8), при финансовой поддержке РФФИ и Правительства Иркутской области, проект № 17-45-388077 р_а, на оборудовании приборного центра коллективного пользования физико-химического ультрамикроанализа ЛИН СО РАН (ЦКП “Ультрамикроанализ”).
Список литературы
Wolska L., Mechlinska A., Rogowska J., Namiesnik J. Sources and fate of PAHs and PCBs in the marine environment // Crit. Rev. Environ. Sci. Technol. 2012. V. 42. P. 1172.
Федеральный закон о ратификации Стокгольмской конвенции о стойких органических загрязнителях от 27 июня 2011 г. http://kremlin.ru/acts/11757. (14.09.2020).
Стокгольмская конвенция. О Стойких Органических Загрязнителях (CO3). С поправками 2017 г. http:// Downloads/UNEP-POPS-COP-CONVTEXT-2017. Russian.pdf. (14.01.2021).
OSPAR Commission. Protecting and conserving the North-East Atlantic and its resources. https://www.ospar.org/convention. (12.12.2020).
IARC-International Agency for Research on Cancer. Polychlorinated and Polybrominated Biphenyls. Lyon, France: IARC Monographs, 2016. V. 107.
Ampleman M.D., Martinez A., De Wall J., Rawn D.F.K., Hornbuckle K.C., Thorne P.S. Inhalation and dietary exposure to PCBs in urban and rural cohorts via congener-specific measurements // Environ. Sci. Technol. 2015. V. 49. P. 1156.
Jennifer P., Houseman E.A., Smit E., Kile M.L. A path analysis of multiple neurotoxic chemicals and cognitive functioning in older US adults (NHANES 1999–2002) // Environ. Health. 2017. V. 16. P. 1.
AMAP, Assessment 2015: Temporal Trends in Persistent Organic Pollutants in the Arctic. Oslo, Norway: Arctic Monitoring and Assessment Program (AMAP), 2016. 71 p.
Hu D., Hornbuckle K.C. Inadvertent polychlorinated biphenyls in commercial paint pigments // Environ. Sci. Technol. 2010. V. 44. P. 2822.
Pomata D., Di Filippo P., Riccardi C., Rossi V., Simonetti G., Sonego E., Buiarelli F. Method optimization for the simultaneous determination of legacy and emerging halogenated flame retardants in particulate matter collected in an electronic waste recycling facility // Int. J. Environ. Anal. Chem. 2019. V. 8. P. 1.
Lohmann R., Northcott G., Jones K. Assessing the contribution of diffuse domestic burning as a source of PCDD/Fs, PCBs, and PAHs to the UK atmosphere // Environ. Sci. Technol. 2000. V. 34. P. 2892.
Ikonomou M., Sather P., Oh J., Choi W., Chang Y. PCB levels and congener patterns from Korean municipal waste incinerator stack emissions // Chemosphere. 2002. V. 49. P. 205.
Fernández P., Grimalt J.O. On the global distribution of persistent organic pollutants // Environ. Anal. Chem. 2003. V. 57. № 9. P. 514.
McLachlan M.S., Undeman E., Zhao F., MacLeod M. Predicting global scale exposure of humans to PCB 153 from historical emissions // Environ. Sci. Proc. Imp. 2018. V. 20. P. 747.
Самсонов Д.П., Кочетков А.И., Пасынкова Е.М., Запевалов М.А. Содержание стойких органических загрязнителей в компонентах уникальной экологической системы озера Байкал // Метеорология и гидрология. 2017. № 5. С. 105.
Горшков А.Г., Кустова О.В., Дзюба Е.В., Захарова Ю.Р., Шишлянников С.М., Хуторянский В.А. Полихлорированные бифенилы в водной экосистеме Байкала // Химия в интересах устойчивого развития. 2017. Т. 25. № 3. С. 269. (Gorshkov A.G., Kustova O.V., Dzyuba E.V., Zakharova Yu.R., Shishlyannikov S.M., Khutoryanskiy V.A. Polychlorinated biphenyls in lake baikal ecosystem // Chem. Sustainable Dev. 2017. V. 25. P. 255.)
Gorshkov A.G., Kustova O.V., Izosimova O.N., Babenko T.A. POPs monitoring system in lake Baikal – Impact of time or the first need? // Limnol. Freshwater Biol. 2018. № 1. P. 43.
Iwata H., Tanabe S., Ueda K., Tatsukawa R. Persistent orgnochlorine residues in air, water, sediments, and soils from the Lake Baikal region, Russia // Environ. Sci. Technol. 1995. V. 29. № 3. P. 792.
Kucklik J.R., Harvey H.R., Ostrom P.H., Ostrom H.E., Baker J.E. Organochlorine dynamics in the pelagic food web of Lake Baikal // Environ. Toxicol. Chem. 1996. V. 15. № 8. P. 1388.
ГОСТ Р 54503-2011 Вода. Методы определения содержания полихлорированных бифенилов. М.: Стандартинформ, 2013. 27 с.
EPA Method 8082. Polychlorinated Biphenyls (PCBs) by Gas Chromatography.
ПНД Ф 14.1:2:3:4.204-04 Методика определения хлорорганических пестицидов и полихлорированных бифенилов в питьевых, природных и сточных водах методом газовой хроматографии. Екатеринбург: СЕРТИМЕТ, 2018. 28 с.
Reddy A.V.B., Moniruzzaman M., Aminabhavi T.M. Polychlorinated biphenyls (PCBs) in the environment: Recent updates on sampling, pretreatment, cleanup technologies and their analysis // Chem. Eng. J. 2019. V. 358. P. 1186.
Megson D., Benoit N.B., Sandau C.D., Chaudhuri Sri R., Long T., Coulthard E., Johnson G.W. Evaluation of the effectiveness of different indicator PCBs to estimating total PCB concentrations in environmental investigations // Chemosphere. 2019. V. 237. Article 124429.
Yoshioka T., Ueda S., Khodzher T., Bashenkhaeva N., Korovyakova I., Sorokovikova L., Gorbunova L. Distribution of dissolved organic carbon in Lake Baikal and its watershed // Limnology. 2002. V. 3. № 3. P. 159.
Домышева В.М., Сороковикова Л.М., Синюкович В.Н., Онищук Н.А., Сакирко М.В., Томберг И.В., Жученко Н.А., Голобокова Л.П., Ходжер Т.В. Ионный состав воды озера Байкал, его притоков и истока реки Ангара в современный период // Метеорология и гидрология. 2019. № 10. С. 77.
РМГ 61-2010 Рекомендации по межгосударственной стандартизации. Государственная система обеспечения единства измерений. Показатели точности, правильности, прецизионности методик количественного химического анализа. Методы оценки. М.: Стандартинформ, 2012. 62 с.
РМГ 76-2014 Рекомендации по межгосударственной стандартизации. Государственная система обеспечения единства измерений. Внутренний контроль качества результатов количественного химического анализа. М.: Стандартинформ, 2015. 116 с.
Larsen B., Cont M., Montanarella L., Platzner N. Enhanced selectivity in the analysis of chlorobiphenyls on a carborane phenylmethylsiloxane copolymer gas chromatography phase (HT-8) // J. Chromatogr. A. 1995. V. 708. № 1. P. 115.
SANTE/11813/2017. Guidance document on analytical control and method validation procedures for pesticide residues and analysis in food and feed. European commission. 21–22 November 2017. Rev.0.
Никонова А.А., Горшков А.Г. Определение полихлорированных бифенилов в природных средах и биологических объектах методом скоростной масс-спектрометрии // Журн. аналит. химии 2012. Т. 67. № 1. С. 74. (Nikonova A.A., Gorshkov A.G. Determination of polychlorinated biphenyls by fast chromatography-mass-spectrometry in environmental and biological objects // J. Anal. Chem. 2012. V. 67. № 1. P. 73.)
Holst von C., Muller A., Bjorklund E., Anklam E. Inhouse validation of a sampled method for the determination of PCBs in food and feeding stuffs // Eur. Food Res. Technol. 2001. V. 213. P. 154.
Gorshkov A.G., Kustova O.V., Tomberg I.V., Zhuchenko N.A., Sakirko M.V., Khutoryansky V.A. Polychlorinated biphenyls in the tributaries of Southern Baikal // Limnol. Freshwater Biol. 2020. № 1. P. 353.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии