

Журнал аналитической химии, 2021, T. 76, № 12, стр. 1089-1099
Распознавание и определение сульфаниламидов методом флуориметрии в ближней ик-области по их влиянию на скорость каталитического окисления карбоцианинового красителя пероксидом водорода
И. А. Степанова a, А. Н. Лебедева a, А. В. Шик a, Е. В. Скоробогатов a, М. К. Беклемишев a, *
a Московский государственный университет имени М.В. Ломоносова, химический факультет
119991 Москва, ГСП-1, Ленинские горы, 1, стр. 3, Россия
* E-mail: mkb@analyt.chem.msu.ru
Поступила в редакцию 16.06.2021
После доработки 21.06.2021
Принята к публикации 21.06.2021
Аннотация
Работа направлена на развитие флуориметрических методов анализа биообъектов с целью расширения круга определяемых низкомолекулярных органических аналитов и сокращения операций пробоподготовки при использовании доступных флуорофоров и реагентов, а также применения одних и тех же флуориметрических систем для качественного и количественного анализа. Предложено использовать катализируемую медью(II) реакцию окисления карбоцианинового флуорофора пероксидом водорода, приводящую к изменению интенсивности флуоресценции в ближней ИК-области (700 нм). Найдено, что несколько органических соединений разной природы ускоряют или замедляют индикаторную реакцию, причем в различной степени при разном времени протекания процесса. В качестве модельных аналитов подробнее рассмотрели восемь сульфаниламидов, которые можно различить на качественном уровне за счет использования кинетического фактора при обработке данных методом главных компонент. На примере фталилсульфатиазола показано, что получение сигнала возможно не только в водном растворе, но и в присутствии гомогената мышц индейки на уровне 0.08–0.5 мМ (sr = 0.09) без использования разделения. Сделано заключение о перспективах развития подобных флуоресцентных платформ.
Флуориметрические методы определения низкомолекулярных органических соединений развиваются по пути прямого определения аналитов и сокращения операций пробоподготовки [1]. Такие методы, как правило, просты, экспрессны и относительно доступны в части аппаратурного оформления. Однако развитию и более широкому использованию флуориметрии органических аналитов препятствует ряд обстоятельств. Для получения селективных зондов в подавляющем большинстве случаев требуется синтез. Синтетические сложности и малая доступность реагентов ограничивают их практическое использование, а способность зонда давать отклик на единственный аналит не позволяет перейти к определению других на той же платформе. Несложно определять флуориметрическими методами типичные тушители, соединения, способные к переносу энергии или электрона или связывающиеся с флуорофором ковалентно [1–4]; для других аналитов флуориметрическое определение проводят косвенно или с использованием дериватизации [5]. Для определения низкомолекулярных органических аналитов недостаточно широко применяется флуориметрия в ближней ИК-области спектра – так называемом первом окне прозрачности биологических тканей (NIR-I): от 650 до 1000 нм [6, 7], в котором собственная флуоресценция биологических объектов и поглощение ими возбуждающего и излученного света минимальны. Для регистрации сигнала обычно используют спектрофлуориметры и реже – фотокамеры [8], в частности, камеры смартфонов для флуоресценции в видимой области [9]. Для регистрации эмиссии в ближней ИК (БИК)-области спектра также целесообразно применять фотографию [10].
Таким образом, очевидно, что актуальна разработка новых флуоресцентных сенсорных платформ, которые позволяли бы получать отклик органических соединений разной природы (в том числе не обладающих флуоресценцией и не взаимодействующих с флуорофорами); измерять сигнал в биоматрицах за счет использования БИК-области спектра; создавать линейки различных сенсоров на единой платформе. Целесообразно также использование сочетания двух сигналов: флуориметрического и фотометрического [11]. Желательно, чтобы разрабатываемые платформы не предусматривали синтез реагентов.
Нами предложен подход к решению перечисленных задач за счет использования реакций окисления красителей, излучающих в ближней ИК-области спектра. Если такая реакция катализируется ионом переходного металла, то по влиянию на ее протекание можно было бы определять соединения-лиганды, что позволило бы расширить круг аналитов, определяемых флуориметрическими методами. Варьирование природы металла-катализатора могло бы привести к расширению круга аналитов. В химическом анализе ранее использовалось немало каталитических редокс-систем [13, 14], однако БИК-флуорофоры в качестве восстановителей не применяли. Впрочем, подбор условий окисления таковых не должен вызывать принципиальных сложностей.
В данной работе обнаружена возможность окисления коммерчески доступного карбоцианинового красителя I пероксидом водорода, катализируемого медью(II) (схема 1 ). Одним из традиционных восстановителей в этой каталитической реакции был гидрохинон [14], а активаторами служили аминосоединения [14, 15]. Изученное нами влияние низкомолекулярных органических соединений (в основном лекарственных веществ) на протекание реакции показало возможность получения сигналов для соединений разной природы.
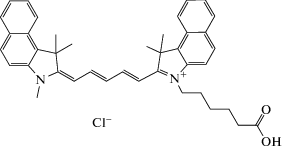
Схема 1 . Структура красителя I (Су5.5 карбоновая кислота).
Цель данной работы – показать перспективы и целесообразность исследования систем предлагаемого типа как с целью распознавания соединений близкой природы, так и для определения отдельных веществ. В качестве определяемых соединений более подробно рассмотрены сульфаниламиды (схема 2 ), применяемые в составе антибактериальных препаратов. Для флуоресцентного определения сульфаниламиды обычно дериватизируют, чаще всего флуорескамином, в сочетании с ВЭЖХ в до- или послеколоночном варианте [5, 16]. Используют также собственную флуоресценцию сульфаниламидов в УФ-области или фотоиндуцированную флуоресценцию [17]. Для определения сульфаниламидов методом флуоресцентного поляризационного иммуноанализа синтезируют трейсеры с производными флуоресцеина [18].
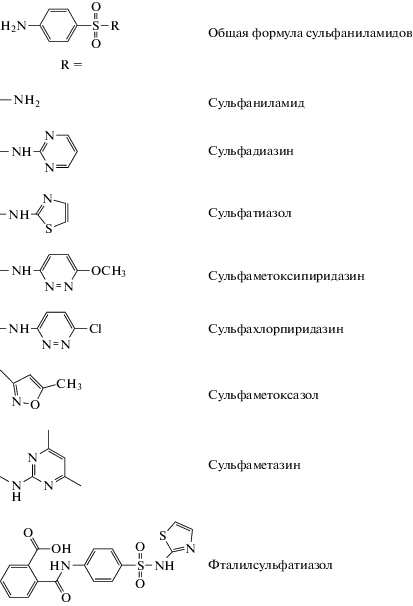
Схема 2 . Структурные формулы изученных сульфаниламидов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы и оборудование. Краситель I (Cy5.5-COOH), CAS No. 1449612-07-0, мол. масса 619.2, приобретенный в ООО “Люмипроб Рус” (Россия), растворяли в 95%-ном этаноле (ООО “Брынцалов-А”, Россия) и получали при этом раствор с концентрацией 1 г/л. В день, когда проводили эксперимент, из него готовили коллоидный раствор путем разбавления водой в 60 раз (0.017 г/л). Использовали пероксид водорода ос. ч., другие вещества имели квалификацию х. ч., ч. д. а. или поступали из Sigma-Aldrich (ФРГ). Растворы модельных аналитов (5 мМ) готовили в 95%-ном этаноле или в воде, дополнительно очищенной на установке Millipore. Медь(II) вводили в виде сульфата, используя 1 × 10–4 М водный раствор.
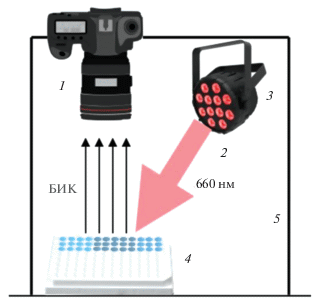
Большинство опытов проводили в 96-луночных флуориметрических планшетах (Thermo Scientific Nunc F96 MicroWell, белые, кат. № 136101). Спектры поглощения в УФ- и видимой областях регистрировали на спектрофотометре СФ-102 (Интерфотофизика, Россия) в кварцевых кюветах длиной 1 см. Спектры флуоресценции получали на спектрофлуориметре “Флюорат-02 Панорама” (Люмэкс, Россия) в кварцевых кюветах длиной 1 или 0.2 см. Флуоресценцию в БИК-диапазоне в 96-луночных планшетах регистрировали с помощью установки (рис. 1), содержащей светодиодный источник (11 красных светодиодов мощностью 3 Вт с максимумом излучения 660 нм) (Minifermer, Москва, Россия) и цифровую фотокамеру (в фотоаппарате Nikon D80 стандартный светофильтр, расположенный перед матрицей, был заменен на светофильтр с пропусканием выше 700 нм). Для количественных измерений получали два кадра с поворотом планшета на 180 градусов для компенсации не вполне равномерного освещения планшета из-за бокового расположения источника света.
Рис. 1.
Установка для регистрации флуоресценции в ближнем ИК-диапазоне: 1 – модернизированный фотоаппарат Nikon D80, 2 – красные светодиоды, 3 – алюминиевый радиатор для отвода тепла, 4 – 96-луночный полистирольный планшет с образцами, 5 – светонепроницаемый кожух.
В качестве модельных аналитов использовали 47 соединений – действующих веществ распространенных лекарств. Эти соединения содержат широкий спектр функциональных групп, имеют надежно установленный состав и коммерчески доступны. Среди изученных соединений можно выделить несколько групп, например аминогликозиды, цефалоспорины, фторхинолоны, сульфаниламиды, что позволяет изучать поведение соединений, сходных по структуре.
Методика эксперимента. Для каталитического окисления красителя I в лунку планшета с помощью дозатора в типичном эксперименте добавляли (конкретные условия указаны в подписях к рисункам; вещества добавляли в лунку в том порядке, в котором они перечислены): 30 мкл буферного раствора (0.05 М водный раствор тетрабората натрия для создания рН 9.2 или 0.1 М ацетатный буферный раствор с рН 3.7), 60 мкл 1 мМ раствора меди(II), 30 мкл 1 М раствора Н2О2, 60 мкл 5 мМ раствора модельного аналита, 60 мкл воды (если раствор аналита приготовлен в этаноле) или 60 мкл этанола (в случае водного раствора аналита) и 60 мкл раствора красителя I (0.017 г/л). Содержание этанола в серии опытов поддерживали постоянным, поскольку от него зависит квантовый выход эмиссии красителя. Считали, что реакция начинается в момент добавления красителя. Заметим, что в боратном буферном растворе медь(II) окисляет пероксид водорода, превращаясь в оксид меди(I) [19]. При высоких концентрациях меди(II) (от 1 мМ) в лунках был заметен рыжеватый осадок Cu2O, который, однако, не мешал измерениям.
За окислением красителя следили по флуоресценции в ближней ИК-области. Интенсивности фиксировали фотографическим методом в режиме отражения в планшете, что давало возможность быстро и одновременно измерять несколько образцов, работать с мутными суспензиями, многократно фиксировать сигнал во времени. Фотографии планшета получали через различные промежутки времени (от 1 мин до 1–2 ч) в БИК-визуализаторе (рис. 1). Фотографии оцифровывали в программе ImageJ, рассчитывая общую интенсивность. Усредненные по каждой лунке интенсивности использовали в качестве сигнала; иногда представляли сигнал как долю от интенсивности на фотографии, полученной сразу после начала реакции. Для хемометрической обработки получали по шесть параллельных результатов в соседних лунках планшета и загружали их в программу Unscrambler X (Camo Software, Норвегия), где обрабатывали методом главных компонент, используя установки по умолчанию.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
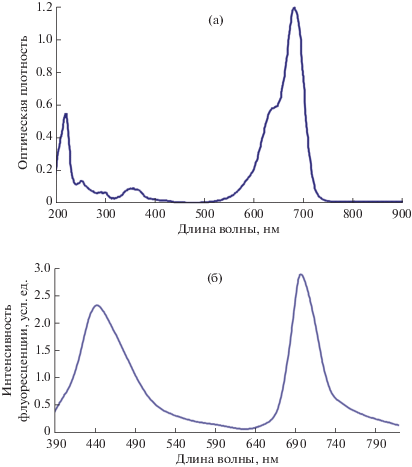
Свойства системы в отсутствие модельных аналитов. Использовали краситель I, поглощающий свет при 660 нм и излучающий в БИК-области (в этаноле максимум находится при 700 нм, рис. 2). Реагент относится к гидрофобным карбоцианинам, которые при разбавлении органического раствора водой образуют прозрачный, очень слабо флуоресцирующий коллоидный раствор наночастиц диаметром порядка 100 нм [10]. Однако при добавлении органического растворителя растворимость карбоцианина и его флуоресценция повышаются. Установлено, что введение 15–25 об. % этанола в водный раствор позволяет наблюдать БИК-флуоресценцию красителя I, удобную для измерений.
Рис. 2.
Спектры поглощения (а) и флуоресценции (возбуждение при 370 нм) (б) красителя I в этаноле (2.7 × 10−6 М).
В присутствии пероксида водорода и солей меди(II) краситель I медленно окисляется, при этом падает интенсивность эмиссии при 710 нм и поглощение при 660 нм. Светопоглощение и флуоресценция изменяются не синхронно: так, при рН 3.7 изменения контрольного сигнала в БИК-диапазоне заметны через 10 мин после начала реакции, а изменения поглощения начинаются только через полчаса. В данной работе мы опирались на измерения БИК-флуоресценции, меньше зависящей от собственной окраски анализируемых объектов.
Влияние модельных аналитов на интенсивность флуоресценции. На скорость окисления красителя I могут влиять различные соединения, в том числе образующие комплексы с ионами меди(II). Для оценки возможности получения отклика модельных аналитов проводили скрининговое исследование в 96-луночном планшете (рис. 3). В нейтральном и слабощелочном растворах сигнал дают немногие соединения, в частности, заметно ускоряют окисление красителя диклофенак, рибофлавин и сульфаниламиды (темные лунки на рис. 3а). В слабокислой среде набор модельных веществ, изменяющих сигнал, расширяется (рис. 3б): ускоряют реакцию также цефотаксим, сульфаниламиды, эритромицин; замедляют – бензилпенициллин, неомицин, гентамицин, метамизол, прокаин, ванкомицин, симвастатин, изониазид, моксифлоксацин, тетрациклин, фенотиазины.
Рис. 3.
Фотографии флуоресцирующих планшетов с системой краситель I–Cu(II)–Н2О2 через 20 мин после начала реакции в присутствии модельных аналитов: 1-я строка – контроль (без аналита), цефтриаксон, цефазолин, цефтазидим, цефотаксим, бензилпенициллин, амикацин, неомицин, фосфомицин, гентамицин, стрептомицин, метамизол; 2-я строка: циметидин, пирацетам, аскорбат, прокаин, ванкомицин, диклофенак, глутатионат, рибофлавин, сульфадиазин, сульфаметоксазол, сульфатиазол, α-гидроксипролин; 3-я строка: 5-гидрокситриптофан, эритромицин, сульфаметазин, тирамин, симвастатин, винпоцетин, меропенем, хлорамфеникол, мебгидролин, изониазид, ампициллин, доксициклин; 4-я строка: тетрациклин, пефлоксацин, энрофлоксацин, ципрофлоксацин, офлоксацин, левофлоксацин, моксифлоксацин, триметоприм, промазин, прометазин, хлорпромазин, перфеназин: (а) – рН 7.4, (б) – рН 3.7.

В присутствии некоторых аналитов интенсивность флуоресценции красителя выше контрольной, т.е. его окисление замедляется. Это заметно, например, для симвастатина (рис. 3а, 3б, ряд 3, поз. 5), что можно объяснить агрегацией: для гидрофобных карбоцианинов характерно тушение эмиссии в водных растворах и ее разгорание при включении красителя в гидрофобные домены различных наноструктур [10, 20]. Симвастатин, как и другие липофильные водорастворимые соединения [21], вероятно, образует мицеллоподобные частицы при отсутствии четкой критической концентрации мицеллобразования. Включение карбоцианина в такие структуры может иметь два следствия: повышение квантового выхода эмиссии из-за перехода в гидрофобное окружение и замедление окисления пероксидом; и то, и другое могло бы объяснить повышение измеряемой интенсивности по сравнению с контрольным опытом.
Как видно из рис. 3, рассматриваемая флуориметрическая система чувствительна к соединениям разной природы. Представляет интерес изучение возможности распознавания аналитов, сходных по структуре.
Влияние сульфаниламидов на флуоресцентный сигнал. Для более подробного рассмотрения выбрали сульфаниламиды (8 соединений). Известно, что эти соединения образуют комплексы с медью(II) [22]. В отсутствие металла-катализатора и пероксида сульфаниламиды не влияют на БИК-флуоресценцию, за исключением фталилсульфатиазола (ФСТ), который в слабокислой среде (рН 3.7) частично тушит эмиссию красителя, в том числе в отсутствие пероксида и катализатора. В боратном буферном растворе ФСТ и сульфатиазол ускоряют каталитическую реакцию окисления красителя (рис. 4); другие соединения (сульфадиазин, сульфаметазин, сульфаметоксипиридазин, сульфахлорпиридазин) реакцию замедляют, давая сигнал выше контрольного (рис. 5). Эти соединения относительно липофильны, а их высокие сигналы можно объяснить теми же причинами, что приведены выше для симвастатина. Фосфатный буферный раствор (рН 7.4) ингибирует окисление красителя (рис. 4в).
Рис. 4.
Зависимость БИК-флуоресцентного сигнала системы пероксид–Cu(II)–фталилсульфатиазол от времени при рН 3.7 в отсутствие (а) и в присутствии ФСТ (0.5 мМ в лунке) (б); (в) – зависимость от рН, полученная через 29 мин после начала реакции. В лунки вводили по 30 мкл буферного раствора и 1 × 10–4 М CuSO4, по 60 мкл: 0.5 М Н2О2, 5 мМ сульфаниламида в этаноле, воду и водный раствор красителя I (0.017 г/л) в перечисленном порядке. Буферные растворы: HCl (рН 2), ацетатные (рН 3–5), фосфатный (рН 7.4), боратные (рН 9.2–10.8).
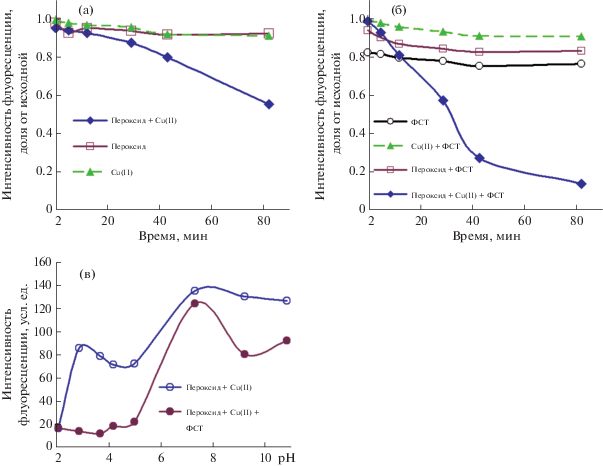
Рис. 5.
Пример фотографии флуоресцирующего планшета в ближнем ИК-диапазоне через 30 мин после начала реакции окисления красителя I пероксидом водорода в присутствии меди(II): столбец 0 – контрольный опыт, 1 – сульфадиазин, 2 – сульфаметоксазол, 3 – сульфатиазол, 4 – сульфаметазин, 5 – сульфаметоксипиридазин, 6 – фталилсульфатиазол, 7 – сульфаниламид, 8 – сульфахлорпиридазин. Концентрации растворов вводимой меди(II) и Н2О2 (М) показаны слева от изображений; в лунки помещали по 30 и 60 мкл этих растворов соответственно, а также 60 мкл модельного раствора аналита (5 мМ), 60 мкл воды или этанола и 30 мкл 0.05 М раствора Na2B4O7 (рН 9.2).

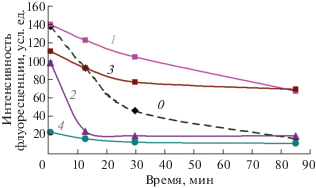
Особенность каталитических редокс-систем – изменение сигнала во времени, что увеличивает продолжительность анализа по сравнению с некаталитическими методами, однако дает возможность наблюдения сигнала через различные промежутки времени, при этом разные аналиты могут проявлять свое влияние в различной степени. Как видно из рис. 5 и 6, концентрационные условия и время наблюдения сигнала можно выбрать так, что сигнал будут давать, например, фталилсульфатиазол (на первых минутах реакции) и сульфатиазол (через 13 мин) – за счет ускорения окисления красителя (уменьшение флуоресценции); через полчаса и далее остальные сульфаниламиды будут давать сигнал, наоборот, выше контрольного за счет замедления реакции. (Перечисленные результаты относятся к концентрации добавляемого раствора меди(II) 1 мМ; при снижении количества катализатора времена увеличиваются: так, для 0.1 мМ раствора меди(II) отличие сигнала остальных сульфаниламидов от контрольного наступит через 1.5 ч).
Рис. 6.
Кинетические кривые для реакции окисления красителя I, катализируемой медью(II), в присутствии некоторых сульфаниламидов: 0 – без сульфаниламида, 1 – сульфаметоксипиридазин, 2 – сульфатиазол, 3 – сульфаниламид, 4 – фталилсульфатиазол. Условия: вводили 60 мкл 0.5 М Н2О2 и 30 мкл 1 мМ раствора сульфата меди(II) (строка “0.001/0.5” на рис. 5); остальные условия см. в подписи к рис. 5.
Идентификация сульфаниламидов в водном растворе. Для оценки возможностей предлагаемой флуориметрической системы провели распознавание сульфаниламидов в растворе. Для каждого из восьми аналитов выполнили по 6 параллельных экспериментов в системе H2O2–Cu(II)–краситель I при рН 3.7 (каждым сульфаниламидом заполнили по шесть лунок планшета). Фиксировали флуоресценцию красителя через различные промежутки времени, выбирая для обработки фотографии, на которых сигналы сульфаниламидов существенно различались (через 2, 10, 15 и 21 мин после начала реакции). Измеренные интенсивности обработали методом главных компонент (ГК). Пример графика счетов приведен на рис. 7 (одинаковыми символами обозначены параллельные опыты с одним сульфаниламидом).
Рис. 7.
(а) График счетов метода главных компонент для реакции H2O2–Cu(II)–краситель I при рН 3.7 в присутствии 8 сульфаниламидов, перечисленных в легенде в координатах 1-й и 3-й главных компонент; (б) некоторые фотографии, использованные для обработки (при временах реакции 2 и 15 мин). Аналиты помещали по 6 лунок в ряд в порядке, перечисленном в легенде: А1–А6 – контроль (без сульфаниламида), А7–А12 – сульфадиазин, В1–В6 – сульфаметоксазол и т.д. вплоть до лунки Е6. Условия реакции – см. подпись к рис. 4.
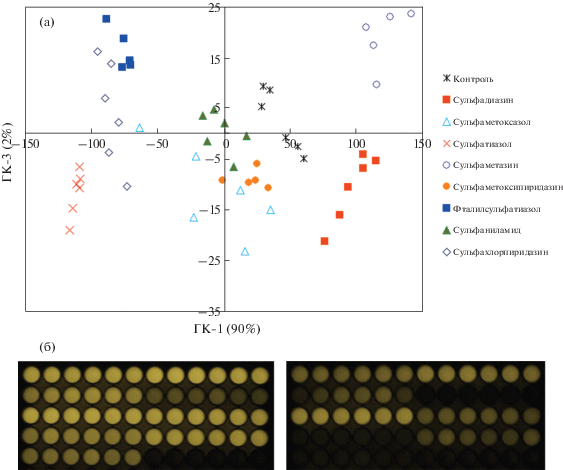
Как видно из рис. 7, разным сульфаниламидам отвечают разные группы точек на графике. Все восемь соединений одновременно различить нельзя, однако любые пары сульфаниламидов разделяются в одной из систем координат: 1–2 ГК, 1–3 ГК (рис. 7) либо 2–3 ГК.
Приведенные результаты указывают на перспективность развития флуоресцентных платформ рассматриваемого типа для решения задач качественного анализа.
Определение фталилсульфатиазола в водном растворе. Изучили возможность использования рассматриваемой флуориметрической системы для наблюдения сигнала ФСТ в водном растворе, а также в присутствии гомогената мышц индейки, обладающей собственной флуоресценцией при 400–600 нм и сильно поглощающей возбуждающее излучение в видимом и УФ-диапазоне. Определение проводили в слабощелочной среде, в которой набор потенциально мешающих аналитов аналитов более узок, чем в кислой (рис. 3). Интенсивность флуоресценции пропорциональна логарифму концентрации ФСТ (рис. 8а) при времени реакции не более 8 мин. Среднее значение относительного стандартного отклонения для этого интервала составило 0.17 (для 4 параллельных опытов). При малых временах реакции (например, 2 мин) есть тенденция к повышению сигнала по сравнению с контрольным опытом, т.е. ФСТ способен не только ускорять, но, как и другие сульфаниламиды, замедлять индикаторную реакцию.
Рис. 8.
Сигнал различных концентраций фталилсульфатиазола (ФСТ) в водном растворе (а) и в присутствии гомогената мыщц индейки (б). На вставках показаны фотографии лунок планшетов с параллельными опытами для приведенных на графиках концентраций ФСТ (на рис. 8а – для кривой 2). Время от начала реакции до измерения: (а): 1 – 2 мин, 2 – 8 мин; (б): 13 мин. Конечные концентрации в лунке планшета: (а): Na2B4O7 5 мМ, Cu(II) 1 × 10–5 М, Н2О2 0.05 М, ФСТ от 0 до 0.3 мМ, этанол 19 об. %; (б): Na2B4O7 5 мМ, Cu(II) 0.2 мМ, Н2О2 0.2 М, гомогенат индейки 3 г/л, ФСТ от 0 до 0.5 мМ; этанол 17 об. %; во всех случаях концентрация красителя 5.5 мкМ.

Для того чтобы наблюдать сигнал ФСТ в присутствии гомогената индейки, концентрации пероксида и меди(II) потребовалось увеличить в 4 и 20 раз соответственно. Полученная зависимость (рис. 8б) линейна в диапазоне концентраций ФСТ 8 × 10–5–5 × 10–4 М (коэффициент корреляции – 0.95, sr = 0.09, усредненное по диапазону концентраций).
Полученные результаты подтверждают, что в режиме отражения можно измерять флуоресценцию мутных суспензий, а поскольку красное и БИК-излучение в заметной степени проникают через биологическую ткань, целесообразно использование БИК-флуорофора [6, 23], позволяющего получать сигнал аналита в таком сложном объекте, как гомогенат мышц индейки, без отделения матрицы.
* * *
Таким образом, на основе каталитической реакции окисления БИК-флуорофора предложена флуориметрическая сенсорная платформа, позволяющая получать сигнал соединений различной природы, в том числе нетипичных для определения флуориметрическими методами. Используемый в работе метод БИК-флуориметрии с фотографической регистрацией сигнала подтверждает свою высокую производительность, удобство и целесообразность использования для определения и обнаружения низкомолекулярных органических аналитов. Предложена индикаторная реакция, позволившая получать сигналы веществ разной природы, различать соединения одного класса, анализировать биологические матрицы, которая к тому же может быть легко модифицирована путем замены красителя, окислителя или катализатора, что создаст предпосылки для дальнейшего расширения круга аналитов и разработки эффективных аналитических методик. Представленный материал свидетельствует о перспективности использования рассматриваемой флуориметрической системы не только для определения/обнаружения индивидуальных аналитов, но и для классификации (дискриминации) объектов, чему будут посвящены отдельные публикации.
Авторы благодарят Алексея Добротворского (photodrom.com) за предоставление БИК-фотоаппаратов и Владислава Орехова за помощь в расчетах по хемометрике.
Работа выполнена при поддержке Российского научного фонда (проект № 20-13-00330).
Список литературы
Demchenko A.P. Introduction to Fluorescence Sensing. Springer, Switzerland, 2015. 818 p.
Verbitskiy E.V., Rusinov G.L., Chupakhin O.N., Charushin V.N. Design of fluorescent sensors based on azaheterocyclic push-pull systems towards nitroaromatic explosives and related compounds: A review // Dyes Pigm. 2020. V. 180. Article 108414. https://doi.org/10.1016/j.dyepig.2020.108414
Chen X., Zhou Y., Peng X., Yoon J. Fluorescent and colorimetric probes for detection of thiols // Chem. Soc. Rev. 2010. V. 39. P. 2120. https://doi.org/10.1039/b925092a
Salem F.B. Spectrophotometric and fluorimetric determination of catecholamines // Anal. Lett. 1993. V. 26. P. 281. https://doi.org/10.1080/00032719308017385
Gehring T.A., Rushing L.G., Thompson H.C., Jr. Determination of sulfonamides in edible salmon tissue by liquid chromatography with postcolumn derivatization and fluorescence detection // J. AOAC Int. 1997. V. 80. P. 751.
Chen C., Tian R., Zeng Y., Chu C., Liu G. Activatable fluorescence probes for “turn-on” and ratiometric biosensing and bioimaging: From NIR-I to NIR-II // Bioconjugate Chem. 2020. V. 31. P. 276. https://doi.org/10.1021/acs.bioconjchem.9b00734
Wu D., Chen L., Lee W., Ko G., Yin J., Yoon J. Recent progress in the development of organic dye based near-infrared fluorescence probes for metal ions // Coord. Chem. Rev. 2018. V. 354. P. 74. https://doi.org/10.1016/j.ccr.2017.06.011
Rukosueva E.A., Dobrolyubov E.O., Goryacheva I.Yu., Beklemishev M.K. Discrimination of whiskies using an “add-a-fluorophore” fluorescent fingerprinting strategy // Microchem. J. 2019. V. 145. P. 397. https://doi.org/10.1016/j.microc.2018.11.002
Liu T., Zhang S., Liu W., Zhao S., Lu Z., Wang Y., Wang G., Zou P., Wang X., Zhao Q., Rao H. Smartphone based platform for ratiometric fluorometric and colorimetric determination H2O2 and glucose // Sens. Actuators B. 2020. V. 305. Article 127524. https://doi.org/10.1016/j.snb.2019.127524
Zakharenkova S.A., Katkova E.A., Doroshenko I.A., Kriveleva A.S., Lebedeva A.N., Vidinchuk T.A., Shik A.V., Abramchuk S.S., Podrugina T.A., Beklemishev M.K. Aggregation-based fluorescence amplification strategy: “Turn-on” sensing of aminoglycosides using near-IR carbocyanine dyes and pre-micellar surfactants // Spectrochim. Acta A. 2021. V. 247. Article 119109. https://doi.org/10.1016/j.saa.2020.119109
Zhou Y., Huang X., Hu X., Tong W., Leng Y., Xiong Y. Recent advances in colorimetry/fluorimetry-based dual-modal sensing technologies // Biosens. Bioelectron. 2021. V. 190. Article 113386. https://doi.org/10.1016/j.bios.2021.113386
Mottola H.A., Perez-Bendito D. Kinetic determinations and some kinetic aspects of analytical chemistry // Anal. Chem. 1994. V. 66. P. 131R.
Crouch S.R., Scheeline A., Kirkor E.S. Kinetic determinations and some kinetic aspects of analytical chemistry // Anal. Chem. 2000. V. 72. P. 53. https://doi.org/10.1021/a1000004b
Долманова И.Ф., Пешкова В.М. Определение микроколичеств меди с использованием каталитической реакции окисления гидрохинона перекисью водорода в присутствии пиридина // Журн. аналит. химии. 1964. Т. 19. С. 297.
Beklemishev M.K., Petrova Yu.Yu., Dolmanova I.F. Sorption-catalytic determination of imazapyr on a copper-containing sorbent // Mikrochim. Acta. 2001. V. 136. № 1–2. P. 35.
Maudens K.E., Zhang G.-F., Lambert W.E. Quantitative analysis of twelve sulfonamides in honey after acidic hydrolysis by high-performance liquid chromatography with post-column derivatization and fluorescence detection // J. Chromatogr. A. 2004. V. 1047. P. 85. https://doi.org/10.1016/j.chroma.2004.07.007
Flores J.L., Fernandez de Cordova M.L., Diaz A.M. Flow-through optosensor combined with photochemically induced fluorescence for simultaneous determination of binary mixtures of sulfonamides in pharmaceuticals, milk and urine // Anal. Chim. Acta. 2007. V. 600. P. 164.
Eremin S.A., Murtazina N.R., Ermolenko D.N., Zherdev A.V., Mart’ianov A.A., Yazynina E.V., Michura I.V., Formanovsky A.A., Dzantiev B.B. Production of polyclonal antibodies and development of fluo-rescence polarization immunoassay for sulfanilamide // Anal. Lett. 2005. V. 38. P. 951. https://doi.org/10.1081/AL-200054059
Miliero F.J., Sharma V.K., Karn B. The rate of reduction of copper(II) with hydrogen peroxide in seawater // Marine Chem. 1991. V. 36. P. 71.
Humphry-Baker R., Graetzel M., Steiger R. Drastic fluorescence enhancement and photochemical stabilization of cyanine dyes through micellar systems // J. Am. Chem. Soc. 1980. V. 102. P. 847.
Madenci D., Egelhaaf S.U. Self-assembly in aqueous bile salt solutions // Curr. Opin. Colloid Interface Sci. 2010. V. 15. P. 109. https://doi.org/10.1016/j.cocis.2009.11.010
Bult A., Uitterdijk J.D., Klasen H.B. Copper(II) complexes of sulfanilamide derivates // Transit. Met. Chem. 1979. V. 4. P. 285.
Reineck P., Gibson B.C. Near-infrared fluorescent nanomaterials for bioimaging and sensing // Adv. Opt. Mater. 2017. V. 5. № 2. 1600446. https://doi.org/10.1002/adom.201600446
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии