

Журнал аналитической химии, 2021, T. 76, № 2, стр. 166-182
Идентификация и определение неионогенных поверхностно-активных веществ методом ультра высокоэффективной жидкостной хроматографии‒масс-спектрометрии высокого разрешения
В. Г. Амелин a, b, *, Д. С. Большаков b
a Владимирский государственный университет имени Александра Григорьевича и Николая Григорьевича Столетовых
600000 Владимир, ул. Горького, 87, Россия
b Федеральный центр охраны здоровья животных
600901 Владимир, мкр. Юрьевец, Россия
* E-mail: amelinvg@mail.ru
Поступила в редакцию 26.04.2020
После доработки 10.05.2020
Принята к публикации 02.06.2020
Аннотация
Предложена методика идентификации и определения неионогенных поверхностно-активных веществ (НПАВ) в природных водах методом ультра-ВЭЖХ с масс-спектрометрическим детектированием высокого разрешения. Установлены основные аналитические характеристики определения наиболее распространённых НПАВ – алкилфенолэтоксилатов и алкилоксиэтоксилатов – по предложенной методике. Рассмотрены основные аддукты полимергомологов определяемых ПАВ, образующихся в условиях ионизации электраспылением. Для извлечения аналитов из образцов природных вод использован метод дисперсионной жидкостно-жидкостной микроэкстракции смесью трихлорметан–ацетонитрил. Рассмотрена возможность определения НПАВ при непосредственном электрораспылении подготовленного образца в масс-спектрометр. Пределы определения ПАВ в выбранных условиях варьировали от 0.1 до 0.5 нг/мл, диапазон определяемых содержаний составил от 1 до 1000 нг/мл. Относительное стандартное отклонение результатов анализа не превышает 0.15. Продолжительность идентификации компонентов проб составила 30 мин, определение обнаруженных аналитов занимает 1 ч.
Подавляющая часть неионогенных поверхностно-активных веществ (НПАВ) представляет собой продукты присоединения оксида этилена к веществам, имеющим подвижный атом водорода (фенолам, спиртам, кислотам, аминам, амидам). Общая формула этих соединений R–Х–(СН2–СН2О)mН, где R – алкильный или алкиларильный радикал; X = –О–, –СОО–, –СONН и др. Общим для этой группы НПАВ является присутствие полиоксиэтиленовой цепи [1].
Причиной появления НПАВ в природных водах являются главным образом сбросы неочищенных сточных вод, некоторое количество НПАВ может поступать с грунтовыми водами, а также с атмосферными осадками. Содержание НПАВ в воде нормируется. Поскольку НПАВ представляют собой большую группу оксиэтилированных соединений различных классов, значения предельно допустимых концентраций (ПДК) для индивидуальных веществ имеют достаточно большой разброс. Например, для водных объектов хозяйственно-питьевого и культурно-бытового назначения ПДК синтанолов и оксиэтиленалкилфенолов составляет 0.1 мг/л, неонолов – 0.1–0.3 мг/л, стеароксов – 0.5–1 мг/л. Для водных объектов рыбохозяйственного назначения ПДК различных синтанолов колеблется от 0.0005 до 0.002 мг/л, неонолов – от 0.0001 до 0.3 мг/л. При определении суммарной концентрации НПАВ в водах условно принято считать ПДК равной 0.1 мг/л [1].
Помимо классических стандартизованных методов группового определения НПАВ [1–3], в последнее время получило распространение использование хроматографических методов анализа с масс-спектрометрическим детектированием (табл. 1).
Таблица 1.
Хроматографические методы определения НПАВ
| Аналиты | Объекты анализа (матрица) | Пробоподготовка | Метод анализа | Метод детектирования | смин | Лите-ратура |
|---|---|---|---|---|---|---|
| Алкилфенолэтоксилаты, оксоалкогольэтоксилаты (этоксилаты жирных спиртов) | Стандартные образцы, модельные растворы природной воды | Вода: упаривание, растворение осадка в хлороформе или тетрахлорметане | ЖХ | МС (электронная ионизация, химическая ионизация изобутаном) | – | [4] |
| Этоксилаты жирных спиртов, нонилфенолэтоксилаты, диэтаноламиды жирных кислот кокосового масла, нонилфеноксимонокарбоксилаты, нонилфенол, октилфенол и линейные алкилбензолсульфонаты | Донные отложения и прибрежные воды | Вода: фильтрование, предварительное концентрирование ТФЭ. Донные отложения: экстракция
УЗ смесью метанол–дихлорметан (7 : 3, об.), ТФЭ |
ЖХ | МС (химическая ионизация при атмосферном давлении/ ионизация электрораспылением) | Вода:
0.01–0.20 мкг/л; донные отложения: 0.5–10 мкг/кг |
[5] |
| Полиэтоксилированные жирные спирты, полиэтоксилированные нонилфенолы, алкилбензолсульфонаты | Поверхностные и сточные воды | ЖЖЭ смесью дихлорметан–изопропанол | ПИА | МС (химическая ионизация при атмосферном давлении; ионная ловушка) | 93–797 пг | [6] |
| Линейные алкилбензолсульфонаты, алкилэтоксисульфаты, алкилсульфаты, нонилфенолполиэтоксилаты, алкогольполиэтоксилаты (полиэтоксилаты жирных спиртов) и их карбоксилированные метаболиты (сульфофенилкарбоновые кислоты и алкилфенолэтоксикарбоксилаты) | Вода и осадки | ТФЭ (вода), жидкостная экстракция под давлением (осадки) | ЖХ | МС (ионизация электрораспылением) | 0.05–0.5 нг/мл (вода), 1–10 нг/г (осадки) | [7] |
| Алкилфенолы, алкилфенолэпоксилаты, алкилфенолэтоксикарбоксилаты, бисфенол А | Сточные воды, природные воды | ТФЭ | ВЭЖХ | МС (ионизация электрораспылением, тройной квадруполь) | 0.1–10 мкг/л (400 мл пробы, 0.5 мл экстракта) | [8] |
| Алифатические этоксилированные спирты, нонилфенолполиэтоксилаты | Сточные, питьевые, речные воды | ТФЭ | ЖХ | МС (ионизация электрораспылением) | 0.0002–0.6 мкг/л | [9] |
| Алкилфенолполиэтоксилаты, этоксилаты жирных спиртов | Осадки сточных вод | Экстракция в точке помутнения | ЖХ | МС | 0.09–0.38 мг/кг | [10] |
| Линейные алкилбензолсульфонаты, алкогольэтоксисульфаты, алкилфенолэтоксилаты, алкогольэтоксилаты, алкилфенолы | Вода объектов окружающей среды | ТФЭ | ЖХ | МС (ионизация электрораспылением) | 40–200 нг/л; 90–440 нг/л; 0.4–2 мкг/л (в зависимости от матрицы) | [11] |
В работе [4] рассмотрено применение метода жидкостной хроматографии с квадрупольным масс-спектрометрическим детектированием (ЖХ–МС) в режиме электронной и химической ионизации и изобутана в качестве реакционного газа для определения НПАВ класса алкилфенолэтоксилатов CnH2n+ 1C6H4O(CH2CH2O)mH и этоксилатов жирных спиртов CnH2n+ 1O(CH2CH2O)mH. Аналитами являлись октилфенолэтоксилат (Тритон Х-100), нонилфенолэтоксилат (Akopal N060) и цетилэтоксилат (Brij 56). Разделение проводили в режиме градиентного элюирования на колонке с привитыми аминопропиловыми группами. В результате получена информация о распределении гомологов и наличии примесей в чистых препаратах. Отмечено, что метод может быть использован для изучения первичной биодеградации НПАВ в воде (на примере модельных растворов загрязненных образцов природной воды) (табл. 1).
Для определения этоксилатов жирных спиртов, нонилфенолэтоксилатов, диэтаноламидов жирных кислот кокосового масла, нонилфеноксимонокарбоксилатов, нонилфенола, октилфенола и линейных алкилбензолсульфонатов в донных отложениях и образцах воды предложено использовать метод ЖХ–МС с химической ионизацией при атмосферном давлении/электрораспылительной ионизацией в сочетании с пробоподготовкой методом твердофазной экстракции (ТФЭ) [5]. Методика позволила установить достаточно высокие концентрации нонилфенолэтоксилата и нонилфенола рядом с местами сброса промышленных и городских сточных вод. Нонилфенол обнаружен в 47% проб воды и в 77% всех проанализированных образцов донных отложений. Концентрации нонилфенола варьировали от 0.15 до 4.1 мкг/л в морской воде и от 8 до 1050 мкг/кг в донных отложениях. Концентрации диэтаноламидов жирных кислот кокосового масла в отложениях (в которых преобладают гомологи С11–С15) варьировали в диапазоне от 30 до 2700 мкг/кг, в морской воде концентрации определяемого компонента составляли до 24 мкг/л. Установлено, что этоксилаты жирных спиртов накапливаются в донных отложениях, и поэтому они были обнаружены во всех проанализированных образцах. Диапазон определяемых содержаний этоксилатов жирных спиртов составил от 37 до 1300 мкг/кг.
Описан экспресс-метод определения анионных (АПАВ), катионных ПАВ и НПАВ в воде методом проточно-инжекционного анализа в сочетании с масс-спектрометрическим детектированием с электрораспылительной ионизацией [6]. Для извлечения и концентрирования аналитов использовали метод жидкостно-жидкостной экстракции (ЖЖЭ) смесью дихлорметан–изопропанол (90 : 10, по объему). Количественный анализ проводили с использованием изотопно меченного триэтоксилированного нонилфенола и дибутилнафталинсульфоната натрия в качестве внутренних стандартов. Анализ осуществляли путем чередования положительного и отрицательного режимов ионизации, что позволило одновременно определить наиболее распространенные ПАВ за короткое время. Пределы обнаружения варьировали от 93 до 797 пг (табл. 1). Метод отличается высокой экспрессностью, поскольку не требует хроматографического разделения подготовленного экстракта. Однако в некоторых случаях для достоверной идентификации аналитов необходим дополнительный анализ методом масс-спектрометрии дочерних ионов.
В работе [7] описана методика одновременного определения в объектах окружающей среды основных АПАВ и НПАВ: линейных алкилбензолсульфонатов, алкилэтоксисульфатов, алкилсульфатов, нонилфенолполиэтоксилатов, алкогольполиэтоксилатов (полиэтоксилатов жирных спиртов) и их карбоксилированных метаболитов (сульфофенилкарбоновых кислот и алкилфенолэтоксикарбоксилатов). Аналиты экстрагировали из проб воды и осадков методом ТФЭ и жидкостной экстракции под давлением. Идентификацию и оценку количественного содержания целевых соединений проводили методом ЖХ–МС с электрораспылительной ионизацией при чередовании положительного и отрицательного режимов. Степень извлечения большинства аналитов изменялась от 70 до 107%. Пределы обнаружения составили 0.05–0.5 нг/мл для воды и 1–10 нг/г для осадков. Разработанную методику использовали для одновременного определения ПАВ в пробах, отобранных из различных водных экосистем. Во всех образцах обнаружены аналиты различного происхождения. Самые высокие концентрации метаболитов ПАВ в воде составили 149.6 мкг/л для сульфофенилкарбоновых кислот и 3.9 мкг/л для алкилфенолэтоксикарбоксилатов.
Предложена методика определения алкилфенолов, алкилфенолэтоксилатов, алкилфенолэтоксикарбоксилатов и бисфенола А в поверхностных водах методом ВЭЖХ с масс-спектрометрическим детектированием (тройной квадруполь) [8]. Для концентрирования проб и очистки полученных экстрактов применяли метод ТФЭ. Для разделения алкилфенолов и алкилфенолэтоксикарбоксилатов использовали градиентный режим элюирования без добавления буферных растворов. Применение системы ацетонитрил–вода в качестве подвижной фазы способствовало увеличению достоверности определения за счет уменьшения числа аддуктов фрагментации, имеющих одинаковые значения m/z. Нонилфенолэтоксилаты (m = 1–17) определяли отдельно в режиме положительной ионизации при использовании подвижной фазы, содержащей ацетат аммония. Проанализированы сбросы предприятий текстильной промышленности, соответствующие стоки с очистных сооружений и воды рек. Среди определяемых веществ нонилфенолэтоксикарбоксилаты и нонилфенолэтоксилаты показали самые высокие концентрации в пробах воды: до 4.5 мкг/л в сточных водах и 3.6 мкг/л в водах рек.
Анализ литературы показал, что наиболее часто используют в промышленности и определяют в различных объектах оксиэтилированные алкилфенолы и оксиэтилированные жирные спирты. Для концентрирования проб различного происхождения и очистки полученных экстрактов используют преимущественно методы ТФЭ и ЖЖЭ, которые характеризуются значительным расходом реагентов и длительностью. Более современным и экологичным методом извлечения загрязняющих веществ различных классов из водных образцов является дисперсионная жидкостно-жидкостная микроэкстракция (ДЖЖМЭ [12–14]. Кроме того, современные системы масс-спектрометрии позволяют проводить непосредственный ввод образца природных вод при ионизации электрораспылением, минуя стадию хроматографического разделения подготовленного экстракта. Новые возможности метода существенно упрощают мониторинговые исследования объектов окружающей среды на содержание НПАВ.
Цель данной работы заключалась в создании и апробации способа идентификации и определении НПАВ в природных водах, основанного на сочетании методов ДЖЖМЭ и ультра-ВЭЖХ (УВЭЖХ)–квадруполь времяпролетной масс-спектрометрии высокого разрешения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Аппаратура. Использовали ультра высокоэффективный жидкостной хроматограф UltiMate 3000 (Thermo Scientific, США) в сочетании с квадруполь-времяпролетным масс-спектрометрическим детектором maXis 4G и электрораспылительную ионизацию в устройстве ionBooster (Bruker Daltonics, Германия). Разделение проводили на колонке ACQUITY UPLC® BEH C18 (30 × 2.1 мм, 1.7 мкм) (Waters, США) в режиме градиентного элюирования.
Применяли весы аналитические Sartorius TE214S специального класса точности с пределом взвешивания 0.1 мг (Sartorius, Германия), центрифугу MPW-260R (MPW Med. Instruments, Польша), микродозаторы Biohit с переменным объемом 10–100 мкл, 100–1000 мкл, 1000–5000 мкл и пределом допускаемой погрешности измерения не более ±2%; фильтры мембранные политетрафторэтиленовые 0.20 мкм диаметром 25 мм (Corning Incorporated, Германия).
Реактивы. Использовали стандартные образцы НПАВ ОП-10 и ОП-7 (ГОСТ 8433-81), Тритон Х-100 (Sigma-Aldrich, США), ОС-20 (ГОСТ 10730-82) и Brij 35 (Sigma-Aldrich, США). Исходные стандартные растворы (1 мг/мл) готовили растворением точной навески препарата в метаноле. Рабочие растворы готовили разбавлением исходного деионированной водой (15–18 МОм см, ОСТ 11 029.003-80). Использовали метанол (Fisher Scientific UK, Великобритания), ацетонитрил, изопропиловый спирт (Scharlab S.L., Испания), муравьиную кислоту, формиат натрия (Sigma-Aldrich, США).
Идентификация и определение. Идентификацию НПАВ по полученным хроматограммам проводили с использованием программного продукта DataAnalysis-4.1, TargetAnalysis (Bruker Daltonics, Германия), составление картины изотопного распределения аналита – с использованием IsotopePattern (Bruker Daltonics, Германия). Неизвестную концентрацию аналита в пробе (cx) рассчитывали методом стандартной добавки по формуле:
сx = сдоб/[(Sx + доб/Sx) – 1],
где сдоб – концентрация добавки в пробе, нг/мл (нг/г); Sx, Sx + доб – площади пиков m/z в исследуемом растворе и в растворе с добавкой аналита соответственно.
Условия хроматографического разделения и детектирования [15]. Подвижная фаза состояла из 0.1%-ной муравьиной кислоты в воде (элюент А) и 0.1%-ной муравьиной кислоты в ацетонитриле (элюент В). Осуществляли градиентное элюирование: 0 мин – 5% В, 0.5 мин – 5% В, 2 мин – 50% В, 5 мин – 100% В, 6 мин – 5% В, 8 мин – 5% В. Скорость потока подвижной фазы 0.4 мл/мин. Оптимальная температура хроматографической колонки 50°С, объем вводимой пробы 50 мкл. Температура термостата автоматического дозатора 10°С.
Использовали ионизацию электроспреем в устройстве ionBooster (Bruker Daltonics, Германия). Установлены следующие оптимальные значения параметров ионизации: напряжение на щите капилляра 400 В, напряжение на капилляре 1000 В, давление газа-распылителя азота 4.76 атм, поток газа-осушителя азота 6 л/мин, температура газа-осушителя азота 200°С, поток газа-испарителя азота 250 л/ч, температура газа-испарителя азота 250°C.
Проводили регистрацию ионов в диапазоне значений m/z 100 до 1200. Для калибровки масс использовали раствор формиата натрия 10 мМ в смеси вода–изопропиловый спирт (1 : 1). Масс-спектр калибранта получали в интервале времен хроматографирования от 9.5 до 10 мин.
Пробоподготовка. Отбирали 10 мл исследуемой пробы, подкисляли ее добавлением 0.1 мл конц. HCl и впрыскивали микрошприцем 1 мл смеси хлороформа (250 мкл) и ацетонитрила (750 мкл); полученную эмульсию встряхивали и центрифугировали 5 мин при 2000 об/мин. Отбирали нижний слой экстракта в пенициллиновый флакон и упаривали досуха в токе азота при 30°С. Сухой остаток растворяли в смеси 50 мкл метанола и 950 мкл воды, перемешивали, раствор фильтровали через мембранный фильтр 0.20 мкм в микрофлакон и хроматографировали.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Аналитические характеристики НПАВ. Среди всего ассортимента НПАВ наиболее широкое распространение в различных отраслях промышленности и народном хозяйстве получили алкилфенолэтоксилаты и алкилоксиэтоксилаты (этоксилаты жирных спиртов). Алкилфенолэтоксилаты представляют собой смесь полимергомологов СnH2n+ 1C6H4O(C2H4O)mH. К ним следует отнести оксиэтилированные полиэфиры ОП-10 и ОП-7. Среди зарубежных аналогов можно выделить Emulgen 931, Noigen EA-190A, IGEPAL, Тритон Х-100 и др. Алкилоксиэтоксилаты – смесь полимергомологов СnH2n+ 1O(C2H4O)mH. Среди отечественных продуктов к ним относятся синтанолы (например, ОС-20). Зарубежные аналоги: Brij 35, Brij 56, Emulgen 105 и др. (схема 1 ).
Данные НПАВ различаются по числу гомологов (n) и по степени этоксилирования (m). Для установления этих различий в работе использовали метод УВЭЖХ в сочетании с квадруполь времяпролетной масс-спектрометрией высокого разрешения. При выбранных оптимальных условиях хроматографического разделения [15] получены и проанализированы хроматограммы и масс-спектры водных растворов НПАВ с концентрацией 100 нг/мл.

Схема 1 . Структурные формулы неионогенных ПАВ.
Оксиэтилированный полиэфир ОП-10. На рис. 1 представлен масс-спектр водного раствора ОП-10, в котором можно выделить две группы сигналов в диапазонах значений m/z 250–550 и 550–900. Такое распределение ионов по молекулярным массам характерно для полимергомологов с различным количеством мономерных звеньев в цепи. Установлено, что для ОП-10 характерны гомологи С7–С11 (аддукты с ионами аммония, калия и натрия) (рис. 2а), степень этоксилирования (m) для преобладающих гомологов составила от 6 до 19. В спектре также обнаруживаются сигналы, соответствующие по значению m/z аддуктам ОП-10 с ионами щелочных (диапазон значений m/z 600–900) и щелочноземельных (диапазон значений m/z 250–550) металлов. Так, в диапазоне значений m/z 550–900 в спектре имеется пик с максимальной интенсивностью при m/z 683.4328, который соответствует аддукту состава [С9Н19С6Н4О(С2Н4О)10Н + Na]+, слева от него расположены сигналы аддуктов с m = 8–9, справа с m = 11–15 и разницей в значении m/z 44, соответствующей оксиэтильному фрагменту (–C2H4O–). В спектре установлено также наличие аддуктов полимергомологов ОП-10 с ионами калия и аммония [С9Н19С6Н4О(С2Н4О)10Н + K]+, [С9Н19С6Н4О(С2Н4О)10Н + NH4]+ (рис. 1).
Рис. 1.
Масс-спектр полимергомологов ОП-10 [С9H19C6H4O(C2H4O)mH + Ca, Mg]2+, [С9H19C6H4O(C2H4O)mH + K, Na, NH4]+ (числом обозначена степень этилирования).

Рис. 2.
Масс-хроматограммы полимергомологов ОП-10 (m = 10) (а) и Тритон Х-100 (m = 10) (б). (а): С7 – [С7Н15С6Н4О(С2Н4О)10Н + NH4]+; C8 – [С8Н17С6Н4О(С2Н4О)10Н + NH4]+; C9(1) – [С9Н19С6Н4О(С2Н4О)10Н + + NH4]+; С9(2) – [С9Н19С6Н4О(С2Н4О)10Н + Nа]+; С9(3) – [С9Н19С6Н4О(С2Н4О)10Н + K]+; C10 – [С10Н21С6Н4О(С2Н4О)10Н + NH4]+; C11 – [С11Н23С6Н4О(С2Н4О)10Н + NH4]+; (б): С8(1) – [С8Н17С6Н4О(С2Н4О)10Н + + NH4]+; C8(2) – [С8Н17С6Н4О(С2Н4О)10Н + Nа]+; C8(3) – [С8Н17С6Н4О(С2Н4О)10Н + K]+; C9 – [С9Н19С6Н4О(С2Н4О)10Н + NH4]+; C10 – [С10Н21С6Н4О(С2Н4О)10Н + NH4]+.

При хроматографическом разделении в условиях градиентного элюирования с увеличением длины углеводородного радикала увеличивается и время удерживания алкилфенолэтоксилатов с 5.5 до 6.2 мин (рис. 2). Однако для аддукта [С10Н21С6Н4О(С2Н4О)10Н + NH4]+ время удерживания составило 5.7 мин, что может свидетельствовать о существовании изомеров, имеющих меньшее время удерживания. Об изомерных формах алкилфенолэтоксилатов свидетельствуют их несимметричные хроматографические пики (рис. 2а), а также данные работы [4], в которой проведено разделение и изучение изомерных форм.
На рис. 3 представлены масс-хроматограммы и масс-спектры двухзарядных ионов [С10H21C6H4O(C2H4O)mH + Са]2+ при максимальной степени этоксилирования m = 15–16. Как видно, времена удерживания этоксилатов с различной степенью этоксилирования различаются и увеличиваются с увеличением степени этоксилирования (рис. 3а). Это может свидетельствовать об образовании аддуктов оксиэтилированных алкилфенолов до ионизации электрораспылением, а именно в исходном растворе или в процессе хроматографирования.
Рис. 3.
Масс-хроматограммы (а) и масс-спектр (б) полимергомолога ОП-10 [С10H21C6H4O(C2H4O)mH + Са]2+ (m = 12–25).

Установлено, что в процессе электраспылительной ионизации этоксилированных алкилфенолов на масс-хроматограммах и в масс-спектрах регистрируются аддукты с ионами аммония, натрия и калия, причем преобладают в основном аддукты с ионом аммония (рис. 4). На рис. 4 представленны масс-спектры полиэфира ОП-10 в виде аддуктов с ионами аммония для полимергомолога С10. На масс-хроматограмме имеются два несимметричных пика, которые могут свидетельствовать о наличии изомерных форм в исходном продукте ОП-10.
Рис. 4.
Масс-хроматограммы (а) и масс-спектр (б) полимергомолога ОП-10 [С10H21C6H4O(C2H4O)mH + NH4]+ (m = 9–20).

Особенностью применяемого приборного обеспечения является возможность непосредственного ввода пробы в масс-спектрометр высокого разрешения (МСВР) с использованием устройства ionBooster (Bruker Daltonics, Германия) (рис. 5).
Рис. 5.
Масс-спектр полимергомолога ОП-10 [С9H19C6H4O(C2H4O)mH + K, Na]+ (m = 6–16), полученный непосредственным вводом раствора с концентрацией 100 нг/мл.
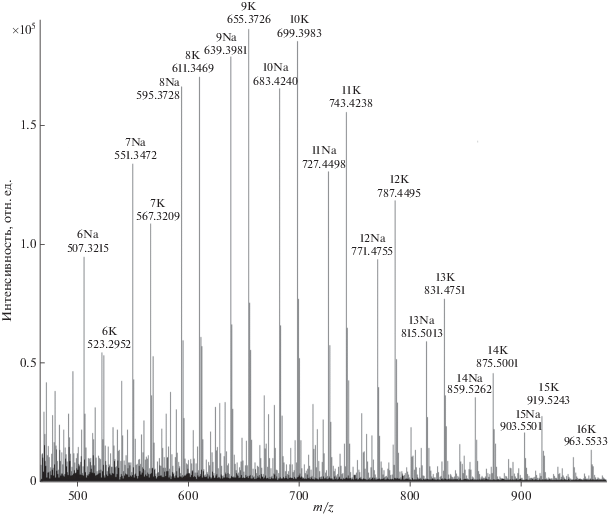
Непосредственное электрораспыление пробы в масс-спектрометр удобно для мониторинговых экспресс-исследований, это позволяет идентифицировать интересующие компоненты без хроматографического разделения подготовленного экстракта. В данных условиях в масс-спектре полимергомологов ОП-10 преобладают однозарядные аддукты с ионами щелочных металлов [M + K(Na)]+.
Оксиэтилированный полиэфир ОП-7. Установлено, что препарат ОП-7 представляет собой смесь полимергомологов от С4 до С7 с преобладанием C6 [С6H13C6H4O(C2H4O)6H + K]+и максимальной степенью этоксилирования m = 6 (рис. 6).
Рис. 6.
Масс-спектр полимергомологов ОП-7 [С6H13C6H4O(C2H4O)mH + K]+ (m = 0–16) и [С4H9C6H4O(C2H4O)mH + + K]+ (m = 2'–16') (а) и масс-хроматограммы полимергомолога ОП-7 [С6Н13С6Н4О(С2Н4О)mН + K]+ (m = 4–15) (б).

На рис. 6б представлены масс-хроматограммы гексилфенолэтоксилата. Как видно, времена удерживания для каждого полимергомолога различаются и составляют от 3.4 до 3.8 мин, что может свидетельствовать об образовании аддуктов уже в исходном растворе, до процесса хроматографирования.
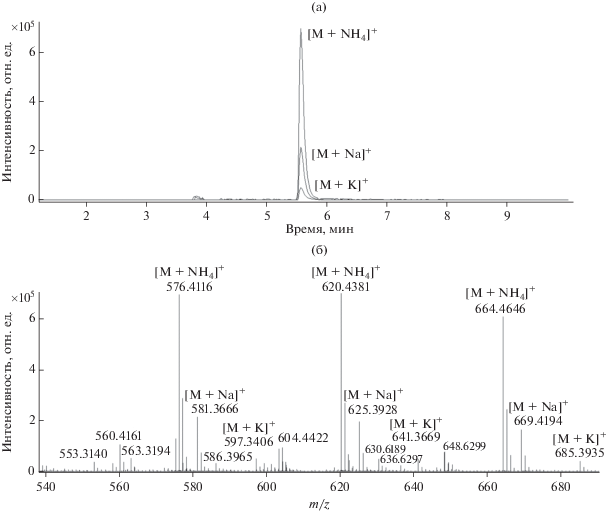
Оксиэтилированный полиэфир Тритон Х-100 представляет собой смесь гомологов С8–С10 (преимущественно С8) с максимальной степенью этоксилирования m = 8–9 (рис. 2б, рис. 7). В спектре полиэфира регистрируются аддукты с ионами натрия, калия и аммония (рис. 8). Как видно из рис. 7, 8, на масс-хроматограммах имеются симметричные пики, что свидетельствует об отсутствии изомеров в препарате Тритон Х-100.
Рис. 7.
Масс-хроматограммы (а) и масс-спектр (б) полимергомолога Тритона Х-100 [С8H17C6H4O(C2H4O)mH + NH4]+ (m = 4–16).

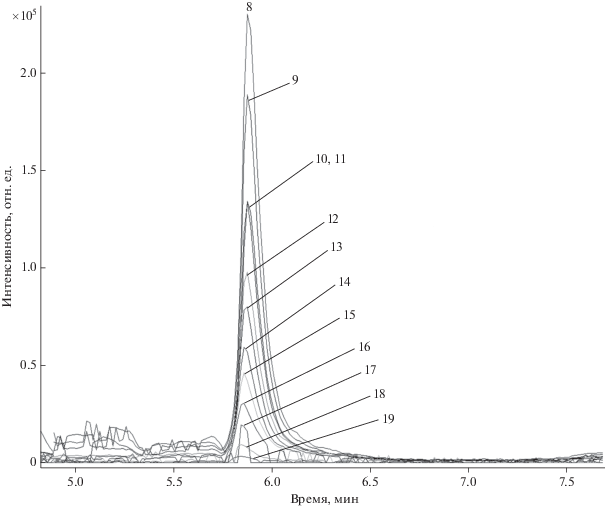
Оксиэтилированные спирты (синтанолы, ОС-20, Brij 35). Данные НПАВ представляют собой этоксилированные спирты с различной степенью этоксилирования. На рис. 9 представленны масс-хроматограммы [С5Н11О(С2Н4О)mН + Na]+ для оксиэтилированного спирта ОC-20. Видно, что степень этоксилирования составляет от 8 до 19. Гомологов и изомеров для данного препарата не обнаружено.
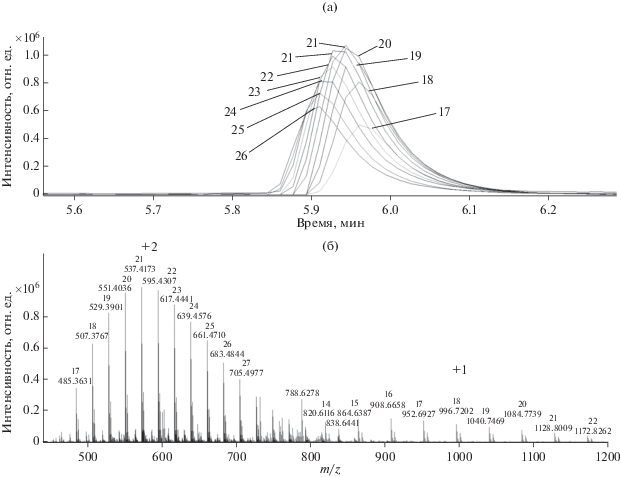
Оксиэтилированный спирт Brij-35 представляет собой лаурилоксиэтоксилат со степенью этоксилирования m = 14–27 (рис. 10). В данном случае отмечено образование как двухзарядных инов [С12Н25О(С2Н4О)mН + 2NH4]2+, так и однозарядных [С12Н25О(С2Н4О)mН + NH4]+, а также аддуктов с ионами натрия и калия.
Рис. 10.
Масс-хроматограммы Brij 35 [С12Н25О(С2Н4О)mН + 2NH4]2+ (а) и масс-спектр [С12Н25О(С2Н4О)mН + + 2NH4]2+, [С12Н25О(С2Н4О)mН + NH4]+ (б) (числом обозначена степень этилирования).
Идентификационные параметры. Картина распределения ионов-аддуктов по молекулярным массам для каждого НПАВ очень индивидуальна и характеризует определенный аналит. Хроматографический профиль НПАВ дает существенную информацию о характере молекулярно-массового распределения гомологов идентифицируемого соединения, наличии изомеров, длине полиоксиэтиленовой цепи (степени этоксилирования). Все эти характеристики служат для подтверждения наличия конкретного соединения в анализируемой пробе (табл. 2), исходя из которых дальнейшую количественную оценку можно проводить по заранее выбранным характеристическим ионам-аддуктам (m/z) (табл. 3, 4). Идентификацию НПАВ проводили по полученным масс-хроматограммам с использованием программы TargetAnalysis-1.3 (Bruker Daltonics, Германия), созданной базы данных (табл. 2) и идентификационным параметрам, которыми служили времена удерживания соединений и точность масс ионов-аддуктов.
Таблица 2.
Идентификационные параметры НПАВ, определяемых методом масс-спектрометрии высокого разрешения
| Ион | Моно-изотопная масса, m/z | Δ, м. д. | tR | смин, нг/мл |
|---|---|---|---|---|
| НПАВ – оксиэтилированные эфиры алкилфенола | ||||
| [С4H9C6H4O(C2H4O)5H + NH4]+ | 388.2694 | 3 | 3.6 | 0.1 |
| [С4H9C6H4O(C2H4O)7H + K]+ | 497.2511 | 3 | 3.6 | 0.1 |
| [С6H13C6H4O(C2H4O)5H + K]+ | 437.2300 | 4 | 3.6 | 0.1 |
| [С8H17C6H4O(C2H4O)9H + NH4]+ | 620.4368 | 4 | 5.6 | 0.1 |
| [С8H17С6Н4О(C2H4O)9Н + Nа]+ | 625.3922 | 3 | 5.6 | 0.1 |
| [С9H19C6H4O(C2H4O)12H + Са]2+ | 394.2294 | 2 | 3.8 | 0.1 |
| [С9H19C6H4O(C2H4O)13H + Са]2+ | 416.2425 | 4 | 3.8 | 0.1 |
| [С9H19C6H4O(C2H4O)12H + Mg]2+ | 386.2406 | 3 | 3.8 | 0.1 |
| [С9H19C6H4O(C2H4O)13H + Mg]2+ | 408.2537 | 5 | 3.8 | 0.1 |
| [С9H19C6H4O(C2H4O)9H + Na]+ | 639.4078 | 3 | 5.8 | 0.2 |
| [С9H19C6H4O(C2H4O)9H + K]+ | 655.3818 | 5 | 5.8 | 0.2 |
| [С9H19C6H4O(C2H4O)9H + NH4]+ | 634.4524 | 4 | 5.8 | 0.2 |
| [С9H19C6H4O(C2H4O)10H + NH4]+ | 678.4787 | 5 | 5.9 | 0.2 |
| [С10H21C6H4O(C2H4O)12H + Na]+ | 785.5021 | 5 | 6.0 | 0.5 |
| [С10H21C6H4O(C2H4O)12H + K]+ | 801.4761 | 5 | 6.0 | 0.5 |
| [С10H21C6H4O(C2H4O)12H + NH4]+ | 780.5467 | 4 | 6.0 | 0.5 |
| [С10H21C6H4O(C2H4O)16H + Са]2+ | 489.3159 | 4 | 3.9 | 0.5 |
| [С10H21C6H4O(C2H4O)15H + Са]2+ | 467.3027 | 5 | 3.9 | 0.5 |
| [С10H21C6H4O(C2H4O)16H + Mg]2+ | 481.3009 | 5 | 3.8 | 0.2 |
| [С10H21C6H4O(C2H4O)15H + Mg]2+ | 459.2878 | 4 | 3.8 | 0.2 |
| НПАВ – оксиэтилированные спирты | ||||
| [С5Н11О(С2Н4О)8Н+Na]+ | 463.2878 | 5 | 5.9 | 0.3 |
| [С12Н25О(С2Н4О)21Н+2NH4]2+ | 573.4083 | 5 | 6.0 | 0.5 |
| [С12Н25О(С2Н4О)21Н+NH4]+ | 1128.7827 | 8 | 6.0 | 0.5 |
Таблица 3.
Аналитические характеристики определения НПАВ в природных водах методом ультра-ВЭЖХ–масс-спектрометрия высокого разрешения
| Аналит | Ион | m/z | Δ, м. д. | tR | смин, нг/мл | сн, нг/мл |
ДОС, нг/мл |
Уравнения градуировочной зависимости | r2 |
|---|---|---|---|---|---|---|---|---|---|
| ОП-10 | [С10H21C6H4O(C2H4O)15H + Са]2+ | 467.3027 | 5 | 3.9 | 0.5 | 1 | 1–1000 | у = 6031.9с + 173476 | 0.9919 |
| [С10H21C6H4O(C2H4O)16H + Са]2+ | 489.3159 | 5 | 3.9 | 0.5 | 1 | 1–1000 | у = 6068.3с + 169442 | 0.9985 | |
| Тритон Х-100 | [С8H17С6Н4О(C2H4O)8Н + NH4]+ | 576.4106 | 2 | 5.6 | 0.3 | 1 | 1–100 | у = (7.7 ± 0.6) × 104с – ‒ (2.3 ± 0.3) × 105 | 0.9978 |
| [С8H17С6Н4О(C2H4O)9Н + NH4]+ | 620.4368 | 3 | 5.6 | 0.3 | 1 | 1–100 | у = (8.4 ± 0.6) × 104с – ‒ (3.3 ± 0.4) × 105 | 0.9969 |
Таблица 4.
Аналитические характеристики определения НПАВ в природных водах методом масс-спектрометрии высокого разрешения
| Аналит | Ион | m/z | Δ, м. д. | смин, нг/мл | сн, нг/мл |
ДОС, нг/мл |
Уравнение градуировочной зависимости | r2 |
|---|---|---|---|---|---|---|---|---|
| ОП-10 | [С10H21C6H4O(C2H4O)15H + Са]2+ | 467.3027 | 5 | 0.5 | 1 | 1–1000 | у = 1577.5с + 50293 | 0.9899 |
| [С10H21C6H4O(C2H4O)16H + Са]2+ | 489.3159 | 5 | 0.5 | 1 | 1–1000 | у = 1386.3с + 45890 | 0.9906 | |
| Тритон Х-100 | [С8H17С6Н4О(C2H4O)9Н + Nа]+ | 625.3922 | 4 | 0.1 | 1 | 1–100 | у = 102.1с – 657.83 | 0.9950 |
| [С8H17С6Н4О(C2H4O)9Н + K]+ | 641.3662 | 5 | 0.1 | 1 | 1–100 | у = 1054.4с – 2231 | 0.9868 |
Определение НПАВ в природных водах. Для извлечения НПАВ из природных вод использовали метод ДЖЖМЭ. В качестве экстрагента использовали хлороформ (250 мкл), в качестве диспергирующего растворителя – ацетонитрил (750 мкл). Условия микроэкстракционного концентрирования аналитов оптимизировали в соответствии с данными работ [12, 13].
В табл. 3 и 4 приведены аналитические характеристики методики определения НПАВ в природных водах методом УВЭЖХ–МСВР и МСВР для характеристических ионов-аддуктов ОП-10 и Тритона Х-100, в качестве которых выбрали полимергомологи С10 и С8 соответственно. Их производные с ионами щелочных и щелочноземельных металлов дают наиболее интенсивные пики в масс-спектре определяемых алкилфенолэтоксилатов.
При апробации методики анализировали природные воды рек Клязьма, Судогда, Колокша и Колпь (Владимирская область). Пробы отбирали на глубине 30 см на расстоянии 1 м от берега в мае 2019 г. На рис. 11 в качестве примера приведена масс-хроматограмма экстракта природной воды из р. Клязьма. Идентифицированы НПАВ класса оксиэтилированных полиэфиров, содержащие индивидуальные соединения с длиной углевородородного радикала С8, С9 и С10 со степенью этоксилирования m = 9, 10 и 16.
Рис. 11.
Масс-хроматограммы экстракта природной воды без добавки (1–8) и с добавкой НПАВ ОП-10 до концентрации 100 нг/мл (1а–8а): 1 – [С10Н21С6Н4О(С2Н4О)15Н + Ca]2+, 2 – [С10Н21С6Н4О(С2Н4О)16Н + Ca]2+, 3 – [С8Н17С6Н4О(С2Н4О)9Н + NH4]+, 4 – [С8Н17С6Н4О(С2Н4О)9Н + Na]+, 5 – [С9Н19С6Н4О(С2Н4О)9Н + NH4]+, 6 – [С9Н19С6Н4О(С2Н4О)10Н + NH4]+, 7 – [С9Н19С6Н4О(С2Н4О)9Н + Na]+, 8 – [С9Н19С6Н4О(С2Н4О)9Н + K]+.
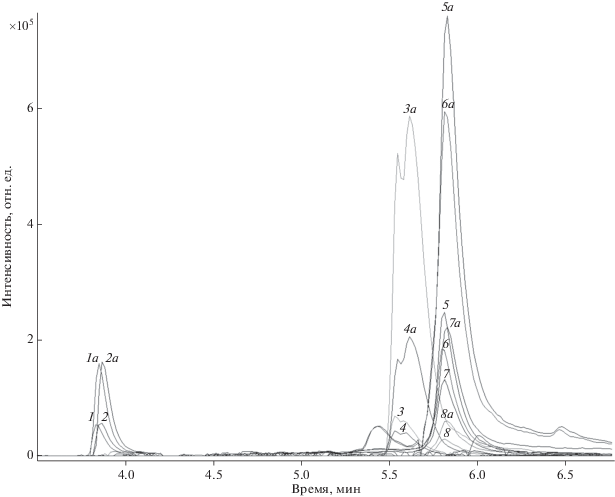
В табл. 5 приведены результаты идентификации НПАВ в различных водах. Отражены относительные (нормированные) концентрации идентифицированных компонентов. В процессе хроматографического разделения и ионизации электрораспылением полимергомологи образуют аддукты с различными ионами, наличие которых позволяет избежать ошибок при идентификации НПАВ.
Таблица 5.
Результаты идентификации НПАВ в природных водах (n = 3, P = 0.95)
| Обнаруженные ионы | Название реки и относительные (нормированные) концентрации | |||
|---|---|---|---|---|
| Колокша | Судогда | Клязьма | Колпь | |
| [С8Н17С6Н4О(С2H4О)9Н + NH4]+ | 2.24 | 2.04 | 1.00 | 1.55 |
| [С8Н17С6Н4О(С2H4О)9Н + Na]+ | 2.68 | 2.11 | 1.00 | 1.04 |
| [C9H19C6H4O(C2H4O)9H + Na]+ | 1.36 | 3.87 | – | 1.00 |
| [C9H19C6H4O(C2H4O)9H + K]+ | 3.54 | 2.99 | – | 1.00 |
| [C9H19C6H4O(C2H4O)9H + NH4]+ | 13.97 | 52.35 | 1.00 | 14.80 |
| [C9H19C6H4O(C2H4O)10H + NH4]+ | 13.88 | 49.39 | 1.00 | 138.62 |
| [C10H21C6H4O(C2H4O)15H + Ca]2+ | 2.57 | 1.45 | 3.03 | 1.00 |
| [C10H21C6H4O(C2H4O)16H + Ca]2+ | 2.63 | 1.40 | 2.18 | 1.00 |
Для количественного анализа использовали метод добавок на уровне 5 нг/мл НПАВ (рис. 11). Полученные результаты представлены в табл. 6.
Таблица 6.
Результаты определения НПАВ в природных водах (n = 3, P = 0.95)
| Обнаруженные НПАВ | Название реки и содержание НПАВ в нг/мл (sr) | |||
|---|---|---|---|---|
| Колокша | Судогда | Клязьма | Колпь | |
| С8Н17С6Н4О(С2Н4О)9Н | 33 ± 8 (0.10) | 15 ± 5 (0.12) | 8 ± 3 (0.15) | 21 ± 6 (0.09) |
| C9H19C6H4O(C2H4O)9H | 143 ± 21 (0.06) | 288 ± 32 (0.04) | 40 ± 9 (0.08) | 140 ± 18 (0.06) |
| C10H21C6H4O(C2H4O)16H | 122 ± 17 (0.05) | 21 ± 5 (0.08) | 63 ± 9 (0.05) | 75 ± 11 (0.15) |
* * *
Таким образом, в анализируемых пробах природных вод обнаружены оксиэтилированные алкилфенолы с длиной углеводородного радикала С8, С9, С10 и степенью этоксилирования m = 9 и 16. Оценка содержаний с использованием метода стандартной добавки позволяет сделать вывод, что обнаруженные НПАВ с углеводородным радикалом С9 и со степенью этоксилирования m = 9 присутствуют в пробах всех рек, кроме р. Клязьма, и НПАВ с углеводородном радикалом С10 и степенью этоксилирования m = 16 в присутствуют р. Колокша в количествах, превышающих нормативные значения ПДК (0.1 мг/л) (рассчитанные значения концентрации НПАВ изменялись от 0.12 до 0.29 мг/л).
Список литературы
РД 52.24.439-2007. Массовая концентрация неионогенных синтетических поверхностно-активных веществ и полиэтиленгликолей в водах. Методика выполнения измерений экстракционно-фотометрическим методом. Ростов н/Д.: Росгидромет, 2007, 24 с.
ПНД Ф 14.1:2.247-07. Количественный химический анализ вод. Методика выполнения измерений массовых концентраций неионогенных синтетических поверхностно-активных веществ (СПАВ) в пробах природных и сточных вод нефелометрическим методом. М.: ФГБУ “ФЦАО”, 2016, 12 с.
ПНД Ф 14.1:2:4.194-2003. Количественный химический анализ вод. Методика измерений массовой концентрации неионогенных поверхностно-активных веществ (НПАВ) в пробах питьевых, природных и сточных вод экстракционно-фотометрическим методом в присутствии анионоактивных ПАВ (АПАВ). М.: ФГБУ “ФЦАО”, 2012, 14 с.
Levsen K., Wagner-Redeker W., Schafer K.H., Dobberstein P. On-line liquid chromatography-mass spectrometry analysis of non-ionic surfactants // J. Chromatogr. A. 1985. V. 323. P. 135.
Petrovic M., Fernandez-Alba A.R., Borrull F., Marce R.M., Mazo E.G., Barcelo D. Occurrence and distribution of nonionic surfactants, their degradation products, and linear alkylbenzene sulfonates in coastal waters and sediments in Spain // Environ. Toxicol. Chem. 2002. V. 21. № 1. P. 37.
Barco M., Planas C., Palacios O., Ventura F., Rivera J., Caixach J. Simultaneous quantitative analysis of anionic, cationic, and nonionic surfactants in water by electrospray ionization mass spectrometry with flow injection analysis // Anal. Chem. 2003. V. 75. P. 5129.
Lara Martin P.A., Gomez-Parra A., Gonzalez-Mazo E. Development of a method for the simultaneous analysis of anionic and non-ionic surfactants and their carboxylated metabolites in environmental samples by mixed-mode liquid chromatography-mass spectrometry // J. Chromatogr. A. 2004. V. 1137. P. 188.
Loos R., Hanke G., Umlauf G., Eisenreich S.J. LC–MS–MS analysis and occurrence of octyl- and nonylphenol, their ethoxylates and their carboxylates in Belgian and Italian textile industry, waste water treatment plant effluents and surface waters // Chemosphere. 2006. V. 66. P. 690.
Crescenzi C., Di Corcia A., Samperi R. Determination of nonionic polyethoxylate surfactants in environmental waters by liquid chromatography/electrospray mass spectrometry // Anal. Chem. 1995. V. 67. P. 1797.
Cantero M., Rubio S., Perez-Bendito D. Determination of non-ionic polyethoxylated surfactants in sewage sludge by coacervative extraction and ion trap liquid chromatography–mass spectrometry // J. Chromatogr. A. 2004. V. 1046. P. 147.
Gomez V., Ferreres L., Pocurull E., Borrull F. Determination of non-ionic and anionic surfactants in environmental water matrices // Talanta. 2011. V. 84. P. 859.
Амелин В.Г., Большаков Д.С., Третьяков А.В. Дисперсионная жидкостно-жидкостная микроэкстракция и твердофазная экстракция при извлечении полярных пестицидов из природных вод и определении их методом мицеллярной электрокинетической хроматографии // Журн. аналит. химии. 2013. Т. 68. № 5. С. 430.
Амелин В.Г., Лаврухин Д.К., Третьяков А.В. Дисперсионная жидкостно-жидкостная микроэкстракция при определении гербицидов – производных мочевины в природных водах методом ВЭЖХ // Журн. аналит. химии. 2013. Т. 68. № 9. С. 908.
Большаков Д.С., Амелин В.Г., Третьяков А.В. Определение гербицидов и их метаболитов в природных водах методом капиллярного зонного электрофореза в сочетании с дисперсионной жидкостно-жидкостной микроэкстракцией и on-line концентрированием // Журн. аналит. химии. 2014. Т. 69. № 1. С. 77.
Амелин В.Г., Большаков Д.С., Андоралов А.М. Скрининг и определение пестицидов различных классов в природной воде без пробоподготовки методом ультра ВЭЖХ–квадруполь-времяпролетной масс-спектрометрии высокого разрешения // Журн. аналит. химии. 2018. Т. 73. № 3. С. 220.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии