

Журнал аналитической химии, 2021, T. 76, № 2, стр. 143-150
Изучение метаболизма нового ноотропного препарата – унифирама методом ультра высокоэффективной жидкостной хроматографии‒масс-спектрометрии высокого разрешения
А. З. Темердашев a, *, М. О. Зорина a, Е. В. Дмитриева a, А. А. Азарян a
a Кубанский государственный университет
350040 Краснодар, ул. Ставропольская, 149, Россия
* E-mail: TemerdashevAZ@gmail.com
Поступила в редакцию 09.06.2020
После доработки 27.06.2020
Принята к публикации 17.08.2020
Аннотация
Рассмотрен метаболизм нового ноотропного препарата – унифирама, который предположительно обладает существенно большей активностью по сравнению с пирацетамом и проходит клинические испытания в качестве средства для улучшения памяти, предотвращения болезни Альцгеймера, синдрома дефицита внимания и различных форм деменции. Предложены структуры двух потенциальных метаболитов унифирама. На основе интерпретации полученных масс-спектров ионов-прекурсоров и ионов-продуктов показано применение методологии нецелевого скрининга с использованием ультра высокоэффективной жидкостной хроматографии–масс-спектрометрии высокого разрешения в целях их обнаружения. Установлено, что определение предполагаемых метаболитов унифирама возможно только с применением ферментативного гидролиза, в то время как минеральный гидролиз приводит к полному разрушению как самого унифирама, так и предполагаемых метаболитов. Использование предложенных условий хроматографирования и масс-спектрометрического детектирования делает возможным обнаружение метаболитов унифирама в течение шести дней после однократного употребления 10 мг вещества.
Унифирам (DM232) (схема 1 ) относится к новому поколению ноотропов, для которых заявленная эффективность существенно превышает широко распространенный пирацетам [1–5]. Минимальная эффективная доза для унифирама в условиях индуцированной скополамином амнезии составляет всего 0.001 мг/кг [1], в то время как для пирацитама это значение составляет 30 мг/кг.
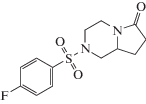
Схема 1 . Структурная формула унифирама.
Помимо влияния унифирама на улучшение памяти, он относится к ряду перспективных препаратов для предотвращения болезни Альцгеймера, синдрома дефицита внимания и различных форм деменции. При проведении реальных испытаний и в клинической практике используют большие дозировки унифирама (0.3–10 мг/кг), в ряде случаев в сочетании с другим перспективным ноотропным препаратом – сунифирамом [2].
Помимо сведений об эффективности препаратов, применяемых для лечения заболеваний центральной нервной системы, а также для улучшения памяти и концентрации внимания, интерес представляет изучение их метаболизма в организме человека.
Среди ноотропов наибольшее распространение и популярность получил пирацетам, который согласно литературным данным практически полностью выводится из организма в нативном виде [6–15]. В отношении унифирама известно, что он выводится в нативном виде частично и имеет очень узкое окно обнаружения [16].
На сегодняшний день данные о потенциальных метаболитах унифирама отсутствуют, что обусловливает актуальность их поиска на основе, в частности, эмпирических и полуэмпирических данных о метаболизме ксенобиотиков в организме человека [17].
Цель настоящей работы – поиск метаболитов унифирама в моче путем нецелевого скрининга с применением ультра высокоэффективной жидкостной хроматографии–масс-спектрометрии высокого разрешения (УВЭЖХ–МСВР), ферментативного гидролиза и метода “разбавил и вколол”.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объект исследования. В эксперименте участвовал доброволец (возраст 21 год), давший информированное устное согласие после ознакомления с планом и условиями исследования. До употребления унифирама образцы мочи собирали в течение двух дней (утром и вечером), после чего доброволец однократно употреблял 10 мг унифирама. Затем образцы мочи собирали в течение 10 дней (утром и вечером). Обязательными условими при выполнении эксперимента являлись отсутствие заболеваний, требующих употребления медикаментов, минимизация факторов стресса и соблюдение диеты на протяжении эксперимента, что позволяло снизить вариативность данных.
Реактивы и реагенты. Стандартный образец унифирама приобретали у “Shanghai Soyoung Biotech. Inc.” (Китай). Метанол “для ВЭЖХ” приобретали у “J.T. Baker” (США). Воду сопротивлением 18.2 МОм см получали с использованием системы Milli-Q Simplicity (Millipore, Франция). Ацетат аммония ч. д. а. и муравьиную кислоту х. ч. для подвижной фазы приобретали у компаний “Вектон” (Россия) и “Acros Organics” (США) соответственно. Гидролиз проб проводили с использованием глюкуронидазы E. Coli и H. Pomatia, приобретенными у компаний “Roche” (Германия) и “Sigma-Aldrich” (США), для минерального гидролиза применяли соляную кислоту х. ч. (Вектон, Россия).
Условия УВЭЖХ–МСВР-анализа. Для поиска вероятных метаболитов использовали квадруполь-времяпролетный масс-спектрометр Bruker MaXis Impact, оснащенный источником электрораспылительной ионизации, соединенный с УВЭ-ЖХ-системой Bruker Elute под управлением программного обеспечения Bruker Compass HyStar 4.1. Полученные данные анализировали с использованием программного обеспечения Bruker Data Analysis 4.4. Для хроматографического разделения применяли аналитическую колонку Bruker Intensity Solo-2 C18 (75 × 2.1 мм, 2.6 мкм), термостатируемую при 40°С и оснащенную предохранительной колонкой с идентичным сорбентом. Условия масс-спектрометрического детектирования и градиентного элюирования приведены в табл. 1, 2.
Таблица 1.
Условия градиентного элюирования
| Время, мин | Подвижная фаза А (5 мМ водный раствор формиата аммония с 1% метанола и 0.05% муравьиной кислоты), % | Подвижная фаза В (5 мМ метанольный раствор формиата аммония и 0.05% муравьиной кислоты), % | Скорость потока подвижной фазы, мл/мин |
|---|---|---|---|
| 0.0 | 96.0 | 4.0 | 0.20 |
| 0.1 | 96.0 | 4.0 | |
| 1.0 | 81.7 | 18.3 | |
| 2.5 | 50.0 | 50 | 0.22 |
| 14.0 | 0.1 | 99.9 | 0.40 |
| 16.0 | 0.1 | 99.9 | 0.48 |
| 16.1 | 96.0 | 4.0 | |
| 19.0 | 96.0 | 4.0 | |
| 19.1 | 96.0 | 4.0 | 0.20 |
| 20.0 | 96.0 | 4.0 |
Таблица 2.
Условия детектирования унифирама и его потенциальных метаболитов
| Параметр | Значение |
|---|---|
| Источник ионизации | Электрораспылительная ионизация |
| Полярность | Положительная |
| Напряжение на капилляре, В | 3500 |
| Напряжение на экстрагирующей линзе, В | 400 |
| Температура источника ионизации, °С | 200 |
| Давление газа-распылителя, МПа | 0.2 |
| Расход газа-осушителя, л/мин | 8 |
| Диапазон сканирования масс, Да | 140–600 |
| Скорость сканирования, Гц | 2 |
| Газ-мишень | Азот |
| Давление газа-мишени, мТорр | 1.5 |
| Энергия соударительной диссоциации, эВ | 20 |
Подготовленные к анализу образцы термостатировали в автоматическом дозаторе при 5°С.
Пробоподготовка. Подготовку проб к анализу проводили путем простого разбавления проб, а также ферментативного и минерального гидролиза, используя ферменты E. coli и H. pomatia. Известно, что после ферментативного гидролиза с использованием E. coli происходит деконъюгация исключительно глюкуронидов, в то время как применение H. pomatia позволяет деконъюгировать как глюкурониды, так и сульфаты благодаря наличию арилсульфатазы, однако эффективность фермента сильно зависит не только от температуры инкубации проб после его добавления, но и от значения рН образца. Так, оптимальное значение рН для фермента E. coli лежит в диапазоне 6.5–6.8, в то время как наибольшая активность H. pomatia достигается при рН 4.5. В обоих случаях для поддержания необходимого значения рН используют буферные растворы – фосфатный и ацетатный соответственно. Для реализации этого подхода к аликвоте пробы мочи объемом 3 мл добавляли 1 мл буферного раствора и 30 мкл фермента, тщательно перемешивали, после чего инкубировали при 50°С в течение 1 ч, охлаждали до комнатной температуры, отбирали 1 мл гидролизата и центрифугировали в течение 10 мин в пробирке типа Эппендорф емк. 1.5 мл при 10 000 об/мин, после чего супернатант переносили в стеклянные виалы для последующего анализа.
Минеральный гидролиз позволяет разрушать практически все возможные виды конъюгатов, что является его несомненным преимуществом, однако ферментативные способы являются гораздо более мягкими и щадящими как по отношению к образцу, так и к прибору (при дальнейшем анализе полученных проб). Минеральный гидролиз проводили следующим образом: аликвоту мочи объемом 3 мл помещали в стеклянную пробирку с завинчивающейся крышкой, добавляли 500 мкл конц. HCl, герметично закрывали, перемешивали, после чего инкубировали при 80°С в течение 1 ч, затем отбирали 1.5 мл гидролизата, переносили в пробирку типа Эппендорф через воронку, заполненную гидрокарбонатом натрия (для нейтрализации кислоты), и центрифугировали в течение 10 мин при 10 000 об/мин с последующим переносом супернатанта в стеклянную виалу для анализа.
Для разбавления проб использовали смесь 0.1%-ный раствор муравьиной кислоты в воде–ацетонитрил (50 : 50, v/v) следующим образом: 200 мкл пробы мочи помещали в пробирку типа Эппендорф и добавляли 800 мкл разбавителя, перемешивали на вортексе в течение 1 мин, после чего центрифугировали в течение 10 мин при 10 000 об/мин.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Учитывая среднюю полярность унифирама, применение обращено-фазовой УВЭЖХ является оптимальным решением. Кроме того, исходя из структуры нативного вещества и базируясь на известных особенностях протекания метаболизма в организме человека, можно предположить структуры некоторых метаболитов (рис. 1).
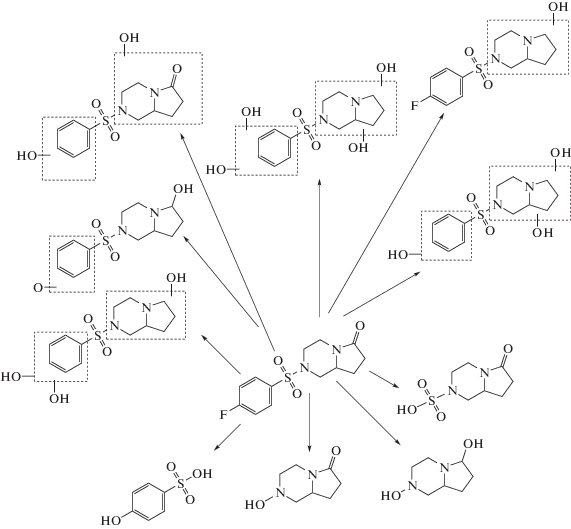
Наши предположения о структурах метаболитов основаны как на описанных в работе [17] закономерностях протекания основных стадий метаболизма ксенобиотиков, так и на расчетах с применением программно-аппаратных комплексов, таких как MetWorks (Thermo Scientific) [18] и GLORY (Hamburg University) [19]. Следует отметить, что, несмотря на постоянное совершенствование подобных программных продуктов, их точность по-прежнему остается относительно невысокой. Кроме того, описанные выше подходы зачастую позволяют сделать предположение лишь о наиболее очевидных метаболитах. В связи с этим применение метода in vitro по-прежнему остается наиболее надежным способом решения подобных задач. Тем не менее применение программных продуктов для прогнозирования путей метаболизма представляет интерес из-за отсутствия в ряде случаев возможности проведения экспериментов с использованием клеточных культур. Однако такой подход требует ряда дополнительных предосторожностей для снижения риска возникновения ложноположительного результата. Среди них – требования к отсутствию пиков эндогенных соединений с близкими к предполагаемым метаболитам значениями параметров удерживания, а также минимизация погрешности определения масс (менее 5 ppm для режима сканирования ионов-прекурсоров и не более 10 ppm в режиме сканирования ионов-продуктов). Дополнительным и немаловажным способом проверки корректности первичных выводов является также оценка изотопного распределения, особенно для таких соединений, как унифирам, имеющих несколько разных гетероатомов в исходной структуре.
Исходя из описанных выше соображений, пробы мочи анализировали до и после употребления унифирама с использованием нескольких режимов сканирования: автоматического выбора ионов-прекурсоров с последующим получением спектра ионов продуктов (auto MS/MS), сканирования полного спектра ионов-прекурсоров (full MS), а также с применением режима широкополосного пропускания ионов с последующей соударительной диссоциацией (bbCID) с привлечением библиотек Target Screener, содержащих спектры ряда ксенобиотиков и их метаболитов и пестицидов для минимизации возникновения ложноположительных результатов.
После анализа хроматограмм (рис. 2) с применением описанных выше критериев удалось выделить два потенциальных метаболита унифирама (рис. 3). Анализируя полученные ионы-прекурсоры и соответствующие им ионы-продукты, можно сделать вывод о том, что образование дефтордигидроксилированного метаболита (М1) происходит указанным на рис. 3 путем, а не за счет замещения атома фтора на гидроксильную группу, о чем свидетельствует наблюдаемая фрагментация (рис. 4, 5). Положение гидроксильных групп в приведенной схеме фрагментации является допущением и приведено в целях удобства расчета масс.
Рис. 2.
Хроматограмма образца мочи (четвертый день после употребления унифирама), содержащего предполагаемые метаболиты, по выделенным протонированным молекулярным ионам (М1 – m/z 313.0853, М2 – m/z 315.0809).



Рис. 5.
Предлагаемая интерпретация зарегистрированного масс-спектра вероятного метаболита унифирама М1 (ТМ – теоретическая масса, НМ – наблюдаемая масса, ООМ – ошибка определения массы).

Исходя из точной массы второго потенциального метаболита и закономерностей протекания метаболизма ксенобиотиков можно сделать вывод о том, что это моногидроксилированный метаболит или сформировавшийся в результате деалкилирования и карбонилирования метаболит. В пользу последнего варианта свидетельствует наблюдаемая фрагментация (рис. 6, 7).

Рис. 7.
Предлагаемая интерпретация зарегистрированного масс-спектра вероятного метаболита унифирама М2 (ТМ – теоретическая масса, НМ – наблюдаемая масса, ООМ – ошибка определения массы).
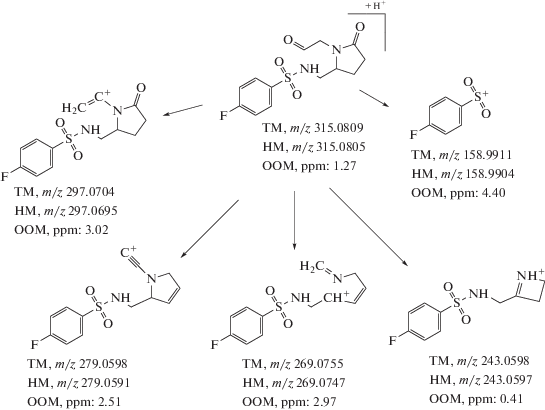
Все предположения базируются на соотнесении наблюдаемых и теоретических масс и их изотопного распределения (табл. 3).
Таблица 3.
Теоретические и наблюдаемые массы унифирама и предполагаемых метаболитов, полученные с применением УВЭЖХ–МСВР с электрораспылительной ионизацией в режиме регистрации положительных ионов
| Компонент | Элементная композиция | Теоретическая масса протонированного иона, m/z | Наблюдаемая масса протонированного иона, m/z | Ошибка определения массы, ppm | Время удерживания, мин |
|---|---|---|---|---|---|
| Унифирам | С13Н15FN2O3S | 299.0860 | 299.0865 | –1.67 | 5.23 |
| М1 | С13Н16N2O5S | 313.0853 | 313.0854 | –0.32 | 3.65 |
| М2 | С13Н15FN2O4S | 315.0809 | 315.0805 | 1.27 | 4.82 |
Полученные результаты справедливы в отношении проб после ферментативного гидролиза. Образцы, подвергшиеся минеральному гидролизу, исключили из рассмотрения ввиду их деградации в процессе инкубирования с соляной кислотой, вероятно, приводившей к разрушению аналитов. В ряде случаев не представлялось возможным обнаружить даже стероидные гормоны, также склонные к деградации в условиях высокотемпературного и продолжительного минерального гидролиза.
К сожалению, зачастую применение масс-спектрометрии не позволяет установить точное положение заместителя (в частности, гидроксильной группы), а достоверное подтверждение возможно исключительно лишь после препаративного выделения предполагаемых метаболитов в достаточных количествах с дальнейшим описанием методами ЯМР- и ИК-спектроскопии (помимо масс-спектрометрии) или путем встречного синтеза предполагаемых метаболитов и сопоставления их параметров удерживания и масс-спектральных характеристик с наблюдаемыми в образце.
Методология нецелевого поиска метаболитов в данном случае построена на постепенном увеличении элюирующей силы подвижной фазы при использовании хроматографического разделения с быстрой регистрацией спектра ионов-прекурсоров и ионов-продуктов. Для повышения экспрессности анализа, а также уменьшения ширины хроматографических пиков помимо градиента по составу подвижной фазы (ПФ) возможно также изменение скорости еe подачи. Подобная плавная развертка градиента позволяет уменьшить количество коэлюирующихся соединений и обеспечить лучшую работу масс-спектрометра в целях нецелевого поиска соединений ввиду меньшего гашения ионизации, для чего в случае масс-спектрометрии высокого разрешения оптимальным выбором является режим auto MS/MS. В случае применения этого режима особое внимание следует уделить критериям выбора ионов-прекурсоров и условиям их динамического исключения. Для проведения экспериментов в рамках поставленной нами задачи было принято решение использовать семь наиболее интенсивных ионов-прекурсоров с их динамическим исключением после трех циклов сканирования и возможностью повторного сканирования при выполнении одного или всех следующих условий: интенсивность иона возрастает до 1 млн имп/с или по прошествии 30 с после последнего сканирования данного иона. Весь цикл сканирования ионов-продуктов и ионов-прекурсоров осуществлялся за 2 с, что при оптимизации метода на быстрое разделение могло бы привести к возникновению ложноотрицательных результатов из-за низкой скорости сканирования, в то время как ее существенное повышение привело бы к значительному падению интенсивности сигналов.
Применение режима сканирования full MS, в свою очередь, часто позволяет добиться максимальной чувствительности для QTOF-систем (ввиду отсутствия потерь в процессе соударительной ионизации), что немаловажно при оценке временных окон обнаружения потенциальных метаболитов после употребления нативного вещества после подтверждения их структуры.
В то же время градиент по скорости подачи подвижной фазы может негативно сказаться на чувствительности анализа, поскольку параметры температуры источника ионизации, напряжения на капилляре и расхода газов на распыление и осушение напрямую зависят от скорости ее потока.
При оптимизации условий качественного и количественного анализа уточнение окон обнаружения, как правило, проводят рутинным для лаборатории методом. В предложенных нами условиях определение нативного унифирама в моче возможно в течение до трех суток с момента его однократного употребления с использованием процедуры разбавления проб мочи. Обнаружение М2 возможно с третьих по пятые сутки, М1 – с третьего по шестой день после применения ферментативного гидролиза.
Исследования проводили при поддержке проектов Госзадания Минобрнауки РФ (проект FZEN-2020-0022) и РФФИ, проект № 19-43-230004 р_а, с участием специалистов ЦКП “Эколого-аналитический центр” Кубанского государственного университета, уникальный идентификатор центра RFMEFI59317Х0008.
Список литературы
Guandalini L., Martino M.V., Di Cesare Mannelli L., Bartolucci G., Melani F., Malik R., Dei S., Floriddia E., Manetti D., Orlandi F., Teodori E., Ghelardini C., Romanelli M.N. Substituted piperazines as nootropic agents: 2- or 3-phenyl derivatives structurally related to the cognition-enhancer DM235 // Bioorg. Med. Chem. Lett. 2015. V. 25. № 8. P. 1700.
Martini E., Norcini M., Ghelardini C., Manetti D., Dei S., Guandalini L., Melchiorre M., Pagella S., Scapecchi S., Teodori E., Romanelli M.N. Design, synthesis and preliminary pharmacological evaluation of new analogues of DM232 (unifiram) and DM235 (sunifiram) as cognition modulators // Bioorg. Med. Chem. Lett. 2008. V. 16. № 23. P. 10034.
Martino M.V., Guandalini L., Di Cesare Mannelli L., Menicatti M., Bartolucci G., Dei S., Manetti D., Teodori E., Ghelardini C., Romanelli M.N. Piperazines as nootropic agents: new derivatives of the potent cognition-enhancer DM235 carrying hydrophilic substituents // Bioorg. Med. Chem. Lett. 2017. V. 25. № 6. P. 1795.
Mondadori C., Ducret T., Häusler A. Elevated corticosteroid levels block the memory-improving effects of nootropics and cholinomimetics // Psychopharmacology. 1992. V. 108. № 1−2. P. 11.
Galeotti N., Ghelardini C., Pittaluga A., Pugliese A.M., Bartolini A., Manetti D., Romanelli M.N., Gualtieri F. AMPA-receptor activation is involved in the antiamnesic effect of DM232 (unifiram) and DM235 (sunifiram) // Naunyn-Schmiedeberg’s Arch Pharmacol. 2003. V. 368. № 6. P. 538.
Mondadori C. In search of the mechanism of action of the nootropics: new insights and potential clinical implications // Life Sci. 1994. V. 55. № 25−26. P. 2171.
Malik R., Gupta R., Srivastava S., Choudhary B.S. Design, synthesis and biological evaluation of selected 3‑[3-(amino) propoxy] benzenamines as acetylcholinesterase inhibitors // J. Biomol. Struct. Dyn. 2016. V. 35. № 11. P. 2382.
Giurgea C., Salama M. Nootropic drugs // Prog. Neuro-Psychopharm. 1977. V. 1. № 3-4. P. 235.
Gouliaev A.H., Senning A. Piracetam and other structurally related nootropics // Brain Res. Rev. 1994. V. 19. № 2. P. 180.
Schindler U. Pre-clinical evaluation of cognition enhancing drugs // Prog. Neuro-Psychopharm. Biol. Psychiatry. 1989. V. 13. P. S99.
Romanelli M.N., Galeotti N., Ghelardini C., Manetti D., Martini E., Gualtieri F. Pharmacological characterization of DM232 (unifiram) and DM235 (sunifiram), new potent cognition enhancers // CNS Drug Rev. 2006. V. 12. № 1. P. 39.
Mondadori C., Petschke F., Häusler A. The effects of nootropics on memory: New aspects for basic research // Pharmacopsychiatry. 1989. V. 22. P. 102.
Mondadori C. The pharmacology of the nootropics; New insights and new questions // Behav. Brain Res. 1993. V. 59. № 1–2. P. 1.
Malik R., Sangwan A., Saihgal R., Jindal D. P., Piplani P. Towards better brain management: Nootropics // Curr. Med. Chem. 2007. V. 14. № 2. P. 123.
Gualtieri F. Unifi nootropics from the lab to the web: A story of academic (and industrial) shortcomings // J. Enzyme Inhib. Med. Chem. 2015. V. 31. № 2. P. 187.
Дмитриева Е.В., Темердашев А.З., Азарян А.А., Гашимова Э.М. Применение УВЭЖХ-МС/МС для определения в моче некоторых анаболических агентов и ноотропов // Аналитика и контроль. 2018. Т. 22. № 1. С. 28.
Peters F.T., Meyer M.R. In vitro approaches to studying the metabolism of new psychoactive compounds // Drug Test. Anal. 2011. V. 3. № 7–8. P. 483.
Wank J., Wagner-Redeker W., Taylor A., Herbold R., Lassahn Paul-Gerhard. High-resolution, accurate mass measurements and metabolite identification: an automated approach using fragment prediction in combination with fragment ion search (FISh) / Thermo Fisher Scientific Application. Note 537, 2016. P. 1.
De Bruyn Kops C., Stork C., Šícho M., Kochev N., Svozil D., Jeliazkova N., Kirchmair J. GLORY: Generator of the structures of likely cytochrome P450 metabolites based on predicted sites of metabolism // Front Chem. 2019. V. 7. № 402. P. 1.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии