

Журнал аналитической химии, 2021, T. 76, № 3, стр. 218-226
Многоэлементный анализ нефти методами АЭС-ИСП и МС-ИСП с использованием микроволновой пробоподготовки
О. Н. Гребнева-Балюк a, *, И. В. Кубракова a, О. А. Тютюнник a, С. Ю. Лапшин a, Д. В. Пряжников a
a Институт геохимии и аналитической химии им. В.И. Вернадского Российской академии наук
119991 Москва, ул. Косыгина, 19, Россия
* E-mail: grebneva@geokhi.ru
Поступила в редакцию 02.03.2020
После доработки 02.09.2020
Принята к публикации 20.09.2020
Аннотация
С целью получения детальной информации об элементном составе нефти разработаны методики АЭС-ИСП- и МС-ИСП-определения широкого круга элементов в нефти после микроволнового разложения образцов. Установлена возможность и выбраны условия микроволнового разложения образцов легкой и сверхвязкой нефти массой до 1000 мг. Исследовано влияние остаточной кислотности и остаточного углерода на формирование аналитического сигнала в АЭС-ИСП и МС-ИСП (высокое разрешение). Показана возможность АЭС-ИСП-определения 20 элементов на уровне n × 10–2–m × 102 мкг/г и МС-ИСП-определения 42 элементов на уровне n × 10–2–m × 102 нг/г.
Данные об элементном составе природных углеводородных систем важны для фундаментальных геохимических исследований, в том числе для понимания механизмов глубинных геохимических процессов, путей переноса и накопления металлов и условий рудообразования [1]. Углеводородные системы играют важную роль в транспорте рудного вещества и формировании рудных залежей: нафтеновые, парафиновые и ароматические углеводороды, входящие в состав мантийных флюидов, устойчивы на больших глубинах (свыше 300 км) в условиях высоких температур и давлений (7000 МПа, 1000°С) и могут участвовать в совместном переносе металлов и органического вещества при минералообразовании [2]. Об этом свидетельствуют находки тяжелых углеводородов в составе газовой фазы из расплавных включений в минералах, а также выявленная пространственная сближенность и закономерная зональность в размещении нефтяных и рудных месторождений [1]. Элементный и изотопный состав углеводородных систем не менее важен для выяснения механизмов биогенного и абиогенного нефтеобразования с целью решения важнейшей геохимической проблемы происхождения нефти и газа [3].
Элементный состав нефти, в первую очередь содержание никеля и ванадия [3–5], характеризует возраст и происхождение нефти, исходные регионы и пути миграции углеводородов; эти данные используются при поиске месторождений нефти [6]. Содержание других элементов позволяет учесть важные прикладные аспекты, связанные с использованием нефти как энергетического ресурса (экологические риски при ее добыче и переработке, эффективность использования в качестве топлива), предопределяет технологию переработки, а также характер добавок, улучшающих свойства нефтепродуктов при транспортировке [7]. Таким образом, желательно, чтобы информация о составе нефти была максимально полной.
В обзорах [7, 8] отмечено, что основными методами определения следовых элементов в природных углеводородных системах являются атомно-абсорбционная спектрометрия с электротермической атомизацией, пламенная атомно-абсорбционная спектрометрия, атомно-эмиссионная спектрометрия с индуктивно связанной плазмой (АЭС-ИСП) и масс-спектрометрия с индуктивно связанной плазмой (МС-ИСП).
Перечисленные методы ориентированы на анализ жидких сред, которые могут быть получены путем разбавления исходного материала органическими растворителями [9–11], а также за счет образования микроэмульсий, стабилизированных поверхностно-активными веществами [12–14], или окислительным разложением органической матрицы [15]. В последнем случае применяют озоление или разложение кислотами [7]. Однако длительность всех этих операций, а также возможные потери или загрязнения, характерные для открытых систем, приводят к необходимости использования закрытых сосудов, обеспечивающих достаточно быстрое протекание процессов, например автоклавов с резистивным нагревом [16] или микроволновых (МВ) систем.
Микроволновую пробоподготовку используют для минерализации различных углеродсодержащих объектов, включая нефти различного состава. Обычно разлагают образцы массой 100–250 мг, что обусловлено значительным давлением, которое возникает в закрытой системе при окислении органических веществ, ограничивающим возможности метода. Добавление к азотной кислоте других окислителей (пероксида водорода) может способствовать разложению, но приводит к дополнительному росту давления на начальном этапе [17–19]. Тем не менее поиск возможностей увеличения массы пробы, обеспечивающего снижение пределов определения и расширение круга аналитов, продолжается. Недавно появились работы [20, 21], в которых сообщается об увеличении массы образца нефти до 1000 мг, что позволило определить редкоземельные элементы (РЗЭ). При этом авторы использовали сложную и дорогостоящую МВ-систему, состоящую из реакционной камеры с пятью негерметичными кварцевыми сосудами объемом 40 мл, давление в которой (до 40 атм) создается напуском аргона. Количественный анализ проводили с использованием квадрупольного масс-спектрометра, дооснащенного ультразвуковым распылителем. Кроме группы РЗЭ, другие элементы не определяли [21].
Снижение пределов определения при анализе нефтепродуктов возможно также за счет концентрирования [22], однако введение дополнительной стадии существенно усложняет анализ и увеличивает его продолжительность.
Цель данной работы – на основе использования новых технических возможностей серийных лабораторных МВ-систем разработать условия пробоподготовки, обеспечивающие определение широкого круга элементов в различных видах нефти многоэлементными спектральными методами с индуктивно связанной плазмой (АЭС-ИСП и МС-ИСП с высоким разрешением).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Оборудование, материалы. Бидистиллированную воду и конц. HNO3 ос. ч. (Химмед, Россия) дополнительно очищали с помощью системы перегонки без кипения BSB-939-IR (Berghof, Германия).
Для МВ-разложения проб использовали лабораторную систему MARS6 (CEM Corp., США) (максимальная выходная мощность 1800 Вт, частота 2.45 ГГц) с сосудами iPrep (объем сосуда 110 мл, максимальная температура 280°С). На рис. 1 приведены полученные нами экспериментальные данные об изменении контролируемых параметров процесса – температуры (Т), мощности (Р) и времени (t).
Рис. 1.
Зависимости температуры (1) и подаваемой мощности (2) от времени при разложении сверхвязкой нефти СВН (масса пробы 0.5 г).

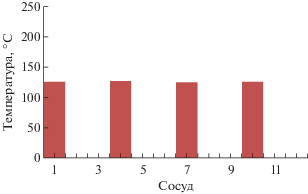
По данным производителя (www.cem.com), применение сосудов нового типа iPrep обеспечивает возможность увеличения массы проб с органической матрицей и повышает полноту окисления органики. Температуру реакционной смеси в каждом сосуде контролировали с помощью системы iWave (дистанционный температурный датчик TempGuard – Light emitting technology), которая обеспечивает контроль температурных условий в каждом сосуде, создает условия и обеспечивает полноту разложения, одинаковые для всех сосудов (стабильность заданной температуры в сосудах подтверждена экспериментально, рис. 2), а также исключает необходимость подсоединения датчиков к сосудам. Это не только удобно для организации рутинного анализа, но и позволяет в ряде случаев снизить его продолжительность при сохранении качества результатов.
Рис. 2.
Воспроизводимость температурных условий при микроволновом разложении нескольких образцов (система iWave).
Для определения элементов использовали АЭС-ИСП-спектрометр IRIS Intrepid II DUO (Thermo Electron Corp., CША) с полупроводниковым CID-детектором. Спектральный диапазон прибора 165–1050 нм. Рабочие параметры, элементы и их спектральные линии приведены в табл. 1.
Таблица 1.
Рабочие условия АЭС-ИСП-спектрометра IRIS Intrepid II DUO
| Параметр | Значение |
|---|---|
| Мощность генератора, Вт | 1150 |
| Скорости плазмообразующего вспомогательного и распылительного потоков соответственно, л/мин | 15, 0.5 и 0.73 |
| Обзор плазмы | DUO (аксиальный + радиальный) |
| Число параллельных измерений | 3 |
| Длины волн определяемых элементов, нм | Al 396.1, Ba 455.4, Ca 317.9, Cd 228.8, Cr 357.8, Cu 324.7, Fe 259.9, K 766.4, Mg 285.2, Mn 257.6, Mo 202.0, Na 818.3, Ni 221.6, Ni 231.6, P 214.9, S 182.0, Sn 283.9, Sr 407.7, Ti 334.9, V 292.4, Zn 213.8 |
МС-ИСП-измерения (элементы и условия их определения приведены в табл. 2) выполняли на спектрометре высокого разрешения с двойной фокусировкой ELEMENT XR (Finnigan MAT., Германия).
Таблица 2.
Рабочие условия МС-ИСП-спектрометра ELEMENT XR
| Параметр | Значение |
|---|---|
| Мощность генератора, Вт | 1200 |
| Скорости плазмообразующего вспомогательного и распылительного потоков соответственно, л/мин | 16, 0.9 и 1.010 |
| Число параллельных измерений | 3 |
| Изотопы: | |
| Режим “низкое разрешение” (R = 300): 7Li, 9Be, 45Sc, 69Ga, 74Ge, 77Se, 85Rb, 88Sr, 89Y, 90Zr, 93Nb, 114Cd, 115In, 121Sb, 139La, 140Ce, 141Pr, 143Nd, 147Sm, 151Eu, 157Gd, 159Tb, 161Dy, 165Ho, 166Er, 169Tm, 172Yb, 175Lu, 180Hf, 181Ta, 184W, 208Pb, 209Bi, 232Th, 238U | |
| Режим “среднее разрешение” (R = 4000): 7Li, 24Mg, 28Si, 45Sc, 51V, 59Co, 60Ni, 63Cu, 66Zn, 69Ga, 74Ge, 75As, 78Se, 90Zr, 93Nb, 105Pd, 110Cd, 115In, 121Sb, 133Cs, 180Hf, 184W, 205Tl, 208Pb, 209Bi, 238U | |
| Режим “высокое разрешение” (R = 10 000): 39K, 75As | |
Для градуировки спектрометров использовали рабочие растворы (0.1, 1.0 и 10 ppm для АЭС-ИСП и 0.1, 1.0 и 10 ppb для МС-ИСП), приготовленные из многоэлементных стандартных растворов с содержанием 100 ppm каждого элемента (High-Purity Standards, США). Рабочие растворы готовили последовательным разбавлением из стандартных растворов путем взвешивания с точностью 0.0001 г.
Остаточную кислотность конечного раствора определяли титрованием 1 М раствором NaOH в присутствии индикатора “метиловый оранжевый”.
Эффективность разложения определяли по остаточной концентрации углерода в растворе методом АЭС-ИСП (С193.0 и С247.8 нм). Для градуировки спектрометра и исследования влияния углерода использовали растворы лимонной кислоты х. ч. (ЛенРеактив, Россия).
Влияние углерода на формирование аналитических сигналов при АЭС-ИСП-определении элементов исследовали на примере модельных растворов с содержанием 0.1 мкг/мл аналитов (Al, Ba, Ca, Cd, Cr, Cu, Fe, K, Mg, Mn, Mo, Na, Ni, P, Sn, Sr, Ti, V, Zn) и 10, 100 и 900 мкг/мл углерода; при МС-ИСП-определении – на примере растворов с содержанием 1 нг/мл аналитов (As, Be, Bi, Ce, Cd, Co, Cs, Dy, Fe, Ga, Ge, Gd, Eu, Er, Hf, Ho, In, K, La, Li, Lu, Mg, Mn, Nb, Nd, Ni, Pr, Rb, Sc, Se, Si, Sm, Sr, Ta, Tb, Th, Tl, Tm, U, V, W, Y, Yb, Zr) и 10, 100 и 900 мкг/мл углерода.
Внутренние стандарты не использовали, так как анализировали не более 10 растворов за один экспериментальный сеанс. Дрейф масс-спектрометра контролировали с помощью дополнительного анализа рабочих стандартных растворов до и после анализа проб.
В качестве исследуемых образцов выбрали нефти месторождений Татарстана с различной вязкостью (легкая – образец (1), сверхвязкая – образец (2)).
Для контроля правильности выбранных условий анализа использовали стандартный образец состава (СОС) Conostan S-21 (SPC Science, Канада) с содержанием 100 мкг/г аналитов (Ag, Al, B, Ba, Ca, Cd, Cr, Cu, Fe, Mg, Mn, Mo, Na, Ni, P, Pb, Si, Sn, Ti, V, Zn). Для получения АЭС-ИСП-результатов методом стандартных добавок к исследуемым образцам нефти перед МВ-разложением путем взвешивания делали добавки 0.03 ± 0.01 или 0.25 ± 0.05 г Conostan-21 c таким расчетом, чтобы общая масса навески не превышала 0.750 г.
Выбор условий микроволнового разложения проб. Разложение проб проводили как минимум для двух одинаковых навесок анализируемых образцов. Одновременно можно выполнять разложение до 12 образцов. Перед отбором пробы образцы нагревали на водяной бане при ≈80°С в течение 30 мин для гомогенизации проб. Далее пробы взвешивали на аналитических весах (с точностью до 0.0001 г) непосредственно в реакционных сосудах.
В сосуд помещали 200–1000 мг образца, добавляли 10 мл конц. HNO3. Полученную смесь выдерживали в вытяжном шкафу в течение 10–15 мин во избежание резкого подъема давления в сосуде на начальной стадии разложения. После этого сосуды собирали и устанавливали в опорный модуль. Модуль размещали на поворотной карусели микроволновой печи.
Температурная программа пробоподготовки описана в табл. 3. В процессе нагрева подаваемая мощность регулируется автоматически: для 1–2 одновременно разлагаемых образцов – 400 Вт, для 3–5 образцов – 800 Вт, для 6 и более образцов – 1600 Вт (тип и количество сосудов распознаются лабораторной системой MARS6 автоматически). Реакционные сосуды открывали по окончании программы после охлаждения до 80°С (для минимизации давления). Полученный в ходе минерализации раствор количественно переводили в объем 25–100 мл бидистиллированной водой.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Ранее нами исследованы некоторые технические решения, эффективные для МВ-подготовки сложных природных объектов. В частности, изучены возможности применения сосудов EasyPrep (с частичным удалением газовой фазы) в сочетании с системой контроля температуры, включающей несколько датчиков (DuoTemp) и обеспечивающей измерение температуры одновременно во всех реакционных сосудах [23]. Показана перспективность таких систем для разложения трудно-вскрываемых минеральных веществ (Т > 260°C), подготовки проб к определению компонентов в виде летучих соединений, разложения в условиях частичного удаления газообразных продуктов реакции, а также одновременного разложения различных по составу и массе проб. На примере определения элементов, образующих летучие соединения (Нg, As, Se и др.), в объектах с органической матрицей установлено, что частичное удаление газовой фазы при сохранении избыточного давления в сосуде не приводит к потерям аналитов [23, 24].
Дальнейшее совершенствование МВ-систем привело к появлению сосудов iPrep, предназначенных в первую очередь для разложения наиболее сложных материалов органической природы, и новой системы контроля температуры во всех сосудах iWave. Эти технические решения апробированы в данной работе на примере подготовки к ИСП-анализу различных видов нефти. С целью расширения круга определяемых элементов, в первую очередь для получения результатов по РЗЭ, которые могут служить маркерами протекания различных природных процессов, применяли метод МС-ИСП с высоким разрешением.
Выбор условий микроволнового разложения проб. Основная задача стадии подготовки пробы – обеспечить условия наиболее эффективного инструментального определения, т.е. приемлемую концентрацию определяемых элементов на фоне минимального количества мешающих компонентов. Современные спектральные методы анализа, в частности АЭС-ИСП и МС-ИСП, предъявляют жесткие требования к составу вводимой пробы (содержанию солей, кислотности раствора и наличию органических соединений). Поэтому при выборе условий пробоподготовки исследовали возможность увеличения массы анализируемой пробы и контролировали параметры, которые напрямую влияют на формирование аналитического сигнала: остаточную кислотность и остаточный углерод.
При выборе параметров МВ-разложения варьировали массу образца, температуру и время МВ-нагрева на примере разложения сверхвязкой нефти как наиболее сложного объекта.
С увеличением навески образца расход кислоты увеличивается и, следовательно, остаточная кислотность снижается (рис. 3) до уровня 0.3 М при разложении 1 г, что дает возможность анализировать получаемый раствор методом АЭС-ИСП с минимальным разбавлением. Для АЭС-ИСП-определения раствор после разложения переводили в колбу емк. 25 мл. При использовании МС-ИСП-спектрометра с высоким разрешением, обеспечивающего существенно более низкие пределы обнаружения элементов, можно применять гораздо бóльшую кратность разбавления: в настоящей работе растворы после разложения разбавляли бидистиллированной водой в 50 раз.

Другим важным параметром является содержание в получаемых растворах остаточного углерода (отношение содержаний углерода до и после разложения). Результаты, полученные при температуре разложения 220°С, показывают, что с увеличением навески увеличивается содержание остаточного углерода (рис. 4). Увеличение температуры с 220 до 240°С приводит к снижению концентрации остаточного углерода.
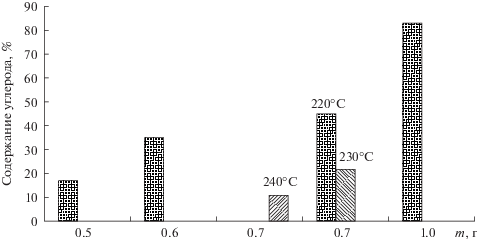
Экспериментально показано, что образец нефти любого состава массой до 1000 мг разлагается полностью. Этому способствуют геометрия сосуда и выбранные условия МВ-разложения. Установлено, что увеличение времени подъема температуры с 25 до 35 мин и времени удерживания температуры с 10 до 20 мин практически не влияет на полноту разложения образцов легкой и сверхвязкой нефти.
В качестве “рабочей” выбрали массу нефти 500 ± 100 мг, которая достаточна для определения большинства элементов; в то же время “мягкие” условия разложения (220–230°С) позволяют увеличить срок службы реакционных сосудов.
АЭС-ИСП- и МС-ИСП-определение элементов. Результаты, полученные при выборе условий МВ-разложения для последующего определения элементов в нефти с применением ИСП, показали, что в растворах после разложения нефти присутствует остаточный углерод, который может влиять на формирование аналитического сигнала, в особенности при МС-ИСП-анализе [7, 25]. Наличие органических соединений в плазме ухудшает еe характеристики, влияющие на точность результатов. В методах с применением ИСП помехи можно разделить на две группы: спектральные и неспектральные. При анализе органических растворов спектральные помехи появляются, как правило, из-за образования побочных продуктов пиролиза матрицы. Неспектральные помехи связаны, во-первых, со спецификой образования аэрозоля, содержащего органические вещества, во-вторых, c процессами транспортировки аналита в плазму (изменение количества), в-третьих, c явлениями, изменяющими тепловые характеристики плазмы.
Влияние остаточного углерода на формирование аналитического сигнала при анализе методами АЭС-ИСП и МС-ИСП изучено на модельных растворах с разными концентрациями лимонной кислоты (10, 100 и 900 мкг/мл в пересчете на углерод). Выбор лимонной кислоты основан на данных [25] об отсутствии существенных различий в отношении матричных эффектов разных углеродсодержащих матриц (глицерин, лимонная кислота, карбонат аммония). При сканировании спектрального диапазона вблизи аналитических линий определяемых элементов показано, что при АЭС-ИСП-определении присутствие углерода (в исследованных концентрациях) не влияет на базовую линию для наиболее важных микроэлементов нефти – Ni, Fe, V. В случаях флуктуации базовой линии использовали коррекцию фона. Присутствие углерода в растворах влияет на формирование аналитического сигнала при АЭС-ИСП-определении некоторых элементов. В частности, наблюдали подавление аналитического сигнала для аналитической линии Ni 221.6 нм (примерно в 5 раз). Объяснений данному экспериментальному факту пока нет. В связи с этим аналитическую линию Ni 221.6 нм не использовали для получения количественных результатов анализа.
В табл. 4 приведены результаты АЭС-ИСП-определения элементов, в том числе полученные методом стандартных добавок с использованием СОС Conostan S-21 (добавки 0.03 ± 0.01 и 0.25 ± ± 0.05 г). Таким образом, показано, что метод АЭС-ИСП в сочетании с МВ-пробоподготовкой позволяет определять в нефти целевые аналиты, в первую очередь, Ni, V и Fe.
Таблица 4.
Результаты (мкг/г) АЭС-ИСП-определение содержания следов элементов в образцах нефти методом АЭС-ИСП (n = 3, Р = 0.95)
| Элемент, длина волны, нм | Предел определения, (ПО)* | Нефть 2 | Нефть 1 |
|---|---|---|---|
| Al 396.1 | 0.4 | 8.8 ± 0.3 | 6.5 ± 0.2 |
| Ba 455.4 | 0.2 | <ПО | <ПО |
| Ca 317.9 | 0.04 | 7.8 ± 0.2 | 7.1 ± 0.2 |
| Cd 228.8 | 0.02 | 1.10 ± 0.05 | <ПО |
| Cr 357.8 | 0.04 | 0.30 ± 0.05 | 0.30 ± 0.05 |
| Cu 324.7 | 0.02 | 0.16 ± 0.01 | <ПО |
| Fe 259.9 | 0.4 | 9.3 ± 0.3 | 1.6 ± 0.1 |
| K 766.4 | 0.2 | <ПО | 2.9 ± 0.1 |
| Mg 285.2 | 0.02 | 0.18 ± 0.01 | <ПО |
| Mn 257.6 | 0.04 | 0.20 ± 0.02 | <ПО |
| Mo 202.0 | 0.05 | 0.50 ± 0.05 | <ПО |
| Na 818.3 | 1.0 | 4.5 ± 0.1 | <ПО |
| Ni 231.6 | 0.06 | 39.0 ± 0.5 | 22.8 ± 0.7 |
| P 214.9 | 1.0 | <ПО | <ПО |
| S 182.0 | 4.0 | 40 ± 1 мг/г | 20.7 ± 0.6 мг/г |
| Sn 283.9 | 0.6 | <ПО | <ПО |
| Sr 407.7 | 0.4 | <ПО | <ПО |
| Ti 334.9 | 0.2 | <ПО | <ПО |
| V 292.4 | 0.2 | 190 ± 5 | 65.5 ± 0.5 |
| Zn 213.8 | 0.1 | <ПО | <ПО |
* За предел определения в табл. 4 и 5 принимали наименьшую концентрацию на градуировочном графике, измеренную в выбранных инструментальных условиях с учетом чистоты реактивов [26].
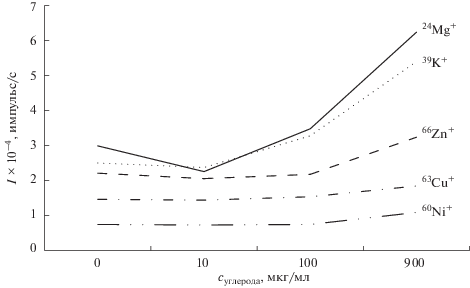
Влияние углерода на формирование аналитического сигнала при МС-ИСП-определении изучали на модельных растворах с содержанием по 1 нг/мл аналитов и 10, 100 и 900 мкг/мл углерода. Показано, что, в отличие от АЭС-ИСП, при МС-ИСП-определении наличие углерода может существенно влиять на аналитические сигналы ряда изотопов. Так, например, для 63Сu+ наблюдали увеличение сигнала на 20%, для 60Ni+ и 66Zn+ – на 30%, для 24Mg+ и 39K+ – на 50% (рис. 5). Очевидно, что содержание остаточного углерода в растворах будет варьироваться при переходе от одного типа нефти к другому, а также при изменении условий разложения. В связи с этим для минимизации случайной погрешности изотопы всех элементов, для которых зафиксировано влияние углерода на формирование аналитического сигнала (подавление или увеличение) более чем на 5%, исключили из количественного анализа.
Рис. 5.
Влияние углерода на интенсивность аналитического сигнала изотопов 24Mg+, 60Ni+, 63Сu+, 39K+ и 66Zn+ (концентрация аналитов 1 нг/мл).
Важно отметить, что углерод практически не влияет на формирование аналитических сигналов РЗЭ – важнейших маркеров, характеризующих происхождение нефти. Это позволяет определять всю группу РЗЭ на уровне низких концентраций без предварительного концентрирования. Результаты МС-ИСП-определения элементов приведены в табл. 5.
Таблица 5.
Результаты (нг/г) МС-ИСП-определения ультраследов элементов в образцах нефти (n = 3, Р = 0.95)
| Изотоп | Предел определения, нг/г | Нефть 2 | Нефть 1 |
|---|---|---|---|
| Режим “низкое разрешение” | |||
| 9Be+ | 0.4 | 7.5 ± 0.5 | 5.2 ± 0.5 |
| 69Ga+ | 0.2 | 60 ± 2 | 21 ± 1 |
| 74Ge+ | 0.1 | 5.3 ± 0.5 | 3.1 ± 0.3 |
| 77Se+ | 5.0 | 55 ± 1 | 17 ± 1 |
| 85Rb+ | 0.4 | 3.3 ± 0.3 | <ПО |
| 88Sr+ | 0.04 | 30 ± 5 | 10 ± 1 |
| 89Y+ | 0.05 | 2.3 ± 0.1 | 1.19 ± 0.04 |
| 90Zr+ | 0.3 | 20 ± 1 | 3.3 ± 0.1 |
| 93Nb+ | 0.4 | <ПО | <ПО |
| 115In+ | 0.08 | 3.9 ± 0.1 | 4.33 ± 0.03 |
| 121Sb+ | 0.5 | 1.50 ± 0.05 | 5.3 ± 0.5 |
| 139La+ | 0.06 | 2.3 ± 0.2 | 1.50 ± 0.05 |
| 140Ce+ | 0.06 | 2.0 ± 0.1 | 1.30 ± 0.01 |
| 141Pr+ | 0.02 | 0.50 ± 0.05 | 0.40 ± 0.04 |
| 143Nd+ | 0.06 | 0.90 ± 0.05 | 0.40 ± 0.03 |
| 147Sm+ | 0.006 | 0.80 ± 0.05 | 0.30 ± 0.05 |
| 151Eu+ | 0.005 | 0.30 ± 0.05 | 0.20 ± 0.03 |
| 157Gd+ | 0.01 | 0.50 ± 0.05 | 0.45 ± 0.05 |
| 159Tb+ | 0.002 | 0.10 ± 0.01 | 0.070 ± 0.005 |
| 161Dy+ | 0.006 | 0.50 ± 0.03 | 0.40 ± 0.01 |
| 165Ho+ | 0.002 | 0.080 ± 0.005 | 0.050 ± 0.005 |
| 166Er+ | 0.006 | 0.26 ± 0.03 | <ПО |
| 169Tm+ | 0.002 | <ПО | <ПО |
| 172Yb+ | 0.008 | 0.20 ± 0.02 | 0.10 ± 0.01 |
| 175Lu+ | 0.004 | 0.050 ± 0.005 | 0.030 ± 0.005 |
| 180Hf+ | 1.0 | <ПО | <ПО |
| 181Ta+ | 2.0 | <ПО | <ПО |
| 184W+ | 8.0 | <ПО | <ПО |
| 209Bi+ | 0.6 | <ПО | <ПО |
| 232Th+ | 0.6 | <ПО | <ПО |
| 238U+ | 0.04 | 0.90 ± 0.05 | 0.20 ± 0. 02 |
| Режим “среднее разрешение” | |||
| 7Li+ | 9.0 | <ПО | <ПО |
| 24Mg+ | 0.8 | 10 ± 1 | 2.5 ± 0.2 |
| 45Sc+ | 0.04 | 0.95 ± 0.05 | 0.60 ± 0.05 |
| 55Mn+ | 8.0 | 250 ± 10 | 85 ± 5 |
| 59Co+ | 0.2 | 28.2 ± 0.7 | 28.4 ± 0.2 |
| 63Cu+ | 1.0 | 40 ± 1 | 12 ± 1 |
| 66Zn+ | 4.0 | <ПО | <ПО |
| 110Cd+ | 0.6 | 1100 ± 100 | 3.0 ± 0.2 |
| 133Cs+ | 0.6 | <ПО | <ПО |
| 205Tl+ | 0.04 | <ПО | 0.7 ± 0.5 |
| Режим “высокое разрешение” | |||
| 75As+ | 6.0 | 70 ± 5 | <ПО |
Важной геохимической характеристикой нефти является вид распределения содержаний элементов, отнесенных к их содержаниям в хондрите С1. Получаемая картина отражает не только условия формирования и особенности геологического материала, но и корректность полученных аналитических данных. Спектры распределения нормализованных к хондриту (с использованием данных работы [27]) содержаний РЗЭ для анализируемых образцов нефти представлены на рис. 6. Видно, что полученные кривые схожи с зависимостями, полученными для нефти в работах [28, 29].

Работа выполнена в рамках темы 0137-2019-0011 при поддержке РФФИ (грант 18-29-06044 мк).
Список литературы
Tissot B.P., Welte D.H. Petroleum Formation and Occurrence. Second Revised and Enlareed Edition. Berlin, Heidelberg, New York, Tokyo: Springer – Verlag, 1984. 720 p.
Буслаева Е.Ю., Новгородова М.И. Элементоорганические соединения в проблеме миграции рудного вещества. М.: Наука, 1989. 152 с.
Пунанова С.А. Микроэлементы нафтидов в процессе онтогенеза углеводородов в связи с нефтегазоностностью. Дис. … докт. геол.-минерал. наук. М.: Институт проблем нефти и газа РАН, 2017. 288 с.
Barwise A.J.G. Role of nickel and vanadium in petroleum classification // Energy Fuels. 1990. V. 4. № 6. P. 647.
Galarraga F., Reategui K., Martïnez A., Martínez M., Llamas J.F., Márquez G. V/Ni ratio as a parameter in palaeoenvironmental characterization of nonmature medium-crude oils from several Latin American basins // J. Pet. Sci. Eng. 2008. V. 61. № 4. P. 9.
Lewan M.D. Factors controlling the proportionality of vanadium to nickel in crude oils // Geochim. Cosmochim. Acta. 1984. V. 48. № 11. P. 2231.
Sánchez R., Todolí J.L., Lienemann C.P., Mermet J.M. Determination of trace elements in petroleum products by inductively coupled plasma techniques: A critical review // Spectrochim. Acta B. 2013. V. 88. № 1. P. 104.
Kishore Nadkarni R.A. Spectroscopic Analysis of Petroleum Products and Lubricants. West Conshohocken, PA, USA: ASTM Int., 2011. 640 p.
Mora J., Todolí J.L., Sempere F.J., Canals A., Hernandis V. Determination of metals in lubricating oils by flame atomic absorption spectrometry using a single-bore high-pressure pneumatic nebulizer // Analyst. 2000. V. 125. P. 2344.
aumette G., Lienemann C.-P., Merdrignac I., Paucot H., Bouyssiere B., Lobinski R. Sensitivity improvement in ICP MS analysis of fuel and light petroleum matrices using a microflow nebulizer and heated spray chamber sample introduction // Talanta. 2009. V. 80. P. 1039
ГОСТ 34242-2017. Нефть и нефтепродукты. Определение никеля, ванадия и железа методом АЭС-ИСП. М.: Стандартинформ, 2017. 20 с.
Souza R.M.S., Meliande A.L.S., da Silveira C.L.P., Aucélio R.Q. Determination of Mo, Zn, Cd, Ti, Ni, V, Fe, Mn, Cr and Co in crude oil using inductively coupled plasma optical emission spectrometry and sample introduction as detergentless microemulsions // Microchem. J. 2006. V. 82. P. 137.
Santelli R.E., Oliveira E.P., de Carvalho M.F.B., Bezerra M.A., Freire A.S. Total sulfur determination in gasoline, kerosene and diesel fuel using inductively coupled plasma optical emission spectrometry after direct sample introduction as detergent emulsions // Spectrochim. Acta B. 2008. V. 63. P. 800.
Santelli R.E., Bezerra M.A., Freire A.S., Oliveira E.P., de Carvalho M.F.B. Non-volatile vanadium determination in petroleum condensate, diesel and gasoline prepared as detergent emulsion using GF AAS // Fuel. 2008. V. 87. P. 1617.
Sugiyama I., Williams-Jones A.E. An approach to determining nickel, vanadium and other metal concentrations in crude oil // Anal. Chim. Acta. 2018. V. 1002. P. 18.
Седых Э.М., Банных Л.Н., Коробейник Г.С., Старшинова Н.П. Определение никеля и ванадия в сырых нефтях методами ЭТААС и АЭС-ИСП после автоклавной минерализации // Заводск. лаборатория. Диагностика материалов. 2010. Т. 76. № 4. С. 4.
dos Anjos S.L., Alves J.C., Rocha Soares S.A., Araujo R.G.O., de Oliveira O.M.C., Queiroz A.F.S., Ferreira S.L.C. Multivariate optimization of a procedure employing microwave-assisted digestion for the determination of nickel and vanadium in crude oil by ICP-OES // Talanta. 2018. V. 178. P. 842.
Pereira J.S.F., Moraes D.P., Antes F.G., Diegl L.O., Santos M.F.P., Guimaraes R.C.L., Fonseca T.C.O., Dressler V.L., Flores E.M.M. Determination of metals and metalloids in light and heavy crude oil by ICP-MS after digestion by microwave-induced combustion // Microchem. J. 2010. V. 96. P. 4.
Yang W., Casey J.F., Gao Y. A new sample preparation method for crude or fuel oils by mineralization utilizing single reaction chamber microwave for broader multi-element analysis by ICP techniques // Fuel. 2017. V. 206. P. 64.
Druzian G.T., Pereira L.S.F., Mello P.A., Mesko M.F., Duarte F.A., Flores E.M.M. Rare earth element determination in heavy crude oil by USN-ICP-MS after using a microwave-assisted single reaction chamber // J. Anal. At. Spectrom. 2016. V. 31. № 6. P. 1185.
Pereira J.S.F., Pereira L.S.F., Mello P.A., Guimarгes R.C.L., Guarnieri R.A., Fonseca T.C.O., Flores E.M.M. Microwave-induced combustion of crude oil for further rare earth elements determination by USN–ICP-MS // Anal. Chim. Acta. 2014. V. 844. P. 8.
Марютина Т.А., Федотов П.С. Жидкостная хроматография со свободной неподвижной фазой в элементном анализе: от нефти до особочистых веществ // Журн. аналит. химии. 2019. № 3. С. 201.
Киселева М.С., Тютюнник О.А., Никулин А.Н., Кубракова И.В. Микроволновая подготовка природных объектов с использованием новых технических решений // Заводск. лаборатория. Диагностика материалов. 2014. Т. 80. № 6. С. 7.
Тютюнник О.А., Гецина М.Л., Торопченова Е.С., Кубракова И.В. Микроволновая пробоподготовка природных объектов к атомно-абсорбционному определению ртути и других токсичных элементов // Журн. аналит. химии. 2013. Т. 68. № 5. С. 420.
Grindlay G., Mora J., de Loos-Vollebregt M., Vanhaecke F. A Systematic study on the influence of carbon on the behavior of hard-to-ionize elements in inductively coupled plasma-mass spectrometry // Spectrochim. Acta B. 2013. V. 86. P. 42.
CITAC/EURACHEM Guide. Guide to Quality in Analytical Chemistry: An Aid to Accreditation, 2002.
Wasson J.T., Kallemeyn G.W. Composition of chondrites // Philos. Trans. Royal Soc. A. 1988. V. 325(1587). P. 535.
Биглов К.Ш., Иванов К.С. Геохимия нефти Зюзеевского месторождения Татарстана / Ежегодник-2008: Тр. ИГГ УрО РАН. 2009. Вып. 156. С. 12.
Калинин Е.П. Геохимическая специфика нефти и ее природа. Обзор // Вестник института геологии Коми научного центра Уральского отделения РАН. 2009. № 1. С. 6.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии