

Журнал аналитической химии, 2021, T. 76, № 3, стр. 253-260
Определение содержания примесей в арсине высокой чистоты методом хромато-масс-спектрометрии
А. Ю. Созин a, *, Т. Г. Сорочкина a, О. Ю. Чернова a, А. П. Котков b, Н. Д. Гришнова b, Д. М. Полежаев b, Г. В. Пушкарев b, С. В. Ермолаев b
a Институт химии высокочистых веществ им. Г.Г. Девятых Российской академии наук
603950 Нижний Новгород, ул. Тропинина, 49, Россия
b АО НПП “Салют”
603950 Нижний Новгород, ул. Ларина, 7, Россия
* E-mail: Sozin@ihps-nnov.ru
Поступила в редакцию 11.03.2020
После доработки 19.04.2020
Принята к публикации 23.10.2020
Аннотация
Методом хромато-масс-спектрометрии исследован примесный состав арсина. Определены примеси постоянных газов, диоксида углерода, гидридов, предельных, непредельных, ароматических углеводородов С1–С6, галогенсодержащих углеводородов, соединений серы, алкилпроизводных арсина и диарсина. Содержание примесей в высокочистом арсине находится на уровне 10–6–10–5 об. %. В арсине после синтеза и во фракциях, выделенных в процессе его ректификационной очистки, концентрации примесей достаточно высоки и лежат в интервале 10–6–0.1 об. %. Пределы обнаружения примесей составили 2 × 10–7–2 × 10–4 об. %.
Высокочистый арсин находит широкое применение для получения полупроводниковых материалов (арсенидов галлия, индия, алюминия), применяющихся в микроэлектронике, лазерной технике, оптоэлектронике гелиоэнергетике, для производства мощных сверхвысокочастотных приборов [1–3], халькогенидных стекол для волоконной оптики [4].
Физико-химические свойства полупроводниковых и оптических материалов существенно зависят от чистоты исходных веществ. Согласно современным требованиям, уровень чистоты арсина, применяемого в микроэлектронике, должен составлять не ниже 99.99994% (ТУ 2114-001-07611801-2010), а содержание в нем примесей, в первую очередь электрически активных, – не выше 10–6–10–5 об. %. Работ, посвященных исследованию примесного состава арсина, в литературе немного. В значительной степени это связано с его высокой токсичностью и в связи с этим предпочтительным анализом получаемых из арсина конечных менее токсичных материалов. Наибольшими аналитическими возможностями определения примесей в арсине обладают методы масс-спектрометрии, ИК-спектроскопии, газовой хроматографии и хромато-масс-спектрометрии [5]. В работах [6, 7] методом масс-спектрометрии в арсине определили примеси толуола, фреонов, ксилола, фосфина с пределом обнаружения 1 × 10–5 об. %. Описано [8, 9] ИК-спектроскопическое исследование содержания воды в арсине с пределом обнаружения 7 × 10–5 об. %. Основными методами анализа арсина, позволяющими определять широкий круг молекулярных примесей с низкими пределами обнаружения, являются газовая хроматография и хромато-масс-спектрометрия [5]. С использованием метода газовой хроматографии определены постоянные газы и диоксид углерода с пределами обнаружения 2 × 10–6–1 × 10–5 об. % [10, 11], летучие неорганические гидриды элементов 3–6 групп периодической системы с пределами обнаружения 2 × × 10–7–1 × 10–4 об. % [12–14], углеводороды С1–С4 с пределами обнаружения 2 × 10–6–1 × 10–56 об. % [13–15]. Во всех работах по газохроматографическому анализу арсина примеси определяли с использованием насадочных колонок и различных селективных детекторов, позволяющих регистрировать с высокой чувствительностью только ограниченный набор примесей. Во многих случаях низкие пределы обнаружения достигнуты за счeт концентрирования и использования проб большого объeма, что не всегда приемлемо в случае анализа высокочистых веществ.
Метод хромато-масс-спектрометрии использовали в основном для идентификации примесей. В работах [16, 17] в арсине установлены постоянные газы, некоторые углеводороды С4–С6, дихлорметан, хлороформ, 1,2-дихлорэтан, гидриды. Пределы их обнаружения составили 2 × 10–7–2 × × 10–5 об. %. В работе [18] этим методом с применением капиллярных адсорбционных колонок идентифицировали более 50 примесных веществ, среди которых постоянные газы, летучие неорганические гидриды, углеводороды С1–С6, хлор- и кислородсодержащие углеводороды, триметилфторсилан, диметилсульфид, карбонилсульфид, толуол, хлорбензол, алкилпроизводные арсина и диарсина.
Хромато-масс-спектрометрический метод является наиболее перспективным для определения примесей в арсине. Его применение в сочетании с капиллярными колонками с различной селективностью даст возможность добиться высокоэффективного хроматографического разделения примесей с близкими свойствами и обеспечить их надежное высокочувствительное определение. Сведения по количественному определению в арсине большинства обнаруженных примесей в литературе отсутствуют.
Цель настоящей работы – разработка методики количественного хромато-масс-спектрометрического определения примесей в арсине высокой чистоты с использованием капиллярных адсорбционных колонок.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исследовали примесный состав арсина, полученного электрохимическим восстановлением мышьяковой кислоты, синтезированной из мышьяксодержащего сырья [19]. Анализировали образцы арсина после синтеза, очищенные низкотемпературной ректификацией, и фракции, выделенные в процессе его очистки.
Для анализа арсина использовали хромато-масс-спектрометр Agilent 6890/MSD 5973N с квадрупольным масс-анализатором. Разработали специальную вакуумную систему дозирования арсина в хромато-масс-спектрометр, предусматривающую возможность применения независимых газовых линий для дозирования арсина разной степени чистоты. Схема установки приведена и подробно описана в работе [18].
Для разделения примесей с невысокими относительно арсина температурами кипения и молекулярными массами использовали капиллярную адсорбционную колонку GS-GasPro 60 м × 0.32 мм с сорбентом модифицированным силикагелем (Agilent Technologies, Inc., США). Для разделения менее летучих примесей использовали колонку с сорбентом политриметилсилилпропином 25 м × × 0.26 мм, df = 0.25 мкм [20]. Условия проведения анализа и разделения примесей, их идентификация описаны в работе [18].
Количественное определение примесей проводили методом внешнего стандарта по площадям хроматографических пиков в режиме селективного ионного детектирования (SIM) по регистрации их единичных ионов, характеризующихся максимальным соотношением сигнал/шум. Значения m/z этих ионов приведены в табл. 1.
Таблица 1.
Концентрации примесей в различных образцах арсина, их регистрируемые массовые числа (m/z) и пределы обнаружения сmin
| Примесь | Концентрация, об. % | m/z | сmin, об. % | ||||
|---|---|---|---|---|---|---|---|
| “исходный” | “легкая” фракция | “тяжелая” фракция | “ректификат” | наши данные |
литературные данные | ||
| N2 (азот) | (9 ± 1) × 10–4 | (2.3 ± 0.2) × 10–5 | (2.6 ± 0.3) × 10–2 | (1.2 ± 0.2) × 10–5 | 28 | 5 × 10–6 | 5 × 10–6 [21, 22] 1 × 10–4 [23] |
| O2 (кислород) | <2 × 10–5 | (1.3 ± 0.1) × 10–4 | <2 × 10–5 | (1.0 ± 0.2) × 10–5 | 32 | 5 × 10–6 | 2 × 10–6 [21] 2.5 × 10–6 [22] |
| Ar (аргон) | (1.0 ± 0.1) × 10–2 | (3.0 ± 0.3) × 10–5 | (3.0 ± 0.3) × 10–4 | (2.0 ± 0.9) × 10–6 | 40 | 9 × 10–7 | 2 × 10–6 [24] |
| CO (угарный газ) | <2 × 10–4 | <2 × 10–4 | <2 × 10–4 | <2 × 10–4 | 12 | 2 × 10–4 | 1 × 10–6 [24] 5 × 10–6 [22, 25] 1 × 10–6 [21] |
| CO2 (углекислый газ) | (2.7 ± 0.2) × 10–2 | 2.14 | (8.8 ± 0.9) × 10–4 | (8 ± 2) × 10–6 | 44 | 1 × 10–6 | 1 × 10–6 [24] 5 × 10–6 [23] 1 × 10–6 [21] 2.5 × 10–6 [22] |
| N2O (оксид азота(I)) | (1.0 ± 0.2) × 10–5 | (2.1 ± 0.3) × 10–3 | <2 × 10–6 | <2 × 10–6 | 44 | 2 × 10–6 | 2 × 10–6 [21] |
| CH4 (метан) | <1 × 10–5 | (2.7 ± 0.3) × 10–4 | <1 × 10–5 | <1 × 10–5 | 15 | 1 × 10–5 | 5.5 × 10–4 [24] 5 × 10–6 [22, 25] 1 × 10–5 [26] |
| C2H2 (ацетилен) | (1.0 ± 0.1) × 10–5 | (5.7 ± 0.6) × 10–4 | <1 × 10–6 | <1 × 10–6 | 26 | 1 × 10–6 | –** |
| C2H4 (этилен) | (7.3 ± 0.7) × 10–1 | 10.9 | (2.8 ± 0.3) × 10–3 | <1 × 10–5 | 27 | 1 × 10–5 | 3 × 10–6 [24] 2 × 10–5 [26] 4 × 10–6 [14] |
| C2H6 (этан) | (7.7 ± 0.8) × 10–4 | (2.1 ± 0.2) × 10–1 | (8.0 ± 0.8) × 10–5 | <2 × 10–6 | 27 | 1 × 10–6 | 5 × 10–6 [22, 27] 2.5 × 10–5 [26] 7 × 10–6 [14] |
| C3H6 (пропилен) | (1.2 ± 0.1) × 10–2 | (7.0 ± 0.7) × 10–5 | (6.3 ± 0.6) × 10–1 | (1.5 ± 0.2) × 10-5 | 41 | 2 × 10–6 | 5 × 10–6 [24] 5 × 10–5 [26] |
| C3H8 (пропан) | (4.0 ± 0.4) × 10–5 | <2 × 10–6 | (3.3 ± 0.3) × 10–3 | <2 × 10–6 | 29 | 2 × 10–6 | 2 × 10–6 [24] 5 × 10–5 [26] 3 × 10–6 [14] |
| C4H6* (бутадиен-1,3) | (9 ± 3) × 10–6 | <1 × 10–6 | (5 ± 2) × 10–6 | <1 × 10–6 | 54 | 1 × 10–6 | – |
| C4H8* (2-метилпропен-1) | (9 ± 3) × 10–5 | <3 × 10–6 | (9 ± 2) × 10–2 | <3 × 10–6 | 41 | 3 × 10–6 | – |
| изо-C4H10 (изобутан) | (1.0 ± 0.1) × 10–4 | (1.0 ± 0.1) × 10–5 | (8.0 ± 0.8) × 10–5 | <3 × 10–6 | 43 | 3 × 10–6 | 1 × 10–6 [24] 2 × 10–6 [14] |
| н-C4H10 (н-бутан) | (1.0 ± 0.1) × 10–4 | (1.0 ± 0.1) × 10–5 | (2.3 ± 0.2) × 10–4 | <2 × 10–6 | 43 | 2 × 10–6 | 1 × 10–6 [24] 4 × 10–5 [26] 2 × 10–6 [14] |
| н-C5H12 (н-пентан) | (1.9 ± 0.5) × 10–5 | <2 × 10–6 | <2 × 10–6 | <2 × 10–6 | 43 | 2 × 10–6 | – |
| C5H12* (2-метилбутан) | <2 × 10–6 | <2 × 10–6 | (1.0 ± 0.3) × 10–5 | <2 × 10–6 | 43 | 2 × 10–6 | – |
| C6H12* (2-метилпентен-1) | <3 × 10–6 | <3 × 10–6 | (9 ± 3) × 10–6 | <3 × 10–6 | 56 | 3 × 10–6 | – |
| C6H14* (2-метилпентан) | <3 × 10–6 | <3 × 10–6 | (9 ± 3) × 10–6 | <3 × 10–6 | 43 | 3 × 10–6 | – |
| n-C6H14 (н-гексан) | <3 × 10–6 | <3 × 10–6 | (1.0 ± 0.2) × 10–5 | <3 × 10–6 | 57 | 3 × 10–6 | – |
| C7H16* (триметилгексан) | <4 × 10–6 | <4 × 10–6 | (3 ± 1) × 10–5 | <4 × 10–6 | 43 | 4 × 10–6 | – |
| C6H6 (бензол) | <1 × 10–6 | <1 × 10–6 | (9 ± 3) × 10–6 | <1 × 10–6 | 78 | 1 × 10–6 | 1 × 10–5 [24] |
| C7H8 (толуол) | <3 × 10–6 | <3 × 10–6 | (1.7 ± 0.5) × 10–5 | <3 × 10–6 | 91 | 3 × 10–6 | 2 × 10–5 [24] |
| SiH4 (силан) | <1 × 10–6 | <1 × 10–6 | <1 × 10–6 | <1 × 10–6 | 30 | 1 × 10–6 | 1 × 10–4 [14, 24
] 1 × 10–6 [22, 28 ] |
| GeH4 (герман) | (1.0 ± 0.1) × 10–5 | (7.0 ± 0.7) × 10–4 | <5 × 10–7 | <5 × 10–7 | 76 | 5 × 10–7 | 1 × 10–4 [14, 24
] 1 × 10–6 [22, 28 ] |
| РН3 (фосфин) | <1 × 10–5 | <1 × 10–5 | (7 ± 2) × 10–5 | <1 × 10–5 | 34 | 1 × 10–5 | 5 × 10–7 [24] 1 × 10–4 [23] 1 × 10–5 [22] 2.5 × 10–6 [21] 1 × 10–6 [14] |
| H2S (сероводород) | (4.0 ± 0.4) × 10–5 | (1.0 ± 0.3) × 10–5 | (4.0 ± 0.4) × 10–5 | <5 × 10–6 | 36 | 5 × 10–6 | 1 × 10–5 [24] 5 × 10–6 [22, 28 ] |
| CH2Cl2 (метиленхлорид) | (5 ± 2) × 10–6 | <1 × 10–6 | (1.5 ± 0.5) × 10–1 | <1 × 10–6 | 49 | 1 × 10–6 | 6 × 10–5 [24] |
| CCl2F2* (дифтордихлорметан) | <7 × 10–7 | <7 × 10–7 | (6 ± 2) × 10–6 | <7 × 10–7 | 85 | 7 × 10–7 | – |
| CH3Cl (хлорметан) | (1.1 ± 0.3) × 10–4 | <1 × 10–6 | (1.7 ± 0.5) × 10–2 | <1 × 10–6 | 50 | 1 × 10–6 | – |
| CHCl3 (хлороформ) | (2.3 ± 0.7) × 10–5 | <8 × 10–7 | (8 ± 2) × 10–2 | <8 × 10–7 | 83 | 8 × 10–7 | 3 × 10–5 [24] |
| C2H2Cl2 (транс-1,2-дихлорэтилен) | <4 × 10–7 | <4 × 10–7 | (1.5 ± 0.5) × 10–6 | <4 × 10–7 | 61 | 4 × 10–7 | – |
| C2HCl3 (трихлорэтилен) | (1.0 ± 0.3) × 10–6 | <4 × 10–7 | (4 ± 1) × 10–1 | <4 × 10–7 | 130 | 4 × 10–7 | – |
| C2H2Cl2 (цис-1,2-дихлорэтилен) | <4 × 10–7 | <4 × 10–7 | (1.0 ± 0.3) × 10–6 | <4 × 10–7 | 61 | 4 × 10–7 | – |
| C2H3Cl (хлорэтилен) | (3.0 ± 0.9) × 10–5 | <6 × 10–7 | (7.1 ± 2.1) × 10–3 | <6 × 10–7 | 62 | 6 × 10–7 | – |
| C2H4Cl2 (1,2-дихлорэтан) | <3 × 10–7 | <3 × 10–7 | (3 ± 1) × 10–6 | <3 × 10–7 | 62 | 3 × 10–7 | 1 × 10–5 [24] |
| С2H5Cl (хлорэтан) | <1 × 10–6 | <1 × 10–6 | (9 ± 2) × 10–6 | <1 × 10–6 | 64 | 1 × 10–6 | – |
| C2H3Cl3 (1,1,2-трихлорэтан) | <2 × 10–6 | <2 × 10–6 | (1.0 ± 0.3) × 10–5 | <2 × 10–6 | 97 | 2 × 10–6 | – |
| C2Cl4 (тетрахлорэтилен) | <2 × 10–7 | <2 × 10–7 | (3 ± 1) × 10–6 | <2 × 10–7 | 166 | 2 × 10–7 | – |
| C3H5Cl* (3-хлорпропен-1) | (4 ± 2) × 10–6 | <1 × 10–6 | (4 ± 2) × 10–2 | <1 × 10–6 | 41 | 1 × 10–6 | – |
| C3H7Cl* (2-хлорпропан) | <1 × 10–6 | <1 × 10–6 | <1 × 10–6 | <1 × 10–6 | 43 | 1 × 10–6 | – |
| C6H5Cl (хлорбензол) | <6 × 10–7 | <6 × 10–7 | (7 ± 2) × 10–6 | <6 × 10–7 | 112 | 6 × 10–7 | – |
| CH3CHO (ацетальдегид) | (1.0 ± 0.3) × 10–4 | <1 × 10–6 | (2.6 ± 0.8) × 10–2 | <1 × 10–6 | 29 | 1 × 10–6 | – |
| (CH3)2O (ацетон) | (8 ± 2) × 10–4 | <8 × 10–6 | (8 ± 2) × 10–1 | <8 × 10–6 | 43 | 8 × 10–6 | – |
| C4H8O* (2-бутанон) | <8 × 10–6 | <8 × 10–6 | (3 ± 1) × 10–5 | <8 × 10–6 | 43 | 8 × 10–6 | – |
| CH3AsH2* (метиларсин) | (2.3 ± 0.7) × 10–2 | <5 × 10–7 | 47 ± 15 | <5 × 10–7 | 90 | 5 × 10–7 | – |
| C2H3AsH2* | (3 ± 1) × 10–4 | <1 × 10–6 | 1.2 ± 0.3 | <1 × 10–6 | 102 | 1 × 10–6 | – |
| (CH3)2AsH* (диметиларсин) | (6 ± 2) × 10–5 | <1 × 10–6 | (3 ± 1) × 10–6 | <1 × 10–6 | 90 | 1 × 10–6 | – |
| C2H5AsH2* (этиларсин) | (2.8 ± 0.8) × 10–3 | <6 × 10–7 | 14 ± 4 | <6 × 10–7 | 106 | 6 × 10–7 | – |
| C3H5AsH2* (пропенарсин) | <8 × 10–7 | <8 × 10–7 | (2.0 ± 0.6) × 10–5 | <8 × 10–7 | 90 | 8 × 10–7 | – |
| C3H7AsH2* (пропиларсин) | (5 ± 1) × 10–5 | <8 × 10–7 | (1.1 ± 0.3) × 10–1 | <8 × 10–7 | 120 | 8 × 10–7 | – |
| CH3AsHC2H3* | (1.3 ± 0.4) × 10–5 | <1 × 10–6 | (9 ± 2) × 10–2 | <1 × 10–6 | 90 | 1 × 10–6 | – |
| CH3AsHC2H5* | (6 ± 2) × 10–5 | <1 × 10–6 | (3 ± 1) × 10–1 | <1 × 10–6 | 90 | 1 × 10–6 | – |
| As2H4* (диарсин) | (1.5 ± 0.5) × 10–4 | <8 × 10–7 | (2.2 ± 0.7) × 10–5 | (3 ± 1) × 10–5 | 150 | 8 × 10–7 | – |
| CH3As2H3* (метилдиарсин) | (1.7 ± 0.5) × 10–5 | <9 × 10–7 | (3 ± 1) × 10–2 | <9 × 10–7 | 150 | 9 × 10–7 | – |
| C2H5As2H3* (этилдиарсин) | <1 × 10–6 | <1 × 10–6 | (9 ± 3) × 10–5 | <1 × 10–6 | 103 | 1 × 10–6 | – |
| (CH3)2SiF2* (диметилдифторсилан) | (1.4 ± 0.4) × 10–4 | <7 × 10–7 | (4 ± 1) × 10–4 | <7 × 10–7 | 81 | 7 × 10–7 | – |
| (CH3)3SiF * (триметилфторсилан) | <1 × 10–6 | <1 × 10–6 | <1 × 10–6 | <1 × 10–6 | 77 | 1 × 10–6 | – |
| CH3SH* (метантиол) | <1 × 10–6 | <1 × 10–6 | (7 ± 2) × 10–6 | <1 × 10–6 | 47 | 1 × 10–6 | – |
| COS* (серооксид углерода) | (8 ± 1) × 10–7 | <5 × 10–7 | (4.0 ± 1.2) × 10–5 | (1.0 ± 0.3) × 10–6 | 60 | 5 × 10–7 | 2.5 × 10–6 [21] 5 × 10–6 [22] |
| (CH3)2S* (диметилсульфид) | <3 × 10–6 | <3 × 10–6 | (2.0 ± 0.7) × 10–5 | <3 × 10–6 | 62 | 3 × 10–6 | – |
| C4H4S* (тиофен) | <1 × 10–6 | <1 × 10–6 | (9 ± 3) × 10–6 | <1 × 10–6 | 84 | 1 × 10–6 | – |
В качестве образцов сравнения использовали аттестованные газовые смеси постоянных газов и углеводородов С1–С6 (соответствуют ТУ 6-16-2956-92). В случае гидридов и ряда хлорсодержащих веществ аттестованные газовые смеси отсутствовали, поэтому соответствующие образцы сравнения готовили самостоятельно [29] статическим разбавлением индивидуальных веществ. Газом-разбавителем служил гелий марки 60 (ТУ 0271-011-45905715-02). Градуировочные смеси готовили в диапазоне парциальных давлений 10–7–10–3 атм (10–5–0.1 об. %). Погрешность их приготовления не превышала 15%.
Концентрацию примеси i (сi) в образцах арсина находили по уравнению:
где pi – установленное парциальное давление примеси, Ргидр – давление пробы арсина.Концентрации веществ, для которых отсутствовали образцы сравнения (алкилпроизводные арсина, диарсин, серосодержащие вещества, некоторые хлорсодержащие углеводороды, ряд изомеров углеводородов С4–С6), определяли с использованием коэффициентов чувствительности детектирования, установленных с использованием их полных сечений ионизации [30–33].
Контрольный опыт проводили дозированием в колонку промывочного газа системы напуска – гелия марки 7.0 (ТУ 0271-001-45905715-02).
Пределы обнаружения примесей по парциальному давлению pmin рассчитывали для режима SIM по утроенному стандартному отклонению сигнала контрольного опыта [34]:
где s (ед. счета) – стандартное отклонение аналитического сигнала контрольного опыта, A (ед. счета/атм) – коэффициент чувствительности детектора к определяемому веществу.Стандартное отклонение сигнала контрольного опыта рассчитывали по колебаниям площади пика, относящегося к времени выхода определяемой примеси:
(3)
$s = \sqrt {\frac{{\sum\limits_{i = 1}^n {{{{\left( {{{B}_{i}} - \bar {B}} \right)}}^{2}}} }}{{n - 1}}} ,$Коэффициенты чувствительности детектирования А определяли экспериментально с применением образцов сравнения, а также рассчитывали с использованием полных сечений ионизации.
Пределы обнаружения примесей сmin рассчитывали из соотношения Pmin и максимального давления анализируемого газа в системе дозирования Pmax = 1.0 атм:
Пределы обнаружения примесей рассчитывали в режиме SIM по ионам, выбранным из их масс-спектров, для которых значение сигнал/шум является максимальным (табл. 1).
Правильность результатов определения подтверждали способом варьирования величины пробы. Для этого сравнивали модуль разности средних значений результатов определения концентраций $\left| {{{{\bar {c}}}_{1}} - {{{\bar {c}}}_{2}}} \right|$ с максимальной погрешностью этой разности ε [35]. Максимальную погрешность разницы результатов ε рассчитывали по формуле:
(5)
$\varepsilon = {{t}_{{P{\text{,}}f}}}{{s}_{{{\text{взв}}}}}\sqrt {\frac{1}{{{{n}_{1}}}} + \frac{1}{{{{n}_{2}}}}} ,$Средневзвешенное стандартных отклонений sвзв рассчитывали по формуле:
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
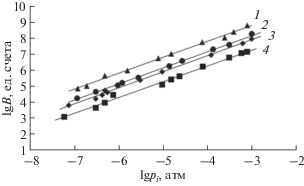
На рис. 1 приведены примеры градуировочных зависимостей для ряда примесных веществ. Видно, что полученные зависимости линейны в исследованной области парциальных давлений примесей.
Рис. 1.
Зависимость логарифма площади хроматографического пика B (ед. счета) от логарифма парциального давления pi (атм) примеси: С2H3Cl (1), GeH4 (2), н-C6H14 (3), CH2Cl2 (4). Уравнения регрессии: (1) lgB = 0.998 lgpi + 11.840, R2 = 0.991; (2) lg B = = 1.011lgpi + 11.237, R2 = 0.999; (3) lgB = 1.010lgpi + + 10.977, R2 = 0.999; (4) lgB = 0.992lgpi + 10.272, R2 = 0.999.
В табл. 1 приведены концентрации примесей в некоторых исследованных образцах арсина, которые в работе [18] отнесены к следующим группам: “исходные” после синтеза, “легкая” и “тяжелая” фракции, отобранные в верхней и нижней частях ректификационной колонны, а также очищенный образец – “ректификат”. Как видно, в исходном образце концентрации примесей находятся на уровне 10–6–0.1 об. %. В ходе очистки в “легкой” фракции концентрируются примеси предельных и непредельных углеводородов С1–С2, оксида азота(I), германа, сероводорода. Их концентрации могут отличаться до трех порядков величины по сравнению с “исходным” образцом. В “тяжелой” фракции концентрируются примеси с более высокими температурами кипения, чем у арсина – предельные, непредельные, ароматические углеводороды С3–С7, хлорсодержащие углеводороды, алкилпроизводные арсина и диарсина. Содержание последних достаточно велико и достигает единиц процентов.
Во всех образцах установлены примеси постоянных газов. Их присутствие во многом связано с возможностью проникновения в систему очистки и перегрузки арсина через полимерные уплотняющие материалы за счет диффузии [36]. Аргон и азот, кроме того, являлись промывочными газами технологического оборудования. Диоксид углерода в ходе очистки концентрируется в “легкой” фракции, несмотря на то, что в соответствии с его температурой кипения он должен переходить в “тяжелую” фракцию. Такое нетипичное поведение этой примеси, вероятно, связано с тем, что при давлении, при котором ведется ректификация, диоксид углерода не сжижается.
Присутствие примесей остальных веществ связано с их поступлением из исходного мышьяксодержащего сырья. Алкилпроизводные арсина и диарсина, а также диарсин могут образовываться в процессе синтеза арсина с участием углеводородов, содержание которых в получаемом арсине велико и достигает 0.1 об. %.
В очищенном образце установлены примеси постоянных газов, серооксида углерода, пропилена, диарсина. Концентрации их не превышают 10–5 об. %. Содержания остальных веществ не превышают пределов обнаружения.
В табл. 1 приведены достигнутые пределы обнаружения примесей. Они рассчитаны для максимального давления пробы арсина 1.0 атм и составили 2 × 10–7–2 × 10–4 об. %. Из табл. 1 видно, что пределы обнаружения находятся на уровне наиболее низких, известных из литературы, а для примесей GeH4, C2H6, C3H6, N2O, COS, H2S, СH2Cl2, СHCl3, С2H4Cl2, С6H6, С7H8 они ниже в 2–100 раз. Для ряда углеводородов С3–С7, галогенсодержащих углеводородов, алкилпроизводных арсина и диарсина пределы обнаружения определены впервые. Достаточно высокие их значения для некоторых кислородсодержащих веществ объясняются размыванием хроматографических пиков, как это показано в работе [18]. Пределы обнаружения этилена и фосфина также достаточно высоки. Это связано с их определением из пробы в 10 раз ниже по величине, чем для остальных веществ, для исключения наложения пика основного компонента [18].
Правильность анализа арсина оценивали методом варьирования величины пробы на примере ряда примесей (табл. 2). Как видно из табл. 2, изменение давления арсина в системе дозирования в 2 раза не приводит к статистически значимым различиям определения концентраций примесей. Различие в результатах определения не превышает максимальной погрешности этой разницы ε [34, 37]. Косвенным подтверждением правильности полученных результатов является соответствие направления концентрирования веществ их летучести при ректификационной очистке арсина.
Таблица 2.
Подтверждение правильности результатов (об. %) анализа арсина методом варьирования величины пробы (n1 = n2 = 5, Р = 0.95)
| Примесь | Давление пробы, атм | sвзв | |с1 – ${{\bar {c}}_{2}}$| | ε | |||
|---|---|---|---|---|---|---|---|
| 1.0 | 0.5 | ||||||
| ${{\bar {c}}_{1}}$ | s1 | ${{\bar {c}}_{2}}$ | s2 | ||||
| N2 | 6.2 × 10–5 | 0.5 × 10–5 | 5.7 × 10–5 | 0.4 × 10–5 | 0.4 × 10–5 | 0.5 × 10–5 | 9.1 × 10–6 |
| Ar | 7.4 × 10–6 | 0.8 × 10–6 | 8.3 × 10–6 | 0.9 × 10–6 | 0.9 × 10–6 | 0.9 × 10–6 | 1.3 × 10–6 |
| CO2 | 1.9 × 10–5 | 0.3 × 10–5 | 1.7 × 10–5 | 0.3 × 10–5 | 0.3 × 10–5 | 0.3 × 10–5 | 0.5 × 10–5 |
| COS | 5.6 × 10–6 | 0.7 × 10–6 | 6.2 × 10–6 | 0.7 × 10–6 | 0.7 × 10–6 | 0.6 × 10–6 | 1.0 × 10–6 |
| H2S | 1.5 × 10–5 | 0.2 × 10–5 | 1.7 × 10–5 | 0.2 × 10–5 | 0.2 × 10–5 | 0.2 × 10–5 | 0.3 × 10–5 |
| C3H6 | 2.5 × 10–5 | 0.4 × 10–5 | 2.2 × 10–5 | 0.3 × 10–5 | 0.4 × 10–5 | 0.3 × 10–5 | 0.6 × 10–5 |
| CH3AsH2 | 5.0 × 10–5 | 1.0 × 10–5 | 6.1 × 10–5 | 1.2 × 10–5 | 1.1 × 10–5 | 1.1 × 10-5 | 5.6 × 10–5 |
| As2H4 | 5.7 × 10–5 | 0.4 × 10–5 | 6.2 × 10–5 | 0.5 × 10–5 | 0.4 × 10–5 | 0.5 × 10–5 | 9.1 × 10–5 |
* * *
Таким образом, методом хромато-масс-спектрометрии исследован примесный состав арсина, в том числе высокочистого. Установлены и определены содержания примесей постоянных газов, диоксида углерода, гидридов, предельных, непредельных, ароматических углеводородов С1–С6, галогенсодержащих углеводородов, соединений серы, алкилпроизводных арсина и диарсина. Достигнутые пределы обнаружения примесей составляют 2 × 10–7–2 × 10–4 об. %. Большинство из них в настоящее время является наиболее низкими. Установлено, что концентрации примесей в высокочистом арсине не превышают 10–7–10–5 об. %. В неочищенных образцах они достаточно высоки и лежат в интервале 10–6–0.1 об. %.
Работа выполнена в соответствии с Программой фундаментальных научных исследований государственных академий наук на 2019–2021 годы, № темы 0095-2019-0001.
Список литературы
Бузынин Ю.Н., Гусев С.А., Данильцев В.М., Дроздов М.Н., Дроздов Ю.Н., Мурель А.В., Хрыкин О.И., Шашкин В.И. Монокристаллические слои GaAs, AlGaAs и InGaAs, полученные методом газофазной эпитаксии их металлоорганических соединений арсенида галлия // Письма в журн. техн. физики. 2000. Т. 26. № 7. С. 64.
Черняев В.Н., Кожитов Л.В. Технология эпитаксиальных слоев арсенида галлия и приборы на их основе. М.: Энергия, 1974. 231 с.
Бланк Т.В., Гольдберг Ю.А. Полупроводниковые фотоэлектропреобразователи для ультрафиолетовой области спектра // Физика и техника полупроводников. 2003. Т. 37. № 9. С. 1025.
Чурбанов М.Ф., Карпов Ю.А., Зломанов П.В., Федоров В.А. Высокочистые вещества. М.: Научный мир, 2018. 994 с.
Крылов В.А. Анализ высокочистых летучих веществ // Журн. аналит. химии. 2002. Т. 57. № 8. С. 790.
Зайков А.А., Зырянов С.М., Пульников И.И., Скорынин Г.М., Власов В.А. Определение содержания газообразных примесей в высокочистом арсине при его очистке на газовых центрифугах // Изв. Томского политехн. ун-та. 2006. Т. 309. № 3. С. 81.
Зайков А.А., Зырянов С.М., Кулинич Ю.А., Пульников И.И., Скорынин Г.М., Власов В.А. Исследование загрязнений, вносимых резиновыми уплотнителями, в очищаемый на газовых центрифугах арсин // Изв. Томского политехн. ун-та. 2007. Т. 310. № 1. С. 137.
Радугин Д.А., Сенников П.Г., Шкрунин В.Е., Яньков С.В. Молекулярное состояние и растворимость воды в арсине // Высокочистые вещества. 1993. № 1. С. 67.
Девятых Г.Г., Сенников П.Г. Спектроскопическое определение и изучение молекулярного состояния примеси воды в высокочистых летучих неорганических веществах // Успехи химии. 1995. Т. 64. № 9. С. 872.
Иванова Н.Т., Вислых Н.А., Воеводина В.В. Аналитический контроль производства гидридов As, P, Si и смесей на их основе // Высокочистые вещества. 1989. № 6. С. 102.
Крылов В.А., Красотский С.Г., Малышев В.М., Салганский Ю.М. Криогенный метод концентрирования примесей водорода, аргона, кислорода и азота при их газохроматографическом определении в летучих неорганических гидридах // Журн. аналит. химии. 2001. Т. 56. № 9. С. 1137.
Ежелева А.Е., Снопатин Г.Е., Малыгина Л.С. Применение пламенно-фотометрического детектора при хроматографическом анализе летучих неорганических гидридов особой чистоты // /Журн. аналит. химии. 1979. Т. 34. № 7. С. 2308.
Ежелева А.Е., Малыгина Л.С., Крылов В.А. Применение фотоионизационного детектора при газохроматографическом определении летучих неорганических гидридов и некоторых органических веществ // Высокочистые вещества. 1987. № 3. С. 214.
Воротынцев В.М., Балабанов В.В., Абдрахманов Р.Р., Малыгина Л.С. Электрохимический синтез особо чистого арсина // Высокочистые вещества. 1993. № 5. С. 22.
Крылов В.А., Чернова О.Ю., Салганский Ю.М., Малыгина Л.С., Котков А.П. Высокочувствительное определение примесей углеводородов в арсине методом реакционной газовой хроматографии /// Журн. аналит. химии. 2003. Т. 58. № 8. С. 841.
Иванова Н.Т., Вислых Н.А., Воеводина В.В. Источник примесей при получении арсина и фосфина // Высокочистые вещества. 1990. № 5. С. 198.
Крылов В.А., Чернова О.Ю., Созин А.Ю., Котков А.П. Хромато-масс-спектрометрический анализ арсина высокой чистоты // Заводск. лаборатория. Диагностика материалов. 2010. Т. 76. № 3. С. 13.
Созин А.Ю., Чернова О.Ю., Сорочкина Т.Г., Котков А.П., Гришнова Н.Д., Полежаев Д.М., Пушкарев Г.В., Буланова А.А. Идентификация примесей в высокочистом арсине методом хромато-масс-спектрометрии // Журн. аналит. химии. 2020. Т. 75. № 5. С. 422.
Турыгин В.В., Смирнов М.К., Березкин М.Ю., Сохадзе Л.А., Степнова Н.П., Томилов А.П., Федоров В.А., Потолоков Н.А. Физико-химические основы получения высокочистых соединений мышьяка из продуктов детоксикации люизита // Неорганические материалы. 2017. Т. 53. № 4. С. 392.
Березкин В.Г., Королев А.А., Хотимский В.С. Политриметилсилилпропин как адсорбент в капиллярной газовой хроматографии // Доклады АН. 2000. Т. 370. С. 200.
http://www.mathesongas.com. (27.02.2020)
https://www.versummaterials.com (27.02.2020)
https://www.praxair.com (27.02.2020)
Девятых Г.Г., Карпов Ю.А., Осипова Л.И. Выставка-коллекция веществ особой чистоты. М.: Наука, 2003. 236 с.
Девятых Г.Г. Гидридный метод получения элементов особой чистоты. Получение и анализ веществ особой чистоты. Горький: ИХ АН СССР, 1974. С. 15.
Девятых Г.Г., Зорин А.Д. Летучие неорганические гидриды особой чистоты. М.: Наука, 1974. 206 с.
Жигач А.Ф., Стасиневич Д.С. Химия гидридов. Л.: Химия, 1969. 676 с.
Девятых Г.Г. О гидридном методе получения элементов особой чистоты // Труды по химии и химической технологии ИХ АН СССР. 1962. Вып. 2. С. 221.
Крылов В.А., Чернова О.Ю., Созин А.Ю. Высокочувствительное хромато-масс-спектрометрическое определение примесей в моногермане высокой чистоты с применением адсорбционной капиллярной колонки с углеродным сорбентом // Заводск. лаборатория. Диагностика материалов. 2016. Т. 82. № 2. С. 23.
Крылов В.А., Созин А.Ю., Зорин В.А., Березкин В.Г., Крылов А.В. Хроматомасс – спектрометрическое определение примесей в изотопно-обогащенном силане высокой чистоты // Масс-спектрометрия. 2008. Т. 4. С. 225.
Fitch W.L. Calculation of relative electron impact total ionization cross sections for organic molecules // Anal. Chem. 1983. V. 55. P. 832.
Mann J. B. Ionization cross sections of the elements calculated from mean-square radii of atomic orbitals // J. Chem. Phys. 1967. V. 46. P. 1646.
Beran J. A., Kevan L. Molecular electron ionization cross sections at 70 eV // J. Phys. Chem. 1959. V. 73. P. 3866.
Золотов Ю.А. Основы аналитической химии. Изд. 2-е.: в 2-х тт. М.: Высшая школа, 2000. Т. 1. 351 с.
Чарыков А.К. Математическая обработка результатов химического анализа. Методы обнаружения и оценки ошибок. Л.: Химия, 1984. 168 с.
Девятых Г.Г., Крылов В.А., Красотский С.Г., Саркисов А.В., Батурина Н.М., Колесников А.Н., Яньков С.В. Влияние газопроницаемости политетрафторэтилена на правильность газохроматографического определения постоянных газов в высокочистых летучих веществах // Высокочистые вещества. 1988. № 6. С. 173.
ГОСТ Р 8.736-2011 Измерения прямые многократные. Методы обработки результатов измерений. Основные положения. М.: Стандартинформ, 2013. 20 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии