

Журнал аналитической химии, 2021, T. 76, № 9, стр. 832-843
Новый сорбент для гидрофильной хроматографии на основе силикагеля, модифицированного по реакции уги
Н. Ю. Чикурова a, А. О. Шемякина a, Д. Э. Брыскина b, В. Н. Нуриев a, А. А. Комаров b, М. А. Статкус a, А. Н. Ставрианиди a, А. В. Чернобровкина a, *
a Московский государственный университет имени М.В. Ломоносова, химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
b ООО “НВЦ Агроветзащита”
129329 Москва, Игарский проезд, 4, стр. 2, Россия
* E-mail: chernobrovkina@analyt.chem.msu.ru
Поступила в редакцию 05.04.2021
После доработки 16.04.2021
Принята к публикации 16.04.2021
Аннотация
Получены новые неподвижные фазы для гидрофильной хроматографии (ГИХ) с функциональными слоями, сформированными по мультикомпонентной реакции Уги. В качестве компонентов реакции использованы ацетон, гликолевая кислота, этиловый эфир изоциануксусной кислоты и 3-аминопропилсиликагель, выступающий также в роли матрицы сорбентов. Изучение влияния растворителя на выход реакции показало, что большая степень покрытия матрицы достигается при проведении реакции в этаноле. Оценку хроматографических свойств новых сорбентов в сравнении с исходной матрицей проводили с использованием тестов Танака для гидрофильных фаз, а также путем изучения удерживания полярных аналитов различных классов. Синтезированные сорбенты продемонстрировали высокую эффективность и селективность при разделении модельных смесей сахаров, аминокислот и водорастворимых витаминов в режиме ГИХ.
Гидрофильная хроматография (ГИХ) – современный метод определения полярных нейтральных и заряженных веществ на полярных неподвижных фазах, обеспечивающий иную селективность по сравнению с традиционно используемой обращенно-фазовой хроматографией (ОФ ВЭЖХ) и позволяющий разделять сильнополярные аналиты, которые практически не удерживаются в режиме ОФ ВЭЖХ и имеют несимметричную форму пиков [1, 2]. Высокие селективность и эффективность в гидрофильной хроматографии достигаются за счет использования полярных сорбентов с разнообразными по строению и природе функциональными группами, среди которых наибольшие гидрофильность и факторы удерживания полярных аналитов обеспечивают амидные неподвижные фазы [2]. Однако в связи с усложнением задач, решаемых ГИХ, создание неподвижных фаз с улучшенной эффективностью и альтернативной селективностью остается одним из основных направлений развития метода.
На текущий момент синтез новых селективных неподвижных фаз для ГИХ требует сложных многостадийных процедур ковалентного модифицирования. В большинстве случаев проводят реакции с функциональными группами, закрепленными на поверхности предварительно модифицированного силикагеля [3–5], однако зачастую для одновременного введения в функциональный слой сорбентов групп различных типов сначала получают соответствующие лиганды и только после этого закрепляют их на поверхности матрицы [6–8].
Перспективным подходом к созданию новых функциональных слоев сорбентов для ГИХ является использование мультикомпонентных реакций, преимуществами которых являются высокий выход продуктов, простота исполнения, возможность получать необходимый продукт в одном реакторе в результате одностадийной или многостадийной реакции без выделения промежуточных продуктов (“one-pot“ реакция), а также разнообразие возможностей в изменении структуры функционального слоя путем варьирования исходных соединений. Одним из примеров таких реакций, позволяющих получать гидрофильные амидные группы, является реакция Уги [9, 10] – мультикомпонентная реакция, включающая взаимодействие аминов, карбонильных соединений, изоцианидов и органических/неорганических кислот. Классическую реакцию Уги можно проводить как в растворе, так и в твердой фазе. Реакция может протекать в водной, водно-органической либо в органической среде, причем выбор растворителя может значительно влиять на выход реакции [9]. Кроме того, селективность реакции в воде уменьшается из-за образования продуктов реакции Пассерини, которая в таких условиях значительно ускоряется. Добавление кислот Льюиса в реакционную смесь может стать благоприятным фактором для повышения выхода реакции [11].
На сегодняшний день в литературе существует всего несколько примеров использования мультикомпонентных реакций, в том числе реакции Уги, для создания новых неподвижных фаз. При этом практически все варианты предполагают предварительное получение лигандов по данной реакции и их последующее закрепление на поверхности матрицы. Так, в работе [6] мультикомпонентную реакцию Уги использовали для создания цвиттер-ионных фрагментов на поверхности сорбентов для ОФ ВЭЖХ и ГИХ. Авторы синтезировали аминофосфонатные цвиттер-ионные лиганды и только после очистки ковалентно закрепили их на поверхности силикагеля. Синтез лиганда по реакции Уги обеспечил высокий выход продукта и возможность легкого управления селективностью получаемых сорбентов. Однако предложенный авторами работы [6] способ синтеза неподвижных фаз является трудоемким и требует большого количества времени из-за необходимости очистки лиганда и последующего модифицирования матрицы.
Один из вариантов успешного использования реакции Уги на твердых носителях – полистирольных смолах – продемонстрировали авторы работы [12]. В данном случае твердый носитель с предварительно введенными функциональными группами выступал в качестве одного из компонентов мультикомпонентной реакции для получения и выделения ряда соединений. Авторы подтвердили успешное протекание реакции и получение соответствующего продукта методом ЯМР-спектроскопии.
Учитывая такие преимущества реакции Уги, как высокий выход, быстрота протекания и мягкие условия проведения (комнатная температура) [9, 10], а также формирование гидрофильных амидных фрагментов в продуктах реакции, можно предположить, что мультикомпонентная реакция Уги может быть простым и перспективным подходом для синтеза новых гидрофильных неподвижных фаз для ГИХ. При этом использование аминированного силикагеля в качестве аминокомпонента в этой реакции обеспечит модифицирование матрицы исключительно необходимыми функциональными группами благодаря легкости отделения возможных побочных продуктов конкурирующей реакции Пассерини, образующихся исключительно в жидкой фазе. Такой способ синтеза неподвижной фазы для ГИХ станет уникальным в своем роде, поскольку в реакции непосредственно будет участвовать аминированная матрица, и образующийся лиганд сразу же будет закрепляться на ней.
Целью данной работы являлся синтез сорбентов на основе аминированного силикагеля с использованием мультикомпонентной реакции Уги, выбор растворителя для повышения степени модифицирования матрицы, изучение полученных сорбентов в режиме ГИХ на примере разделения полярных веществ различных классов и оценка возможностей их применения для анализа реальных объектов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приборы и материалы. Для синтеза сорбентов в качестве матрицы использовали силикагель с привитыми аминопропильными радикалами Диасфер-110-Амин (БиоХиммак СТ, Россия) со сферическими частицами диаметром 5 мкм, 1.70% N. Применяли ацетон ч. д. а. (ХимМед, Россия), гликолевую кислоту ч. д. а. (Panreac, Испания), этанол ч. д. а. (Лабтех, Россия), метанол ч. д. а. (Лабтех, Россия).
Для синтеза этилового эфира изоциануксусной кислоты использовали оксихлорид фосфора oc. ч. (Элма-Хим, Россия) напрямую из вскрытой ампулы без дополнительной очистки; этил-N-формилглицинат 98%, синтетический [13, 14]; дихлорметан х. ч. (Компонент-Реактив, Россия), перегоняли над свежим оксидом фосфора(V); триэтиламин х. ч. (Acros, Россия), перегоняли над гидроксидом кальция.
Для изучения свойств сорбентов использовали толуол х. ч. (Компонент-Реактив, Россия); аденозин, >99.0%; урацил, >99.0%; теобромин, >98.0%; теофиллин, >98.0%; уридин, >98.0%; видарабин моногидрат, >98.0%; 5-метилуридин, >98.0%; 2'-деоксиуридин, >98.0%; N, N, N-триметилфенил-аммоний хлорид, >98.0%; тозилат натрия, >90.0% (TCI, Япония).
Сахара: D-(+)-рибоза, 98%; D-(+)-глюкоза, 99.5%; D-(+)-фруктоза, 99.9%; D-(+)-лактоза моногидрат, >98%; D-(+)-мальтоза моногидрат, >98%; D-(+)-сахароза, >98%; L-(+)-рамноза моногидрат, >98%; D-(+)-ксилоза, >98%; D-(+)-раффиноза пентагидрат (TCI, Япония).
Витамины: никотинамид, 99.7%; кислота никотиновая (Sigma-Aldrich, США). Рибофлавин (B2), 99%; пиридоксин гидрохлорид (B6), 99%; цианокобаламин (В12) ч. д. а.; аскорбиновая кислота (витамин C), 99%; тиамин (В1), >96% (TCI, Япония).
Аминокислоты: D,L-фенилаланин х. ч.; L-пролин х. ч.; D,L-серин х. ч.; L-изолейцин х. ч.; L-лейцин х. ч.; L-метионин х. ч. (Serva, Германия); L-аспарагин х. ч.; β-аланин х. ч; глицин х. ч. (Merck, Германия).
Прочие реактивы: ацетонитрил, “HPLC gradient grade”; кислота уксусная ледяная, 99.5%; кислота ортофосфорная, 85% х. ч., хлорид калия ч. д. а. (Panreac, Испания); этанол ч. д. а.; ацетат аммония ч. (Лабтех, Россия); ацетон ч. д. а. (ХимМед, Россия); 1,4-диоксан ч. д. а. (Компонент-Реактив, Россия).
Для изучения хроматографических свойств сорбентов использовали систему ВЭЖХ Dionex Ultimate 3000 (Thermo Scientific, США), состоящую из двухканального насоса высокого давления, автоматической системы ввода пробы, термостата для колонок и детектора на диодной матрице. Регистрацию хроматограмм осуществляли с помощью программного пакета Chromeleon 7 (Thermo Fisher Scientific, США). Для масс-спектрометрического детектирования применяли тройной квадрупольный масс-анализатор Qtrap 5500 (AB Sciex, Канада), оснащенный источником электрораспылительной ионизации (ЭРИ). Данные обрабатывали с помощью программного обеспечения Analyst (version 1.5, AB Sciex, Канада). Система ВЭЖХ состояла из изократического ВЭ-ЖХ-насоса, шестиходового крана-дозатора и рефрактометрического детектора (Agilent Technologies, США). Регистрацию хроматограмм осуществляли с помощью программного пакета ChemStation (Agilent Technologies, США). Жидкостной хроматограф Shimadzu Prominence состоял из следующих блоков: насоса высокого давления LC-20AD с четырехканальным градиентным смесителем на стороне низкого давления, дегазатора DGU-20A5R, управляющего модуля CBM-20A, ручного инжектора Rheodyne 7725i, детектора по светорассеянию ELSD-LT II. Сжатый воздух для детектора получали с помощью компрессора Jun-Air OF302-25B. Давление сжатого воздуха для детектора составляло 350 кПа, температура нагрева испарителя +40°C, усиление детектора k = 6 выбирали, исходя из высоты пиков на хроматограмме в ходе эксперимента. Объем пробы – 20 мкл.
В работе использовали стальные колонки размером 100 × 3 мм, заполнение которых осуществляли при помощи насоса Knauer K-1900 (Knauer, Германия).
Условия масс-спектрометрического детектирования. Для разделения сахаров на колонке С-Ум использовали двухкомпонентную смесь растворителей, состоящую из деионизованной воды и ацетонитрила, при 30°C со скоростью потока 0.4 мл/мин. Элюирование проводили в градиентном режиме: 0–4.5 мин 85–83% CH3CN; 4.5–11 мин 63% CH3CN, 11–19 мин 85% CH3CN. Сахара детектировали в режиме ЭРИ-МС/МС-регистрации отрицательных ионов. Напряжение на капилляре –4500 В; температура источника ионов 350°C; давление газа-распылителя (N2) 50 psi; газа-осушителя (N2) 50 psi; газа-завесы (N2) 20 psi; входной потенциал –10 В; потенциал на выходе из ячейки соударений –15 В.
Витамины разделяли, используя двухкомпонентную смесь растворителей, состоящую из 20 мМ буферного раствора формиата аммония с рН 3.7 (A) и ацетонитрила (B). Скорость потока 0.4 мл/мин, температура термостата колонки 25°C. Условия градиентного элюирования: 0–6 мин 98% CH3CN, 6–25 мин 98–5% CH3CN, 25–30 мин 5% CH3CN, 30–38 мин 98% CH3CN. Витамины детектировали в режиме ЭРИ-МС/МС-регистрации отрицательных ионов. Напряжение на капилляре 4500 В; температура источника ионов 350°C; давление газа-распылителя (N2) 50 psi; газа-осушителя (N2) 50 psi; газа-завесы (N2) 20 psi; входной потенциал 10 В; потенциал на выходе из ячейки соударений 15 В.
Специфичные параметры детектирования, а именно потенциал декластеризации (ДП) и энергию соударений (ЭС), подбирали для каждого аналита по отдельности (табл. 1). Время регистрации каждого ионного перехода 50 мс.
Таблица 1.
Масс-спектрометрические параметры, выбранные для детектирования сахаров и витаминов
| Соединение | Полярность | ДП, В | Ион предшест-венник, m/z | Фрагментный ион, m/z | ЭС, В |
|---|---|---|---|---|---|
| B1 | + | 70 | 265 | 122.1 | 21 |
| 265 | 144.1 | 19 | |||
| B2 | + | 70 | 377.2 | 172.1 | 53 |
| 377.2 | 243.1 | 31 | |||
| Никотинамид | + | 70 | 123 | 96.2 | 27 |
| 123 | 79.9 | 29 | |||
| B6 | + | 70 | 169.8 | 134.1 | 29 |
| 169.8 | 152.1 | 19 | |||
| Никотиновая кислота | + | 70 | 124 | 78 | 31 |
| 124 | 80 | 29 | |||
| B12 | + | 70 | 678.5 | 147.1 | 35 |
| 678.5 | 359.3 | 35 | |||
| Glu/Fru | – | –30 | 179.1 | 119.1 | –10 |
| 179.1 | 89.1 | –20 | |||
| 179.1 | 59.1 | –25 | |||
| Suc/Mal/Lac | – | –40 | 341.3 | 59.1 | –55 |
| 341.3 | 179.1 | –18 | |||
| Xyl/Rib | – | –30 | 149.1 | 59.1 | –25 |
Синтез этилового эфира изоциануксусной кислоты проводили в соответствии со схемой 1 [13]:
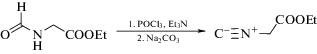
Схема 1 . Схема синтеза этилового эфира изоциануксусной кислоты.
К раствору 19.7 г (0.15 моль) перегнанного этилового эфира N-формилглицина [14] и 37.5 г (0.37 моль) триэтиламина в 150 мл дихлорметана добавляли по каплям при 0°С 23.0 г (0.15 моль) оксихлорида фосфора(V). Смесь перемешивали в течение 1 ч, поддерживая температуру около 0°С. Медленно прибавляли раствор 30 г (0.28 моль) безводного карбоната натрия в 120 мл воды при энергичном перемешивании при 20–25°С. Полученную смесь перемешивали 30 мин при комнатной температуре. После разделения фаз водную фазу разбавляли в 2.5 раза и экстрагировали двумя порциями дихлорметана по 80 мл. Объединeнные органические фазы промывали насыщенным раствором хлорида натрия, высушивали над карбонатом калия, растворитель отгоняли на роторном испарителе без нагревания. Остаток перегоняли в вакууме водоструйного насоса, собирая фракцию, кипящую при 79–81°С (10 мм рт. ст.). Получили 9.8 г (56%) практически бесцветной жидкости с неприятным запахом, желтеющей при стоянии. По данным ЯМР 1H в течение более года состав продукта практически не меняется (мольная чистота ≥95%), если хранится в стеклянной посуде при –20°С (в атмосфере сухого аргона). Спектры ЯМР 1Н и 13С подтвердили структуру полученного соединения. ЯМР 1Н (CDCl3; 400 МГц; δ, м.д.; J, Гц): 4.28 (2Н, кв, 3J = 7.15 Гц, CH3СН2О), 4.22 (2Н, с, СН2N), 1.31 (3Н, т, J = 7.15 Гц, СН3CH2O). ЯМР 13С (CDCl3, 100 МГц, δ, м.д.): 163.76, 160.86, 62.60, 43.34, 13.81.
Синтез сорбентов и заполнение хроматографических колонок. Сорбенты С-Ум (в метаноле) и С‑Уэ (в этаноле) получали ковалентным модифицированием полярных фрагментов матрицы – аминопропилсиликагеля “Диасфер-амин” по реакции Уги в разных растворителях: метаноле и этаноле, соответственно (схема 2 ). Строго соблюдали порядок добавления реагентов. К 1.2 г аминопропилсиликагеля (1) добавляли 95 мкл ацетона (2), 142 мкл этилового эфира изоциануксусной кислоты (lgP = 0.91) (3) и 0.1 г гликолевой кислоты (4). Реакционную смесь перемешивали в течение 24 ч со скоростью 250–300 об/мин. Продукт реакции отфильтровывали на фильтре со стеклянным пористым дном, промывали 100 мл дистиллированной воды и 80 мл ацетонитрила.
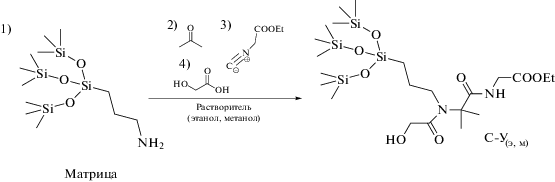
Схема 2 . Схема синтеза и предполагаемая структура неподвижных фаз С-Ум (в метаноле) и С-Уэ (в этаноле).
Для заполнения колонок использовали смесь 10 мл 0.1 М раствора KCl, 8 мл этанола и 2 мл 1,4-диоксана в качестве раствора для приготовления суспензии сорбентов, а дегазированный ацетонитрил – в качестве подвижной фазы. Заполнение колонки проводили при давлении 250–300 бар.
Анализ реальных объектов. Вино Gran Marques Reserva предварительно разбавляли в два раза; 0.3 г кофе Nescafe Gold Cappuccino сначала растворяли в 100 мл воды и затем также разбавляли в два раза. Пробоподготовка включала очистку пробы путем твердофазной экстракции на картридже Chromabond® C18 ec f (силикагель, модифицированный октадецильными группами, объем 3 мл, масса адсорбента 500 мг, Macherey-Nagel, Германия) при помощи вакуумного манифолда. Картридж предварительно кондиционировали 10 мл ацетонитрила и 5 мл деионизованной воды. К полученным экстрактам добавляли ацетонитрил для достижения соотношения вода–ацетонитрил согласно составу подвижной фазы.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В настоящей работе для получения неподвижных фаз по мультикомпонентной реакции Уги использовали 3-аминопропилсиликагель (сорбент С), ацетон, гликолевую кислоту и этиловый эфир изоциануксусной кислоты. Как отмечено выше, мультикомпонентные реакции предоставляют широкий спектр возможностей для варьирования структуры получаемых продуктов путем замены одного или нескольких компонентов реакции. На начальном этапе работы основной задачей было добиться лучшей степени покрытия матрицы функциональным слоем фиксированной структуры. С учетом литературных данных о влиянии органического растворителя на выход реакции синтез проводили в метаноле и в этаноле. В результате получили сорбенты, условно обозначенные как С-Ум (в метаноле) и С-Уэ (в этаноле). Для оценки успешности синтеза хроматографические характеристики полученных сорбентов сравнивали с таковыми для матрицы (сорбента С – 3-аминопропилсиликагеля). Сравнение осуществляли с помощью теста Танака для гидрофильных неподвижных фаз [15, 16] и на примере разделения модельных смесей сахаров, водорастворимых витаминов и аминокислот.
Тест Танака. Исходя из величин фактора удерживания уридина k(U) (табл. 2), отвечающего за оценку гидрофильных взаимодействий, сорбенты, полученные по реакции Уги, демонстрируют меньшие величины фактора удерживания уридина k(U) по сравнению с матрицей, что, по-видимому, связано с введением гидрофобных фрагментов в структуру функционального слоя. Это согласуется с величинами параметров гидрофобности Ханша lgP, рассчитанных с помощью Epiweb 4.1 для аминопропильного радикала и структурного фрагмента, полученного после реакции Уги, и составляющих 0.34 и 0.51 соответственно. В соответствии с результатами теста, представленными в табл. 2, значения большинства параметров для сорбента С-Ум близки и незначительно отличаются от таковых для сорбента С (матрицы). В случае сорбента С-Уэ существенное уменьшение наряду с k(U) наблюдали для метиленовой α(СН2), гидроксильной α(ОН) и анионообменной селективности α(АX). Полученные данные позволяют предположить, что проведение реакции в этаноле обеспечивает больший выход по сравнению с метанолом и, следовательно, приводит к увеличению степени покрытия матрицы новым функциональным слоем.
Таблица 2.
Величины коэффициентов селективности теста Танака
| Параметр | С (матрица) | С-Ум | С-Уэ |
|---|---|---|---|
| k(U) | 3.95 | 3.78 | 2.68 |
| α(CH2) | 1.49 | 1.55 | 1.11 |
| α(OH) | 2.09 | 2.30 | 1.54 |
| α(V/A) | 1.36 | 1.37 | 1.30 |
| α(CX) | 0.06 | 0.05 | 0.33 |
| α(AX) | 14.31 | 15.01 | 11.87 |
| α(Tb/Tp) | 0.72 | 0.77 | 0.72 |
При этом отмечено, что селективность по отношению к стереоизомерам α(V/A) и катионообменная селективность α(СX) практически не изменяются для обоих синтезированных сорбентов по сравнению с матрицей, а величины α(Tb/Tp) < 1 подтверждают основную природу всех сорбентов. Незначительное увеличение α(СX) для сорбента С-Уэ по сравнению с матрицей может быть вызвано большим фактором удерживания гидрофобного катиона и меньшим – полярного урацила, являющимися тестовыми аналитами для расчета этого параметра, вследствие снижения гидрофильности поверхности при модифицировании по реакции Уги.
Сахара являются удобными модельными аналитами для изучения свойств и возможностей гидрофильных сорбентов в отношении разделения полярных нейтральных веществ. В удерживание сахаров вносит вклад не только распределение в адсорбированном на полярной неподвижной фазе приповерхностном слое воды, но также и адсорбционный механизм, и образование водородных связей с ОН-группами [17–19].
Показано, что при модифицировании матрицы по реакции Уги времена удерживания сахаров уменьшаются, что согласуется с уменьшением гидрофильности поверхности по результатам теста Танака. Однако при этом значительно увеличивается эффективность (рис. 1), что может быть следствием уменьшения вклада сопротивления массопереносу при уменьшении толщины функционального слоя, в том числе приповерхностного слоя воды (в котором реализуется распределение) в результате создания структурных фрагментов по реакции Уги на поверхности аминопропилсиликагеля. Более того, селективность для пары мальтоза/лактоза, обладающих одинаковым значением параметра гидрофобности Ханша (lgP = –5.46, Epiweb 4.1), существенно увеличилась для сорбентов С-Ум и С-Уэ по сравнению с матрицей С (рис. 1), что обеспечило возможность разрешения этой пары сахаров. Коэффициенты селективности αMal/Lact составили 1.15–1.20. В целом для новых сорбентов селективность по другим сахарам несколько уменьшилась, однако благодаря увеличению эффективности при модифицировании аминопропилсиликагеля по реакции Уги удалось разделить девять сахаров за 23 мин при составе подвижной фазы CH3CN–Н2O (85 : 15, по объему).
Рис. 1.
Шкалы селективности сахаров относительно рибозы (слева), диаграмма эффективности определения сахаров (справа). Подвижная фаза: CH3CN–H2O (85 : 15, по объему); скорость потока 1 мл/мин; рефрактометрическое детектирование: 1 – рибоза, 2 – ксилоза, 3 – арабиноза, 4 – фруктоза, 5 – манноза, 6 – глюкоза, 7 – сахароза, 8 – мальтоза, 9 – лактоза.
Поскольку использование рефрактометрического детектора позволяет работать только в изократическом режиме, далее в работе использовали испарительный детектор по светорассеянию, что дало возможность осуществлять градиентное элюирование, тем самым увеличив количество разделяемых углеводов в модельной смеси. Так, на сорбенте С-Ум, демонстрирующем лучшую эффективность по сахарам, за 20 мин было достигнуто разделение модельной смеси 10 сахаров, включая трисахарид раффинозу, факторы удерживания которого слишком высоки в изократическом режиме элюирования. Соотношение воды и ацетонитрила в подвижной фазе изменяли в интервале 14–27 и 86–73% соответственно. Сорбент С-Ум продемонстрировал приемлемую форму пиков (коэффициенты асимметрии 1.0–2.1) и хорошее разрешение для смеси сахаров. Хроматограмма и условия элюирования приведены на рис. 2, рассчитанные хроматографические параметры – в табл. 3.
Рис. 2.
Хроматограмма модельной смеси сахаров. Условия: сорбент С-Ум, подвижная фаза H2O–CH3CN; скорость потока 1 мл/мин; режим градиентного элюирования: 0–5.5 мин 14% H2O, 6.5–12.5 мин 22% H2O, 13.5–26.5 мин 27% H2O, 28.5–32.5 мин 14% H2O, детектирование по светорассеянию (размеры колонки 100 × 3 мм): 1 – рибоза, 2 – ксилоза, 3 – арабиноза, 4 – фруктоза, 5 – манноза, 6 – глюкоза, 7 – сахароза, 8 – мальтоза, 9 – лактоза, 10 – раффиноза.
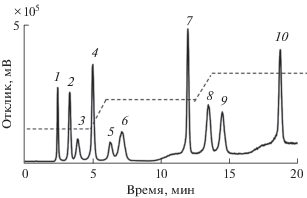
Таблица 3.
Хроматографические параметры разделения сахаров
| Сахар | k' | Rs | As | N, тт/м |
|---|---|---|---|---|
| Рибоза | 1.7 | – | 2.1 | 11 000 |
| Ксилоза | 2.6 | 1.8 | 1.2 | 8000 |
| Арабиноза | 3.3 | 1.6 | 1.6 | 9000 |
| Фруктоза | 4.4 | 1.7 | 1.7 | 18 000 |
| Манноза | 5.8 | 1.5 | 1.0 | 22 000 |
| Глюкоза | 6.7 | 1.2 | 1.2 | 14 000 |
| Сахароза | 12.1 | 2.1 | 1.3 | – |
| Мальтоза | 13.6 | 1.2 | 1.4 | – |
| Лактоза | 14.7 | 1.1 | 1.2 | – |
| Раффиноза | 19.4 | 1.4 | 1.6 | – |
Условия: сорбент С-Ум, подвижная фаза H2O–CH3CN; скорость потока 1 мл/мин; режим градиентного элюирования (см. рис. 4), детектирование по светорассеянию.
Аминокислоты. В качестве модельных объектов для изучения свойств и возможностей синтезированных сорбентов использовали также аминокислоты, представляющие класс полярных цвиттер-ионных аналитов. Для разделения смесей аминокислот в качестве подвижной фазы выбрали фосфатный буферный раствор, не поглощающий электромагнитное излучение при длине волны 210 нм, используемой для детектирования аминокислот [20]. При выборе условий элюирования содержание буфера снизили до 1 мМ для обеспечения растворимости фосфата в водно-органической среде, долю водной фазы варьировали в пределах от 10 до 15%. Для синтезированных сорбентов оптимальная доля воды составила 15%. Повышение рН элюента до 6.5 – вблизи изоэлектрических точек большинства аминокислот – привело к увеличению их факторов удерживания и повышению селективности.
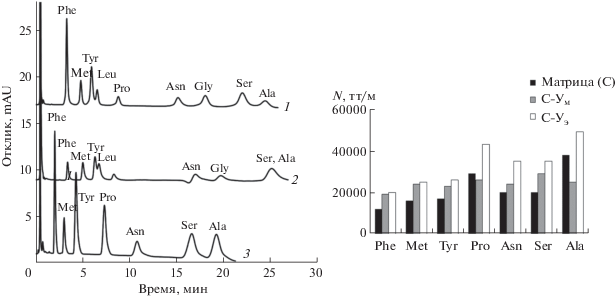
Для сорбентов С-Ум и С-Уэ увеличилось время удерживания аминокислот по сравнению с сорбентом С, а также возросла эффективность по аминокислотам (рис. 3). Несмотря большую гидрофобность полученных сорбентов, удерживание цвиттер-ионных аминокислот на них заметно возросло по сравнению с матрицей, вероятно, благодаря введению гидрокси- и амидных групп в функциональный слой сорбентов, что позволило увеличить число разделяемых аналитов. Однако, несмотря на большие величины факторов удерживания аминокислот на сорбенте С-Ум, на нем не удалось достичь разрешения пары серин/аланин. Сорбент С-Уэ продемонстрировал лучшую селективность по парам тирозин/лейцин и серин/аланин, лучшую симметрию пиков и большую эффективность по отношению к аминокислотам. Таким образом, можно сделать вывод, что модифицирование аминопропилсиликагеля по реакции Уги с выбранными реагентами в этаноле приводит к получению сорбента с лучшими хроматографическими характеристиками для веществ цвиттер-ионной природы, таких как аминокислоты, вероятно, вследствие большего выхода реакции в этаноле по сравнению с метанолом. Хроматограммы модельных смесей аминокислот представлены на рис. 3.
Рис. 3.
Хроматограммы модельной смеси аминокислот (слева), диаграмма эффективности определения аминокислот (справа). Сорбенты: С-Уэ (1), С-Ум (2), С (матрица) (3); подвижная фаза CH3CN–1 мМ фосфатный буферный раствор с pH 6.5 (85 : 15, по объему); скорость потока 1 мл/мин; УФ-детектирование при 210 нм.
Водорастворимые витамины. Различия в гидрофильности и разнообразие физико-химических свойств делает водорастворимые витамины интересными модельными аналитами для изучения свойств сорбентов в режиме ГИХ. В составе подвижной фазы в этом случае рационально использовать ацетатно-аммонийный буферный раствор, обладающий высокой растворимостью в присутствии ацетонитрила. Это позволяет работать в градиентном режиме элюирования, а также повышать концентрацию элюирующего иона при наличии электростатических взаимодействий заряженных форм аналитов с неподвижной фазой и высокой ионообменной емкости сорбентов.
Исходя из данных [21], для разделения соединений данного класса использовали 100 мМ ацетатно-аммонийный буферный раствор и ацетонитрил в качестве подвижной фазы; детектирование проводили при длине волны УФ-детектора 270 нм. С целью повышения экспрессности для разделения смеси семи витаминов использовали градиентное элюирование. На первом этапе выбирали условия разделения слабоудерживаемых витаминов. Чтобы добиться хорошего разделения смеси витаминов, для каждого сорбента выбирали pH буферного раствора (5.4–5.8) и долю водной фазы в элюенте в диапазоне 8–12%. Оптимальное содержание водной фазы для всех сорбентов составило 10%, а pH буферного раствора – 5.4, так как при pH 5.8 не удалось обеспечить полного разрешения витаминов B1 и B2. Далее выбирали условия градиентного элюирования витамина B12, а также никотиновой и аскорбиновой кислот, имеющих большие факторы удерживания на сорбентах, характеризующихся высокой анионообменной селективностью согласно тесту Танака (табл. 2). Путем повышения содержания ацетатно-аммонийного буферного раствора в подвижной фазе в диапазоне 20–28% удалось добиться полного разрешения пиков и экспрессного разделения этих витаминов на полученных сорбентах при содержании буферного раствора 28% в качестве второй ступени градиентного режима.
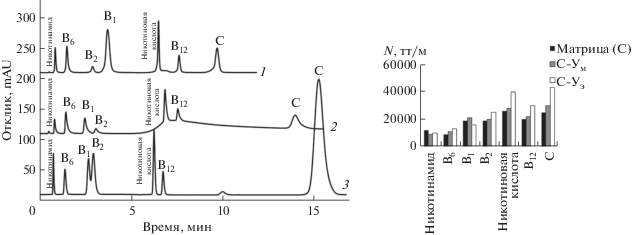
Как видно из рис. 4, для обоих сорбентов, полученных по реакции Уги, улучшается разрешение пары витаминов B1/B2 (Rs = 1.8 для С-Ум) по сравнению с матрицей, а на сорбенте С-Уэ наблюдается иная селективность – обратный порядок элюирования данной пары (Rs = 2.0 для С-Уэ). Увеличение фактора удерживания положительно заряженного тиамина согласуется с уменьшением анионообменной селективности α(AX) сорбента С-Уэ и увеличением α(СX). Уменьшение гидрофильности и анионообменной селективности данного сорбента привело к уменьшению удерживания витаминов кислотной природы – аскорбиновой и никотиновой кислот – и более экспрессному разделению семи витаминов. Кроме того, сорбент С-Уэ продемонстрировал лучшую симметрию пиков (As от 1.0 до 1.2) и большую эффективность по витаминам – до 45 000 тт/м (рис. 4). Хроматограммы модельной смеси витаминов представлены на рис. 4.
Рис. 4.
Хроматограммы модельной смеси витаминов (слева), диаграмма эффективности определения витаминов (справа, получена в изократическом режиме элюирования). Сорбенты: С-Уэ (1), С-Ум (2), С (матрица) (3); подвижная фаза: 100 мМ ацетатно-аммонийный буферный раствор с pH 5.4–CH3CN; градиентное элюирование: 0–4.5 мин 10% буферного раствора, 5.5–16 мин 28% буферного раствора; скорость потока 1 мл/мин; УФ-детектирование при 270 нм.
Выбор условий разделения в режиме гидрофильной хроматографии с масс-спектрометрическим детектированием (МС). При определении полярных органических соединений методом ГИХ в сочетании с МС-детектированием необходимо учитывать не только хроматографические параметры разделения, но и условия ионизации аналитов. Ионизация электрораспылением – ключевая стадия в МС-детектировании, во время которой образуются заряженные молекулы аналитов, обычно их протонированные и депротонированные формы, и осуществляется их перенос из фазы мелкого аэрозоля в газовую фазу. На этой стадии состав подвижной фазы и ее pH влияют не только на форму существования молекул аналитов в элюате, но и на распыление этого элюата в виде аэрозоля вследствие увеличения или уменьшения поверхностного натяжения. Компоненты буферных растворов могут конкурировать с молекулами аналитов при ионизации, служить переносчиками заряда, образовывать заряженные кластеры с молекулами растворителей и т.д. Таким образом, необходимо выбрать наиболее подходящий состав подвижной фазы, найдя компромисс между наилучшими условиями для хроматографического разделения и для электрораспылительной ионизации разделяемых соединений.
Для разделения и определения сахаров в варианте ВЭЖХ-МС использовали стандартную подвижную фазу, состоящую из ацетонитрила и воды [22]. Профиль градиента подбирали таким образом, чтобы, увеличивая содержание воды в подвижной фазе, добиться оптимальной формы пиков дисахаридов – глюкозы, мальтозы и лактозы (рис. 5а).
Рис. 5.
Хроматограмма модельной смеси сахаров (а) и витаминов (б) на сорбенте С-Ум. (а): Подвижная фаза – деионизованная вода–ацетонитрил; режим градиентного элюирования: 0–4.5 мин 15% воды, 4.5–9.0 мин c 17 до 37% воды, 9.0–9.1 мин с 37 до 17% воды; МС-детектирование; скорость потока 0.4 мл/мин; температура колонки 30°C; 1 – рибоза, 2 – ксилоза, 3 – фруктоза, 4 – глюкоза, 5 – сахароза, 6 – мальтоза, 7 – лактоза. (б): Подвижная фаза – 20 мМ формиат аммония (pH 3.7)–ацетонитрил: 0–6 мин 2% буферного раствора, 6–25 мин 2–95% буферного раствора, 25–30 мин 5% буферного раствора, 30–38 мин 2% буферного раствора; скорость потока 0.4 мл/мин, температура колонки 25°C; МС-детектирование.
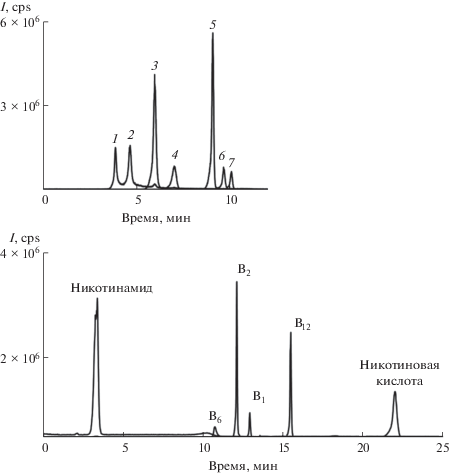
Для разделения и определения витаминов в варианте ВЭЖХ-МС выбирали подходящий состав подвижный фазы, варьируя концентрацию буферного раствора (формиата аммония) от 10 до 30 мМ. Ухудшение ионизации витаминов при увеличении концентрации формиата аммония от 10 до 20 мМ компенсировалось улучшением формы их хроматографических пиков, а при переходе к концентрации 30 мМ интенсивность сигналов снижалась, поэтому было решено далее использовать 20 мМ буферный раствор. Дальнейшее повышение концентрации нецелесообразно из-за ухудшения отклика МС-детектора, а кроме того, невозможно полностью подавить удерживание по ионообменному механизму, как это происходит при использовании фосфатного буферного раствора, несовместимого с МС-детектированием. По этой причине приняли решение понизить pH, добавив муравьиную кислоту к раствору формиата аммония до pH 3.7, а его содержание в составе подвижной фазы в градиентном режиме ли-нейно увеличивали от 2 до 95%. Полученная хроматограмма модельной смеси витаминов группы B представлена на рис. 5б.
Анализ реальных объектов. Для проверки применимости сорбента С-Ум для определения сахаров в реальных объектах провели анализ вина Gran Marques Reserva и растворимого кофе Nescafe Gold Cappucсino. Определяемые углеводы в исследуемых образцах идентифицировали сопоставлением их времен удерживания со временами удерживания компонентов в стандартных растворах. Количественную оценку содержания каждого аналита проводили на основе линейных градуировочных зависимостей площади пика от концентрации. Градуировочные зависимости для десяти сахаров строили с использованием стандартных растворов в диапазоне концентраций 50–800 мг/л, выбранном исходя из предполагаемого содержания в объектах анализа. Правильность предложенного подхода к определению сахаров проверяли методом введено–найдено, воспроизводимость – путем повторного введения образцов.
В результате качественного анализа в красном сухом вине обнаружили фруктозу и глюкозу, а в растворимом кофе Nescafe Gold Cappucсino – сахарозу и лактозу, что характерно для сладкого напитка с добавлением молока. В пробу вина вводили добавки девяти сахаров по 294 мг/л каждого, а в пробу растворимого кофе – добавки десяти сахаров по 100 мг/л. Большое количество глюкозы в вине мешает определению маннозы, поэтому добавку маннозы не вводили. Хроматограммы разбавленного вина и кофе на колонке С-Ум представлены на рис. 6; результаты количественного анализа и проверки правильности методом введено–найдено, а также характеристики полученных градуировочных зависимостей и пределы обнаружения сахаров приведены в табл. 4. Найденные количества сахаров согласуются с введенными, что подтверждает применимость полученного сорбента и способа определения сахаров к анализу реальных объектов.
Рис. 6.
Хроматограммы вина Gran Marques Reserva (сплошная линия), вина с добавками (пунктирная линия) (а); кофе Nescafe Gold Cappucсino (сплошная линия), кофе с добавками (пунктирная линия) (б). Сорбент С-Ум; подвижная фаза вода–CH3CN; скорость потока 1 мл/мин; режим градиентного элюирования. (а): 0–5.5 мин 12% воды, 6.5–12.5 мин 20% воды, 13.5–26.5 мин 25% воды, 28.5–32.5 мин 12% воды; (б): 0–5.5 мин 14% воды, 6.5–12.5 мин 22% воды, 13.5–26.5 мин 27% воды, 28.5–32.5 мин 14% воды; детектирование по светорассеянию: 1 – рибоза, 2 – ксилоза, 3 – арабиноза, 4 – фруктоза, 5 – манноза, 6 – глюкоза, 7 – сахароза, 8 – мальтоза, 9 – лактоза, 10 – раффиноза.
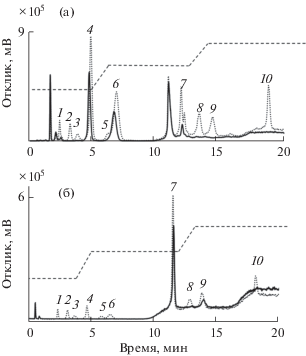
Таблица 4.
Метрологические характеристики определения сахаров (диапазон линейности градуировочных зависимостей 50–800 мг/л)
| Вещество | cмин, мг/л | sr* | r | a × 10–3** | b × 10–5** | Найдено, мг/л | Найдено после введения 294 мг/л***, мг/л | Найдено после введения 100 мг/л****, мг/л |
|---|---|---|---|---|---|---|---|---|
| Рибоза | 7 | 0.03 | 0.996 | 5.77 | 3.50 | – | 280 ± 40 | 104 ± 5 |
| Ксилоза | 8 | 0.04 | 0.995 | 8.13 | 5.06 | – | 220 ± 70 | 100 ± 5 |
| Арабиноза | 20 | 0.16 | 0.992 | 3.53 | 2.34 | – | 250 ± 40 | 120 ± 20 |
| Фруктоза | 5 | 0.05 | 0.995 | 13.36 | 8.01 | 630 ± 60*** | 880 ± 60 | 100 ± 5 |
| Манноза | 35 | 0.21 | 0.991 | 5.02 | 3.73 | – | – | 110 ± 10 |
| Глюкоза | 20 | 0.22 | 0.989 | 10.98 | 8.09 | 600 ± 50*** | 800 ± 100 | 110 ± 10 |
| Сахароза | 20 | 0.06 | 0.998 | 14.06 | 4.80 | 340 ± 30**** | 310 ± 20 | 420 ± 30 |
| Мальтоза | 26 | 0.25 | 0.995 | 11.11 | 4.85 | – | 220 ± 60 | 120 ± 20 |
| Лактоза | 25 | 0.30 | 0.994 | 9.23 | 3.45 | 120 ± 20**** | 220 ± 60 | 210 ± 30 |
| Раффиноза | 30 | 0.04 | 0.998 | 13.83 | 2.47 | – | 260 ± 30 | 100 ± 20 |
* * *
Таким образом, изучены новые неподвижные фазы, синтезированные по мультикомпонентной реакции Уги с использованием матрицы 3-аминопропилсиликагеля, ацетона, гликолевой кислоты и этилового эфира изоциануксусной кислоты в разных растворителях (этанол, метанол). Показано, что большая степень покрытия матрицы достигается при проведении реакции в этаноле. Улучшение разделяющей способности новых сорбентов по отношению к различным классам полярных аналитов по сравнению с исходной матрицей свидетельствует о перспективности предложенного подхода для создания новых неподвижных фаз для ГИХ. Дальнейшие работы в данной области будут направлены на повышение степени покрытия матрицы функциональным слоем, а также варьирование структуры получаемого слоя для управления селективностью получаемых сорбентов.
Работа выполнена при поддержке Российского научного фонда, грант № 20-13-00140.
Список литературы
Buszewski B., Noga S. Hydrophilic interaction liquid chromatograpy (HILIC) – A powerful separation technique // Anal. Bioanal. Chem. 2012. V. 402. P. 231.
Чернобровкина А.В., Смоленков А.Д., Шпигун О.А. Гидрофильная хроматография – перспективный метод определения полярных веществ // Лаборатория и производство. Т. 4. № 4. С. 76.
Moni L., Ciogli A., Acquarica I., Dondoni A., Gasparrini F., Marra A. Synthesis of sugar-based silica gels by copper-catalysed azide–alkyne cycloaddition via a single-step azido-activated silica intermediate and the use of the gels in hydrophilic interaction chromatography // Chem. Eur. J. 2010. V. 16. P. 5712.
Guo Z., Jin Y., Liang T., Liu Y., Xu Q., Liang X., Lei A. Synthesis, chromatographic evaluation and hydrophilic interaction/reversed-phase mixed-mode behavior of a “Click-cyclodextrin” stationary phase // J. Chromatogr. A. 2009. V. 1216. P. 257.
Shen A., Guo Z., Yu L., Cao L., Liang X. A novel zwitterionic HILIC stationary phase based on ‘‘thiol-ene’’ click chemistry between cysteine and vinyl silica // Chem. Commun. 2011. V. 47. P 4550.
Gargano A.F.G., Leek T., Lindner W., Lämmerhofer M. Mixed-mode chromatography with zwitterionic phosphopeptidomimetic selectors from Ugi multicomponent reaction // J. Chromatogr. A. 2013. V. 1317. P. 12.
Ferreira C.C., Gama M.R., Silva G.S., Pereira A.W., Collins C.H., Jardim I.C.S.F. Synthesis and evaluation of a pentafluorobenzamide stationary phase for HPLC separations in the reversed phase and hydrophilic interaction modes // J. Sep. Sci. 2018. V. 41. P. 1.
Kotoni D., d’Acquarica I., Ciogli A., Villani C., Capitani D., Gasparrini F. Design and evaluation of hydrolytically stable bidentate urea-type stationary phases for hydrophilic interaction chromatography // J. Chromatogr. A. 2012. V. 1232. P. 196.
Ugi I., Dömling A. Multicomponent reactions with isocyanides // Angew. Chem. Int. Ed. 2000. V. 39. P. 3168.
Миронов М.А., Бабаев Е.В. Параллельная реакция Уги в студенческих практикумах Урала и Москвы // Рос. хим. журн. (Журн. Рос. хим. об-ва им. Д.И. Менделеева). 2009. Т. 53. № 5. С. 139.
Krasavin M., Parchinsky V., Shumsky A., Konstantinov I., Vantskul A. Proline-like b-turn mimics accessed via Ugi reaction involving monoprotected hydrazines // Tetrahedron Lett. 2010. V. 51. P. 1367.
Strocker A., Keating T., Tempest P., Armstrong R. Use of a convertible isocyanide for generation of Ugi Reaction derivatives on solid support: Synthesis of α-acylaminoesters and pyrroles // Tetrahedron Lett. 1996. V. 37. P. 1149.
Hoppe D., Schöllkopf U. Synthesen mit a-metallierten Isocyaniden XIII. Neue Synthese von 2-Imidazolinen // Liebigs Ann. Chem. 1972. Bd. 763. S. 1.
Титце Л., Айхер Т. Препаративная органическая химия. М.: Мир, 2004. С. 161.
Dolci M., Chromatographic Characterization of Stationary Phases for Hydrophilic Interaction Liquid Chromatography. Runcorn, Cheshire, UK: Thermo Fisher Scientific, 2013.
Kawachi Y., Ikegami T., Takubo H., Ikegami Y., Miyamoto M., Tanaka N. Chromatographic characterization of hydrophilic interaction liquid chromatography stationary phases: Hydrophilicity, charge effects, structural selectivity, and separation efficiency // J. Chromatogr. A. 2011. V. 1218. P. 5903.
Попов А.С., Максимов Г.С., Смоленков А.Д., Шпигун О.А., Чернобровкина А.В. Новые сорбенты для гидрофильной хроматографии на основе силикагеля, ковалентно модифицированного полиэтиленгликолем // Вестн. Моск. ун-та. Сер. 2. Химия. 2021. Т. 62. № 2. С. 164.
Fu Q., Liang T., Li Z., Xu X., Ke Y., Jin Y., Liang X. Carbohydrate separation by hydrophilic interaction liquid chromatography on a ‘click’ maltose column // Carbohydr. Res. 2013. V. 379. P. 13.
Jenkins K. HILIC separation of carbohydrates using BEH amide particle technology // J. Chromatogr. Today. 2015. V. 8. P. 14.
Themelis T., Gotti R., Gatti R. A novel hydrophilic interaction liquid chromatography method for the determination of underivatized amino acids in alimentary supplements // J. Pharm. Biomed. Anal. 2017. V. 145. P. 751.
Karatapanis A.E., Fiamegos Y.C., Stalikas C.D. HILIC separation and quantitation of water-soluble vitamins using diol column // J. Sep. Sci. 2009. V. 32. P. 909.
Sarvin B.A., Seregin A.P., Shpigun O.A., Rodin I.A., Stavrianidi A.N. A novel strategy for isolation and determination of sugars and sugar alcohols from conifers // J. Chromatogr. B. 2018. V. 1092. P. 138.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии