

Журнал аналитической химии, 2021, T. 76, № 9, стр. 844-849
Разделение рацемата пентанола-2 на хиральной неподвижной фазе на основе гомохиральных кристаллов NiSO4⋅6H2O, полученных в условии созревания виедмы
Ю. Ф. Шарафутдинова a, *, А. Ш. Ганиева a, В. Ю. Гуськов a
a Башкирский государственный университет
450076 Уфа, ул. Заки-Валиди, 32, Россия
* E-mail: ms.shaihitdinova94@gmail.com
Поступила в редакцию 26.03.2021
После доработки 31.03.2021
Принята к публикации 01.04.2021
Аннотация
Для разделения рацемата пентанола-2 предложена новая хиральная неподвижная фаза на основе гомохиральных кристаллов α-NiSO4⋅6H2O, полученных в условии созревания Виедмы. Из хроматограмм разделения рацемата пентанола-2 определены времена удерживания и параметры разделения. Разделение происходит в диапазоне концентраций 4–11.3 мг/мл (элюент – смесь н-гептана и хлороформа). Достоверность разделений подтверждена поляриметрическим и газохроматографическим с масс-спектрометрическим детектированием анализом фракций каждого пика. Показано, что параметры разделения не зависят от скорости элюирования. При увеличении объемной доли хлороформа элюирующая сила возрастает, а критерий разделения и коэффициент селективности снижаются. Последний варьировался от 1.09 до 1.32. Максимальная эффективность разделения составила 2000 тт/м. Предложенная неподвижная фаза может быть применена для полупрепаративного разделения энантиомеров.
Проблема разделения рацемических веществ возникла с тех пор, как Пастер обнаружил молекулярную асимметрию органических соединений в 1848 г. [1]. Целью разделения рацемата является как анализ энантиомерного состава, так и полупрепаративное и препаративное выделение оптически чистого изомера, поскольку стереохимические характеристики структуры сильно влияют на биологическую активность органических соединений. Методы разделения рацемических спиртов делятся на две основные группы: (1) разделение путем синтеза и разделения диастереоизомеров и (2) разделение рацемических спиртов путем энантиоселективной кристаллизации, а также хроматографически [2].
Кристаллизация с образованием хиральных (энантиоморфных) кристаллов возможна как для изначально хиральных, так и для ахиральных молекул. В случае последних получаются право- и левовращающие кристаллы с вероятностью 50 : 50. Кондепуди [3–6] показал, что при автокаталитической кристаллизации хлората натрия (ахиральное вещество, кристаллизующееся в энантиоморфной пространственной группе) данное правило может быть нарушено. Чистые хиральные кристаллы хлората натрия можно получить в условиях интенсивного размешивания раствора. Эта концепция спонтанного разделения энантиоморфных кристаллов заключалась в решающей роли вторичного зародышеобразования: растворенное вещество кристаллизуется на поверхности существующего материнского кристалла, а вновь добавленный материал принимает хиральность поверхности.
Виедма [7] в 2005 г. предложил механизм такого спонтанного нарушения симметрии при сопровождающейся размешиванием кристаллизации: в процессе размешивания происходят разбиение мешальником достигшего критического размера кристалла на более мелкие той же хиральности (вторичная нуклеация) [8] и последующий автокаталитический процесс Франка [9] в сочетании с Оствальдским созреванием [10]. Виедма показал, что получение чистых энантиоморфных кристаллов возможно и при высокой скорости первичной нуклеации как за счет создаваемого стеклянными шариками “эффекта мельницы” [7], так и за счет температурного градиента при отсутствии размешивания [11]. Данный метод получения энантиоморфных кристаллов назван “созреванием Виедмы” и успешно применен для получения многих энантиоморфных кристаллов, в том числе и из растворов рацематов [12–17].
Созревание Видемы делает возможным использование энантиоморфных кристаллов для хирального разделения. Соаи с соавт. [18] обнаружили способность поверхности энантиоморфных кристаллов ахиральных молекул к асимметрическому катализу реакции диизопропилцинка с пиримидин-5-карбальдегидом. Одним из таких кристаллов был гексагидрат сульфата никеля. Для полученных группой Соаи в 2015 г. кристаллов α-NiSO4⋅6H2O зафиксирован круговой дихроизм при 390 нм. Обнаружено, что кристаллы с положительным круговым дихроизмом приводят к образованию (S)-пиримидилалконола, а с отрицательным – (R)-пиримидилалконола.
Таким образом, кристаллы α-NiSO4⋅6H2O, полученные в условиях созревания Виедмы, могут быть применены и для разделения энантиомеров. В настоящей работе адсорбент на основе таких кристаллов использовали для разделения рацемата пентанола-2 на энантиомеры.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве адсорбента использовали семиводный сульфат никеля (98%, Кыштымский медеэлектролитный завод, Кыштым, Россия, CAS 10101-98-1). В качестве адсорбента-носителя кристаллов α-NiSO4⋅6H2O применяли силикагель (Chemapol, Прага, Чешская республика, размер частиц (5–40) мкм). Для приготовления растворов NiSO4⋅6H2O использовали высокоочищенную воду с проводимостью 10 мкс/м, полученную на деионизаторе воды ДВ-10UV (Цветхром, Дзержинск, Россия).
Гомохиральные кристаллы α-NiSO4⋅6H2O получали по следующей методике: в насыщенный при 40°С раствор NiSO4⋅7H2O добавляли 15 г стеклянных шариков для создания “эффекта мельницы”, затем включали размешивание со скоростью 1300 об/мин. Раствор испарялся до тех пор, пока было возможно осуществлять перемешивание. Осадок отфильтровывали и высушивали в эксикаторе над свежепрокаленным CaCl2.
Для нанесения α-NiSO4⋅6H2O на поверхность силикагеля последний заливали водой, нагревали до 50°С, после чего вносили навеску α-NiSO4⋅6H2O. Количество модификатора составляло 10% от массы абсорбента-носителя. Суспензию размешивали в течение 15 мин при 100 об/мин. Затем температуру понижали до 40°С со скоростью 1°С/10 мин. Суспензию размешивали в течение дня, далее раствор отфильтровывали и готовый адсорбент высушивали в эксикаторе. После этого адсорбент дополнительно сушили в атмосфере азота в течение часа.
Полученным сорбентом наполняли стальную колонку длиной 25 см и внутренним диаметром 4.6 мм. Колонку заполняли суспензией модифицированного адсорбента в октаноле-1, далее через колонку элюировали ацетонитрил со скоростью потока 10 мл/мин. Процедуру повторяли до плотного заполнения всей колонки.
Исследование проводили методом высокоэффективной жидкостной хроматографии в нормально-фазовом режиме на хроматографе Perkin Elmer series 200 (Perkin Elmer, США) с УФ-детектором. Объем петли инжектора составлял 20 мкл, рабочая длина волны (250–265) нм, скорость элюирования (0.1–0.6) мл/мин. Обработка хроматограмм выполнялась в программе МультиХром версия 2.4 для Аквилона.
В качестве элюентов выбрали н-гептан (>99.5%, Экос-1, Москва, Россия, CAS 142-82-5) и хлороформ х. ч. (Экос-1, CAS 67-66-3). Элюенты дополнительно очищали согласно общепринятым методикам [19]. Гептан экстрагировали конц. H2SO4 (объемное соотношение гептан–H2SO4 9 : 1). Хлороформ встряхивали с конц. H2SO4, промывали водой до нейтральной реакции среды, сушили над CaCl2 и перегоняли. Объемная концентрация хлороформа в смеси гептан–хлороформ варьировалась от 10 до 40%. В качестве объекта исследования использовали пентанол-2 (98%, Sigma-Aldrich, Милуоки, США, CAS 60-32-29-7).
Для установления факта хирального разделения во время элюирования отбирали фракции, соответствующие первому и второму пикам. Далее фракции концентрировали и исследовали методами поляриметрии и газовой хроматографии с масс-спектрометрическим детектированием. Значения угла вращения плоскости поляризованного света каждой из фракций анализировали на автоматическом поляриметре Atago AP-300 (Atago, Япония). Использовали кювету толщиной 100 мм и объемом 5 мл. Рабочая длина волны составляла 589 нм. Содержимое фракций анализировали на газовом хроматографе Agilent 6890 с масс-спектрометрическим детектором Agilent 5973 на колонке DB-624 (длина 30 м, внутренний диаметр 0.25 мм). Использовали следующий режим программирования температуры: 50°C в течение 3 мин; подъем температуры со скоростью 10°C/мин до 100°C; подъем температуры со скоростью 30°C/мин до 220°C, далее изотерма в течение 3 мин. Температура испарителя 250°C, переходной линии 260°C, постоянное давление 58.513 кПа, деление потока 1 : 80. Режим масс-спектрометра: электронная ионизация, 70 эВ, сканирование в диапазоне (29–350) а. е., 5 скан/с. Образец (0.2 мкл) вводили микрошприцом.
Из экспериментальных хроматограмм рассчитывали времена удерживания, коэффициенты селективности и критерии разделения.
Коэффициент селективности энантиомеров рассчитывали по формуле:
Здесь и ниже слабее удерживающийся энантиомер обозначен как R-энантиомер, сильнее удерживающийся – как S-энантиомер.
Для расчета критерия разделения R по причине неполного разделения пиков использовали критерий разделения K2:
где h1 и h2 – высоты первого и второго пиков соответственно, hmin – минимум на кривой элюирования между пиками. В этом случае критерий разделения R рассчитывали из следующего соотношения:РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
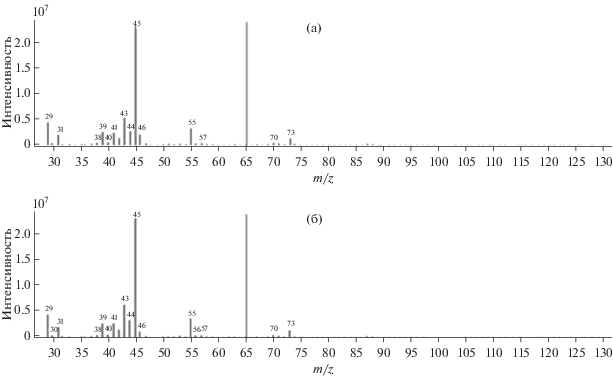
При исследовании адсорбции пентанола-2 на неподвижной фазе, модифицированной α‑Ni-SO4⋅6H2O, происходит разделение рацемата на два пика. Газохроматографический с масс-спектрометрическим детектированием анализ показал, что помимо элюента в обеих пробах содержится только одно вещество (рис. 1). Расшифровка масс-спектров по характеристическим ионам как для первой, так и для второй фракции показала, что данным веществом является пентанол-2. Поляриметрический анализ показал, что для фракции, соответствующей первому пику на хроматограмме, угол вращения плоскости поляризованного света составил +0.11°, а в случае второй фракции –0.11°. Таким образом, очевидно, что два пика на хроматограмме иллюстрируют разделение энантиомеров пентанола-2, причем первый пик на хроматограмме соответствует R-пентанолу, а второй – S-пентанолу.
В табл. 1 приведены параметры разделения пентанола-2 элюированием смесью гептан–хлороформ в соотношении 80 : 20. Как видно, при варьировании скорости потока от 0.1 до 0.6 мл/мин фактор селективности меняется от 1.29 до 1.32. Следовательно, скорость потока не влияет на селективность разделения. Хроматограммы разделения приведены на рис. 2, 3.
Таблица 1.
Значения времен удерживания tR энантиомеров пентанола-2, а также критериев разделения K2, R и коэффициентов селективности α на изучаемом адсорбенте при различных скоростях элюирования (с = = 10 мг/мл, объемное соотношение гептан–хлороформ 80 : 20)
| Скорость потока, мл/мин | tR, мин | α | K2 | R | |
|---|---|---|---|---|---|
| R | S | ||||
| 0.1 | 53.51 | 69.25 | 1.29 | 3.29 | 0.55 |
| 0.15 | 36.01 | 46.32 | 1.29 | 3.48 | 0.56 |
| 0.2 | 27.04 | 35.66 | 1.32 | 4.5 | 0.61 |
| 0.3 | 18.18 | 23.63 | 1.30 | 3.14 | 0.53 |
| 0.4 | 13.69 | 17.76 | 1.30 | 3.66 | 0.57 |
| 0.5 | 11.24 | 14.64 | 1.30 | 3.81 | 0.58 |
| 0.6 | 7.82 | 10.35 | 1.32 | 2.7 | 0.50 |
Рис. 2.
Разделение пентанола-2. Концентрация 10 мг/мл, элюент гептан–хлороформ в соотношении 80 : 20, скорость потока 0.2 мл/мин (α = 1.32, R = 0.61).
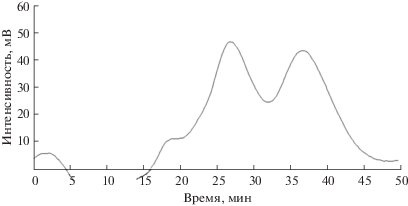
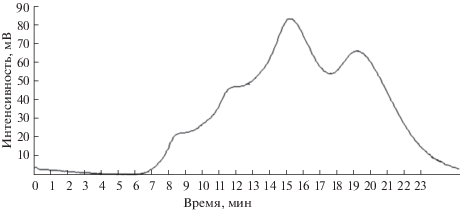
Рис. 3.
Разделение пентанола-2. Концентрация 10 мг/мл, элюент гептан–хлороформ в соотношении 80 : 20, скорость потока 0.5 мл/мин (α = 1.30, R = 0.58).
Изучена зависимость коэффициента селективности и критерия разделения от концентрации рацемата (табл. 2). Показано, что данные величины растут с увеличением концентрации пентанола-2. При концентрации 3 мг/мл и ниже селективность пропадает и разделения не происходит. При концентрации 11.3 мг/мл наблюдается разделение с высоким коэффициентом селективности (α = 1.36), однако уменьшение площадей пиков по сравнению с таковыми при концентрации 10 мг/мл свидетельствует о частичной необратимой адсорбции пентанола-2 на поверхности неподвижной фазы. Это явление подтверждает появление при увеличении элюирующей силы растворителя (увеличение объемной доли хлороформа с 20 до 30%) пиков адсорбировавшегося пентанола-2 при анализе холостой пробы. При объемном соотношении гептан–хлороформ (80 : 20) в области концентраций от 3 до 10 мг/мл параметры удерживания, а также все характеристики разделения воспроизводятся.
Таблица 2.
Значения времен удерживания tR энантиомеров пентанола-2, а также критериев разделения K2, R и коэффициентов селективности α на изучаемом адсорбенте при различных концентрациях рацемата (cкорость потока 0.5 мл/мин, объемное соотношение гептан–хлороформ 80 : 20)
| Концентра-ция, мг/мл | tR, мин | α | K2 | R | |
|---|---|---|---|---|---|
| R | S | ||||
| 3 | 9.08 | – | – | – | – |
| 4 | 9.54 | 11.8 | 1.09 | 2.66 | 0.49 |
| 5 | 10.2 | 12.96 | 1.28 | 2.72 | 0.50 |
| 10 | 11.33 | 14.75 | 1.30 | 2.79 | 0.51 |
| 11.3 | 13.65 | 18.63 | 1.36 | 4.48 | 0.61 |
Отсутствие разделения рацемата при концентрациях 3 мг/мл и ниже обусловлено механизмом хирального разделения. Применяемые энантиоморфные кристаллы состоят из ахиральных соединений. Хиральность таких кристаллов проявляется на надмолекулярном уровне. Механизм хирального разделения на кристаллах α-NiSO4⋅6H2O изучен нами ранее [20]. При хиральном разделении на поверхности с надмолекулярной хиральностью распознавание возникает только при формировании адсорбированного слоя энантиомеров, что происходит только при достаточно больших концентрациях разделяемых веществ. При низких концентрациях аналитов молекулы адсорбируются на достаточно большом расстоянии друг от друга, что делает латеральные взаимодействия невозможными. Поверхность с надмолекулярной хиральностью неспособна распознавать отдельные молекулы энантиомеров. Это объясняет то, что при разделении рацемата пентанола-2 зафиксирована нижняя граница энантиоселективности.
Изучены параметры разделения при различных объемных соотношениях гептан–хлороформ. Скорость потока и концентрация пентанола-2 составляли 0.5 мл/мин и 10 мг/мл соответственно. Из табл. 3 видно, что при увеличении объемной доли хлороформа параметры разделения ухудшаются. Так, при объемном соотношении 80 : 20 α = 1.30, R = 0.60, а при объемном соотношении 60 : 40 коэффициент селективности α и критерий разделения R снижаются до 1.19 и 0.44 соответственно. При этом уменьшается и продолжительность анализа за счет возрастания элюирующей силы растворителя. Эффективность разделения варьировалась от 350 до 2000 тт/м.
Таблица 3.
Значения времен удерживания tR энантиомеров пентанола-2, а также критериев разделения R и коэффициентов селективности α на изучаемом адсорбенте при различных объемных соотношениях гептан–хлороформ (cкорость потока 0.5 мл/мин, с = 10 мг/мл)
| Соотношение гептан–хлороформ | tR, мин | α | K2 | R | |
|---|---|---|---|---|---|
| R | S | ||||
| 80 : 20 | 11.33 | 14.75 | 1.30 | 3.81 | 0.58 |
| 70 : 30 | 10.2 | 12.96 | 1.22 | 2.31 | 0.46 |
| 60 : 40 | 6.26 | 7.45 | 1.19 | 2.07 | 0.46 |
Достигнутая энантиоселективность близка к таковой на другой полученной на основе созревания Виедмы неподвижной фазе для ВЭЖХ на основе 3,4,9,10-перилентетракарбоновой кислоты [21]. Полученные критерии разделения также отличаются незначительно. Однако преимуществом предлагаемой неподвижной фазы является возможность разделения больших концентраций рацемата в идентичных условиях.
Таким образом, на новой хиральной неподвижной фазе на основе энантиоморфных кристаллов α-NiSO4⋅6H2O достигнуто разделение рацемата пентанола-2 с максимальным фактором селективности 1.32. Разделение выполнено в области высоких концентраций (от 4 до 10 мг/мл), что позволит в дальнейшем применять данную хиральную неподвижную фазу для полупрепаративного разделения рацематов.
Работа выполнена за счет гранта Российского научного фонда (проект № 19-73-10079).
Список литературы
Pasteur L. Recherches sur les relations qui peuvent exister entre la forme crystalline, la composition chimique et le sens de la polarisation rotatoire // Ann. Chim. Phys. 1848. V. 24. P. 442.
Klyashchitskii B.A., Shvets V.I. Resolution of racemic alcohols into optical isomers // Russ. Chem. Rev. 1972. V. 41. № 7. P. 592.
Kondepudi D.K., Kaufman R.J., Singh N. Chiral symmetry breaking in sodium chlorate crystallizaton // Science. 1990. V. 250. P. 975.
Kondepudi D.K., Bullock K.L., Digits J.A., Yarborough P.D. Enantiomorphic symmetry breaking in crystallization of molten sodium chlorate // J. Am. Chem. Soc. 1995. V. 117. P. 401.
Kondepudi D.K., Nelson G.W. Chiral symmetrybreaking in nonequilibrium chemical-systems–time scales for chiral selection // Phys. Lett. A. 1984. V. 106. P. 203.
Kondepudi D.K., Nelson G.W. Weak neutral currents and the origin of biomolecular chirality // Nature. 1985. V. 314. P. 438.
Viedma C. Chiral symmetry breaking during crystallization: complete chiral purity induced by nonlinear autocatalysis and recycling // Phys. Rev. Lett. 2005. V. 94. № 6.
Kondepudi D.K., Digits J., Bullock K. Studies in chiral symmetry breaking crystallization I: The effects of stirring and evaporation rates // Chirality. 1995. V. 7. P. 62.
Frank F.C. On spontaneous asymmetric synthesis // Biochim. Biophys. Acta. 1953. V. 11. P. 459.
Noorduin W.L., Meekes H., Bode A.A.C., Van Enckevort W.J.P., Kaptein B., Kellogg R.M., Vlieg E. Emergence of a single solid chiral state from a nearly racemic amino acid derivative // Cryst. Growth Des. 2008. V. 8. P. 1675.
Viedma C., Cintas P. Homochirality beyond grinding: deracemizing chiral crystals by temperature gradient under boiling // Chem. Commun. 2011. V. 47. P. 12786.
Sogutoglu L.-C., Steendam R.R.E, Meekes H., Vlieg E., Rutjes F.P.J.T. Viedma ripening: A reliable crystallisation method to reach single chirality // Chem. Soc. Rev. 2015. V. 44. P. 6723.
Viedma C., McBride J.M., Kahr B., Cintas P. Enantiomer-specific oriented attachment: formation of macroscopic homochiral crystal aggregates from a racemic system // Angewandte Chemie. 2013. V. 52. P. 10545.
Sivakumar R., Kwiatoszynski J., Fouret A., Nguyen T.P.T., Ramrup P., Cheung P.S.M., Cintas P., Viedma C., Cuccia L.A. Directing the Viedma ripening of ethylenediammonium sulfate using “Tailor-made” chiral additives // Crystal Growth and Design. 2016. V. 16. P. 3573.
Sivakumar R., Askari M.S., Woo S., Madwar C., Ottenwaelder X., Bohle D.S., Cuccia L.A. Homochiral crystal generation via sequential dehydration and Viedma ripening // CrystEngComm. 2016. V. 18. P. 4277.
Baglai I., Leeman M., Kellogg R.M., Noorduin W.L. A Viedma ripening route to an enantiopure building block for Levetiracetam and Brivaracetam // Org. Biomol. Chem. 2019. V. 17. P. 35.
Baglai I., Leeman M., Wurst K., Kaptein B., Kellogg R.M., Noorduin W.L. The Strecker reaction coupled to Viedma ripening: A simple route to highly hindered enantiomerically pure amino acids // Chem. Commun. 2018. V. 54. P. 10832.
Matsumoto A., Ozawa H., Inumaru A., Soai K. Asymmetric induction by retgersite, nickel sulfate hexahydrate, in conjunction with asymmetric autocatalysis // New J. Chem. 2015. V. 9. P. 6742.
Рудаков О.Б., Селеменев В.Ф. Физико-химические системы сорбат-сорбент-элюент в жидкостной хроматографии. Воронеж: РИЦ ВГУ, 2003. С. 242.
Gus’kov V.Yu., Allayarova D.A., Garipova G.Z., Pavlova I.N. Supramolecular chiral surface of nickel sulfate hexahydrate crystals and its ability to chirally recognize enantiomers by adsorption data // New J. Chem. 2020. V. 44. P. 17769.
Гуськов В.Ю., Гайнуллина Ю.Ю., Утеева Ж.Д., Мусабиров Д.Э. Применение хиральной неподвижной фазы на основе 3,4,9,10-перилентетракарбоновой кислоты для разделения энантиомеров в условиях газовой и жидкостной хроматографии // Журн. аналит. химии. 2020. Т. 75. С. 537.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии