

Журнал аналитической химии, 2021, T. 76, № 9, стр. 797-803
Сравнение аналитических возможностей различных схем хроматомембранной газовой экстракции
О. В. Родинков a, *, А. С. Бугайченко a, Л. Н. Москвин a
a Санкт-Петербургский государственный университет, Институт химии
198504 Санкт-Петербург, Университетский просп., 26, Петродворец, Россия
* E-mail: o.rodinkov@spbu.ru
Поступила в редакцию 07.04.2021
После доработки 17.04.2021
Принята к публикации 18.04.2021
Аннотация
Сопоставлены аналитические возможности различных схем относительного перемещения потоков анализируемой водной фазы и газа-экстрагента при осуществлении хроматомембранной газовой экстракции. Показано, что в тех случаях, когда эффективность системы возрастает с увеличением скорости потока газа-экстрагента (анализ в режиме online), предпочтительнее прямоточная схема, а в случае анализа предварительно отобранных проб – двухмерная схема.
Газовая экстракция относится к важнейшим методам пробоподготовки при определении летучих органических соединений в водных средах [1, 2]. Она лежит в основе парофазного анализа (ПФА) – метода получения информации о соcтаве конденсированной фазы на основании анализа контактировавшей с ней газовой фазы [3]. В современном понимании ПФА не ограничивается только анализом газа-экстрагента и включает различные варианты дополнительного концентрирования аналитов после их газоэкстракционного извлечения [4]. В зависимости от способа осуществления газовой экстракции различают статические, динамические и проточные варианты ПФА [4, 5]. Традиционные статический и динамический варианты парофазного анализа включают, как правило, ряд рутинных процедур, которые относительно плохо поддаются автоматизации. В рамках современной тенденции автоматизации всей процедуры анализа особую актуальность приобретают проточные варианты ПФА. В них извлечение аналитов происходит при одновременном движении потоков анализируемой жидкости и газа-экстрагента через массообменное устройство [6, 7]. Применение проточных вариантов ПФА позволяет также предотвратить образование артефактов в процессе хранения и транспортировки проб в лабораторию и увеличивает точность анализа за счет исключения или уменьшения действия человеческого фактора на результаты анализа.
Новые возможности для развития проточного ПФА открыл предложенный Л.Н. Москвиным хроматомембранный массообменный процесс [8] и реализуемая в рамках этого процесса хроматомембранная газовая экстракция (ХГЭ) [9]. При осуществлении ХГЭ массообмен между потоками водной и газовой фазой происходит в гидрофобной бипористой матрице из политетрафторэтилена с двумя типами открытых пор, различающимися по размерам. По крупным макропорам перемещается водная фаза, а по микропорам – газ-экстрагент, который подается в массообменный слой (бипористую матрицу) и выводится из него через гидрофобные непроницаемые для водной фазы микропористые мембраны. Поток газа-экстрагента с выделенными из водной фазы летучими компонентами подается либо в дозатор газового хроматографа и анализируется, либо пропускается через сорбционную колонку для газоадсорбционного концентрирования аналитов с их последующей термодесорбцией.
В ХГЭ хроматографический механизм межфазного обмена между жидкой и газовой фазой сочетается с мембранным процессом введения и выведения фаз из зоны массообмена. Подобное сочетание обеспечивает, с одной стороны, высокую эффективность разделения, свойственную хроматографическим методам, а с другой, – возможность разделения в непрерывном или легко автоматизируемом дискретном режиме.
Теоретически направление потоков обменивающихся фаз через массообменный слой может быть любым. Однако практическое значение имеют три схемы относительного перемещения фаз: прямоточная (потоки жидкой и газовой фазы перемещаются в одном направлении); двухмерная (потоки фаз перемещаются под углом 90 град) и противоточная (потоки перемещаются навстречу друг другу) (рис. 1). Закономерности указанных схем ХГЭ подробно изучены, однако непосредственное сравнение их возможностей ранее не проводили.
Рис. 1.
Прямоточная (а), противоточная (б) и двухмерная (в) схемы хроматомембранной газовой экстракции: 1 и 3 – вход и выход потока газа-экстрагента, 2 и 4 – вход и выход потока водной пробы, 5 – массообменный слой, 6 – мембраны.
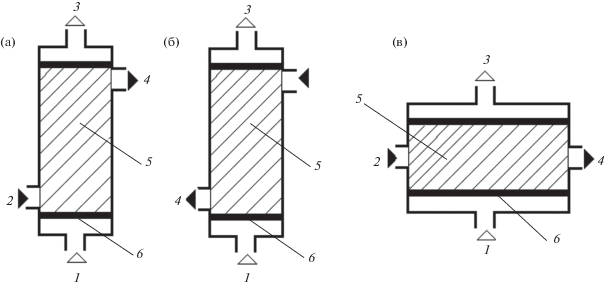
Цель настоящей работы – сравнение аналитических возможностей различных схем хроматомембранной газовой экстракции применительно к определению летучих органических соединений в водных растворах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исследования проводили в хроматомембранных ячейках, конструкция которых позволяла непрерывно пропускать потоки водной пробы и газа-экстрагента в одном, противоположном и перпендикулярном направлениях. Массообменный слой в ячейке состоял из бипористого политетрафторэтилена (ПТФЭ), который получали по известной методике [10]. Исходным материалом для изготовления служил полимеризационный порошок ПТФЭ марки Ф-4ПН-20 производства ООО “Галополимер” (Пермь, Россия). Размеры макропор слоя составляли (50–200) мкм, микропор – (0.5–5) мкм. Хроматомембранные ячейки для прямоточной и противоточной ХГЭ имели массообменный слой цилиндрической формы диаметром 16 мм и высотами 20 и 16 мм (ячейки 1 и 2 соответственно). Ячейки для двухмерного варианта имели массообменный слой в виде параллелепипеда, их габариты (50 × 20 × 10) мм (ячейка 3) и (20 × 10 × 10) мм (ячейка 4). Первой указана длина, за которую принимали протяженность массообменного слоя по направлению движения водной фазы, второй – ширина – протяженность массообменного слоя перпендикулярно движению потоков водной и газовой фаз, третьей – высота – протяженность слоя по направлению движения газовой фазы. Массообменный слой был ограничен с двух противоположных сторон микропористыми ПТФЭ-мембранами марки МФ-ФМ 400 L/G толщиной 300 мкм со средним диаметром пор 0.5 мкм производства НПО “Экофильтр” (Обнинск, Россия).
Анализируемую жидкость вытесняли из пробоотборной емкости и подавали в ячейки под действием избыточного давления газа, создаваемого регулятором давления, входящим в состав формирователя газовых потоков производства “Хроматэк” (Россия). Скорость потока газа-экстрагента через ячейку (WG) устанавливали с помощью регулятора расхода того же формирователя и проверяли с помощью мыльно-пленочного расходомера. Расход жидкости (WL) измеряли с помощью мерного цилиндра и секундомера. Водные растворы с заданной концентрацией тестовых веществ, квалификации которых была не ниже ч., готовили объемно-объемным способом.
Определение тестовых веществ в потоке газа-экстрагента на выходе из ячейки выполняли с помощью газового хроматографа Кристалл-5000.2 (Хроматэк, Россия), снабженного пламенно-ионизационным детектором и капиллярной колонкой BPX-1 (10 м × 0.53 мм × 2.65 мкм) со 100%-ным диметилполисилоксаном в качестве неподвижной фазы. Для этого поток газа-экстрагента направляли в дозирующую петлю (1 мл) автоматического обогреваемого крана-дозатора. Температурные режимы работы хроматографа выбирали в зависимости от природы тестовых веществ.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Условия осуществления хроматомембранной газовой экстракции. Одновременное движение жидкости по макропорам массообменного слоя, а газа-экстрагента – по микропорам возможно тогда, когда в любой точке массообменного слоя, в частности на входе и на выходе из него, выполняется условие:
(1)
${{P}_{{\text{G}}}} < {{P}_{{\text{L}}}} < {{P}_{{\text{G}}}} + \left| {{{P}_{{\text{с}}}}} \right|,$В прямоточной схеме одна из фаз на входе в массообменный слой, где ее давление максимально, и на выходе из этого слоя, где ее давление минимально, контактирует с другой фазой, имеющей ту же направленность градиента давления. В соответствии с неравенством (1) допустимые градиенты давлений фаз ΔР (разность давлений на входе и выходе) определяются неравенством:
В случае противоточной и двухмерной схем одна из фаз на входе в массообменный слой контактирует с другой фазой, находящейся на выходе из него, и допустимые градиенты давлений должны отвечать более жесткому условию, значительно ограничивающему потоки обменивающих фаз через ячейку:
Критерии выбора размеров макропор в ХГЭ те же, что и при выборе размеров частиц сорбентов и носителей в хроматографии. В обоих случаях приходится искать компромисс между увеличением эффективности массообмена и уменьшением проницаемости массообменного слоя при уменьшении размеров частиц. Для решения большинства аналитических задач диапазон размеров макропор (100–200) мкм можно принять в качестве оптимального. Поскольку в ХГЭ межфазный обмен происходит на границе микро- и макропор, микропоры выступают лишь в роли транспортных каналов для прохождения газовой фазы и их радиус не оказывает значимого влияния на эффективность массообмена. Поэтому оптимальный радиус микропор соответствует максимальному расходу газовой фазы. Увеличение радиуса микропор, с одной стороны, ведет к увеличению проницаемости, а с другой, – к уменьшению Рс и, следовательно, к уменьшению допустимой величины ΔРG. Зависимость расхода газа от радиуса микропор проходит через максимум в диапазоне от 0.5 до 2 мкм, который можно принять в качестве оптимального.
Применение бипористых сред с оптимальными размерами макро- и микропор позволяет реализовывать ХГЭ в достаточно широких пределах изменения скоростей потоков обеих фаз (табл. 1). Как видно из табл. 1, в отличие от других схем, в случае прямоточной схемы увеличение расхода водной фазы позволяет увеличивать и расход газа-экстрагента через хроматомембранную ячейку.
Таблица 1.
Предельные расходы воздуха (WG) через хроматомембранные ячейки в зависимости от расхода воды (WL) для различных схем хроматомембранной газовой экстракции
| WL, мл/мин | WG, мл/мин | ||
|---|---|---|---|
| прямоточная ячейка 1 |
противоточная ячейка 1 |
двухмерная ячейка 4 |
|
| 5.0 | 260 | 50 | 90 |
| 10.0 | 340 | 35 | 60 |
| 15.0 | 380 | 20 | 35 |
Для объективного сравнения аналитических возможностей различных схем ХГЭ необходимо учитывать режимы ее осуществления. Если параметры процесса ХГЭ остаются неизменными во времени, в том числе концентрации аналитов на входе в ячейку, то через определенный интервал времени становятся постоянными и концентрации аналитов в потоке газа-экстрагента на выходе из ячейки. Такой режим процесса принято называть стационарным. На практике стационарному режиму соответствует традиционный лабораторный анализ предварительно отобранных проб, а также анализ в режиме online с медленно изменяющимися концентрациями аналитов в потоке анализируемой пробы.
При изменении каких-либо параметров процесса, в частности, при быстром изменении концентраций аналитов на входе в ячейку, концентрации аналитов на выходе из нее изменяются не мгновенно, а постепенно приближаются к своему новому стационарному значению. Такой режим принято называть переходным [11]. Исследование закономерностей переходного режима актуально для анализа в режиме online, когда аналиты непрерывно извлекаются из потока анализируемой среды непосредственно на пробоотборной линии.
Стационарный режим хроматомембранной газовой экстракции. Хроматографический принцип межфазного обмена при осуществлении ХГЭ позволил использовать тарелочную модель для адекватного описания всех схем этого процесса [12–14]. Для прямоточной схемы ХГЭ эффективность массоoбмена соответствует одной эквивалентной теоретической тарелке. При этом зависимость концентрации аналита в потоке газа-экстрагента на выходе из хроматомембранной ячйки cG от его концентрации в жидкой фазе на входе в нее c0 определяется только коэффициентом распределения аналита в системе жидкость–газ (K) и соотношением объемных скоростей потоков (расходов) газа-экстрагента (WG) и жидкой пробы (W0) через ячейку [12]:
(2)
${{c}_{{\text{G}}}} = {{c}_{{\text{0}}}}\frac{{\text{1}}}{{{{\text{1}} \mathord{\left/ {\vphantom {{\text{1}} K}} \right. \kern-0em} K} + {{{{W}_{{\text{G}}}}} \mathord{\left/ {\vphantom {{{{W}_{{\text{G}}}}} {{{W}_{{\text{0}}}}}}} \right. \kern-0em} {{{W}_{{\text{0}}}}}}}},$В прямоточной схеме, если длина массообменного слоя больше высоты, эквивалентной теоретической тарелке (ВЭТТ), то ее изменение не влияет на концентрацию аналитов в выходящем из ячейки потоке газа-экстрагента cG. Учитывая, что типичные значения ВЭТТ в ХГЭ не превышают (5–10) мм, для реализации прямоточной схемы могут быть использованы достаточно миниатюрные ячейки.
В случае противоточной схемы массообменный слой может рассматриваться по аналогии с обычной хроматографической колонкой как ряд последовательно соединенных эквивалентных теоретических тарелок, характеризующихся определенной ВЭТТ [13]. Для этой схемы справедливо:
(3)
${{c}_{{\text{G}}}} = {{c}_{0}}K\frac{{{{{\left[ {{{{{W}_{0}}} \mathord{\left/ {\vphantom {{{{W}_{0}}} {\left( {{{W}_{{\text{G}}}}K} \right)}}} \right. \kern-0em} {\left( {{{W}_{{\text{G}}}}K} \right)}}} \right]}}^{{ - N}}} - 1}}{{{{{\left[ {{{{{W}_{0}}} \mathord{\left/ {\vphantom {{{{W}_{0}}} {\left( {{{W}_{{\text{G}}}}K} \right)}}} \right. \kern-0em} {\left( {{{W}_{{\text{G}}}}K} \right)}}} \right]}}^{{ - N - 1}}} - 1}},$Для описания двухмерной схемы ХГЭ предложена модель двухмерных теоретических тарелок, в основе которой лежит представление о массообменном слое как о прямоугольной матрице (рис. 2), состоящей из совокупности двухмерных тарелок [14]. Эти тарелки характеризуются ВЭТТ по направлению движения потока жидкой фазы и ВЭТТ по направлению движения газовой фазы. В рамках этой модели справедливо:
Рис. 2.
Тарелочная модель двухмерной схемы хроматомембранной газовой экстракции: 1 – поток водной пробы, 2 – поток газа-экстрагента. N и M – число тарелок по направлению движения потоков водной пробы и газа-экстрагента соответственно.
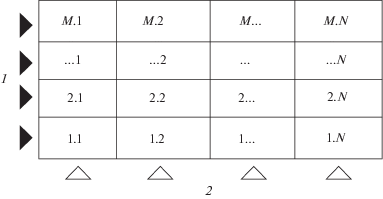
Основным недостатком рассмотренной модели, основанной на представлении о двухмерных теоретических тарелках, является громоздкое математическое описание, ограниченное к тому же целочисленными значениями N и М. Для описания двухмерной схемы ХГЭ представляет интерес известная [15] модель перекрестной экстракции (рис. 3).
Рис. 3.
Перекрестная модель двухмерной схемы ХГЭ: 1 – поток водной пробы, 2 – поток газа-экстрагента. N – число тарелок по направлению движения потоков отдающей фазы пробы.
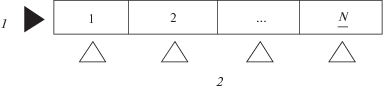
Для того, чтобы использовать модель перекрестной экстракции, необходимо принять, что протяженность тарелки в направлении движения потока газа-экстрагента равна протяженности массообменного слоя и соответственно М = 1. Ранее показано [14], что при прочих равных условиях увеличение высоты массообменного слоя Н приводит к увеличению концентрации аналита в потоке газа-экстрагента, однако представленная на рис. 3 модель этого не учитывает. В этой связи логично принять, что
где N и N – число тарелок в направлении движения потока отдающей фазы в перекрeстной и тарелочной моделях соответственно. В рамках перекрестной модели получаем:Общие закономерности различных схем ХГЭ, независимо от абсолютных значений коэффициентов распределения аналитов и расходов пробы и газа-экстрагента через ячейку, отражают зависимости величины (cG/c0K) от соотношения W0/(WGK) = Y. Экспериментальные и рассчитанные по уравнениям (2)–(4) зависимости для прямоточной, противоточной и двухмерной схем приведены на рис. 4.
Рис. 4.
Закономерности прямоточной, противоточной и двухмерной схем хроматомембранной газовой экстракции. Сплошные линии – зависимости, рассчитанные по уравнению (3) (1 – N = 1, 3 – N = 3, 4 – N = 10) и по уравнению (4) (2 – N = 5, M = 2); значки – экспериментальные данные. 5, 6 – прямоточная схема (ячейка 1); 7, 8 – двухмерная схема (ячейка 3); 9, 10 – противоточная схема (ячейка 1). 5, 7, 9 – бензол; 6, 8, 10 – хлороформ. I – область полного извлечения, II – область частичного извлечения и насыщения, III – область равновесного насыщения (для кривой 4).
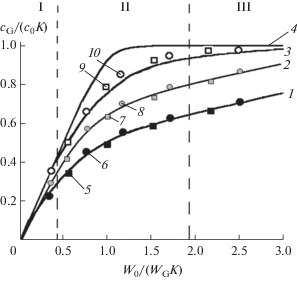
Анализ рассматриваемых зависимостей позволяет, выделить три области осуществления ХГЭ (см. рис. 4): режим полного извлечения аналитов из потока пробы в поток экстрагента (I), когда с достаточной для практики точностью можно принять:
область частичного извлечения или насыщения (II) и область равновесного насыщения (III) потока газа-экстрагента выделяемым аналитом, когда можно принять:С точки зрения аналитического применения наибольший интерес представляют области полного извлечения и равновесного насыщения, которые обеспечивают минимальную погрешность анализа, поскольку в этом случае результаты анализа в минимальной степени подвержены влиянию изменений условий эксперимента. Диапазоны указанных областей зависят от выбранной схемы ХГЭ. Недостатком прямоточной схемы являются узкие диапазоны соотношения расходов фаз, при которых реализуются полное извлечение и равновесное насыщение. Как следует из уравнения (2), выполнение соотношения (5) с погрешностью менее 1 отн. % будет достигнyто, если W0/WG < 100K, а соотношения (6) – если W0/WG > 100K. В случае противоточной и двухмерной схем указанные режимы легко реализуются соответственно при W0/WG < 2K и W0/WG > 2K.
Положительный эффект от увеличения числа тарелок в противоточной схеме постепенно ослабевает по мере увеличения N. Поскольку увеличение длины массообменного слоя приводит к пропорциональному увеличению инерционности экстракционной системы, применение громоздких хроматомембранных ячеек становится нерациональным. С учетом величины ВЭТТ оптимальная длина массообменного слоя ХМЯ для противоточной схемы составляет 20–30 мм, при которой обеспечивается N ≈ (3–4).
Аналогичные тенденции проявляются и для двухмерной схемы ХГЭ. Как указано выше, к повышению концентрации выделяемого компонента в экстрагенте приводит не только увеличение N, но и М, т.е. увеличение как длины, так и высоты массообменного слоя. При прочих равных условиях, в частности, при одном и том же объеме массообменного слоя, максимальные значения сG достигаются при равном числе М и N. Принимая во внимание, что ВЭТТ в направлении потока водной пробы приблизительно в пять раз больше, чем ВЭТТ в направлении потока газа-экстаргента, оптимальное соотношение длины и высоты массообменного слоя составляет 5 : 1.
При лабораторном анализе, когда быстродействие экстракционной системы не имеет принципиального значения, оптимальной является двухмерная схема ХГЭ. Эта схема, в отличие от противоточной, позволяет варьировать соотношения расходов фаз в гораздо более широком диапазоне и, в отличие от прямоточной схемы, дает возможность достигать равновесного насыщения при гораздо меньшем соотношении W0/WG.
Переходный режим. Закономерности этого режима имеют принципиальное значение при контроле состава потока анализируемой среды в режиме online. Установлено [16], что переходный режим ХГЭ адекватно описывается уравнением:
(7)
${{c}_{{\text{G}}}}\left( t \right) = c_{{\text{G}}}^{{{\text{кон}}}}--(c_{{\text{G}}}^{{{\text{кон}}}}--c_{{\text{G}}}^{{\text{0}}}){{e}^{{ - t/}}}^{\tau },$На основании тарелочной теории получено следующее выражение для постоянной времени хроматомембранной ячейки [12]:
Таким образом, к повышению быстродействия хроматомембранной ячейки (уменьшению ее постоянной времени) приводит уменьшение размеров массообменного слоя, увеличение расходов фаз, снижение удельной поверхности носителя и повышение температуры, обусловливающие снижение адсорбции из газовой фазы. При одинаковых размерах и структуре массообменного слоя прямоточная схема ХГЭ обеспечивает максимальное быстродействие, поскольку при ее реализации можно использовать в несколько раз более высокие расходы обменивающихся фаз через ячейку (см. табл. 1). По этой же причине двухмерная схема обладает большим быстродействием по сравнению с противоточной. Динамика изменения концентрации аналита (на примере этилацетата) в потоке газа-экстрагента после изменения его концентрации в потоке водного раствора с 5 до 50 мг/л для различных схем ХГЭ приведена на рис. 5.
Рис. 5.
Изменение концентрации этилацетата в потоке газа-экстрагента после ее увеличения в потоке пробы в 10 раз. W0 = 10 мл/мин: 1 – прямоточная схема, ячейка 2, WG = 250 мл/мин; 2 – двухмерная схема, ячейка 4, WG = 60 мл/мин; 3 – противоточная схема, ячейка 2, WG = 40 мл/мин.
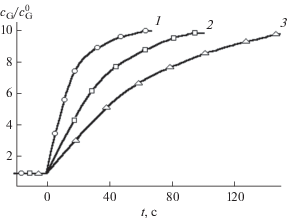
Миниатюрность ячеек и высокая скорость межфазного обмена обеспечивают высокое быстродействие и выгодно отличают ХГЭ от барботирования, где новые стационарные значения сG устанавливаются спустя несколько минут после изменения концентрации в анализируемой среде [17]. При осуществлении ХГЭ постоянная времени для веществ с температурой кипения до 100°С не превышает одной минуты. Негативное влияние адсорбции на быстродействие можно уменьшить, повышая температуру процесса (табл. 2). Кроме увеличения быстродействия, повышение температуры приводит также к значительному (в 3–4 раза) увеличению концентраций аналитов в потоке газа-экстрагента [18].
Список литературы
Poole C., Mester Z., Miró M., Pedersen-Bjergaard S., Pawliszyn J. Extraction for analytical scale sample preparation (IUPAC Technical Report) // Pure Appl. Chem. 2016. V. 88. № 7. P. 649.
Urbanowicz M., Zabiegala B., Namiesnik J. Solventless sample preparation techniques based on solid- and vapoour-phase extraction // Anal. Bioanal. Chem. 2011. V. 399. P. 277.
Иоффе Б.В. О терминологии газохроматографических методов анализа, основанных на внеколоночных фазовых равновесиях и распределениях // Журн. аналит. химии. 1981. Т. 36. № 8. С. 1663.
Родинков О.В., Бугайченко А.С., Москвин Л.Н. Статический парофазный анализ и его современное состояние // Журн. аналит. химии. 2020. Т. 75. № 1. С. 3. (Rodinkov O.V., Bugaichenko A.S., Moskvin L.N. Static head space analysis and its status // J. Anal. Chem. 2020. V. 75. № 1. P. 1.)
Витенберг А.Г., Иоффе Б.В. Газовая экстракция в хроматографическом анализе. Л.: Химия, 1982. 279 с. (Ioffe B.V., Vitenberg A.G. Head-space analysis and related methods in gas chromatography. N.Y.: Wiley Inters, 1984. 276 p.)
Lara-Gonzalo A., Sanchez-Uria J.E., Segovia-Garcia E., Sanz-Medel A. Crirical comparison of automated purge and trap and solid-phase microextraction for routine determination of volatile organic compounds in drinking waters by GC−MS // Talanta. 2008. V. 74. P. 1455.
Saridara C., Brukh R., Mitra S. Development of continuous on-line purge and trap analysis // J. Sep. Sci. 2006. V. 29. № 3. P. 446.
Moskvin L.N., Chromatomembrane method for the continuous separation of substances // J. Chromatogr. A. 1994. V. 669. P. 81.
Moskvin L.N., Rodinkov O.V. Continuous chromatomembrane headspace analysis // J. Cromatogr. A. 1996. V. 725. P. 351.
Преображенский Б.К., Калямин А.В., Лилова О.М., Москвин Л.Н. Зернистый пористый фторопласт для хроматографии. Способ получения и свойства // Радиохимия. 1968. Т. 10. № 3. С. 375.
Шишкин И.Ф. Теоретическая метрология. М.: Издательство стандартов, 1991. С. 165.
Родинков О.В. Закономерности непрерывной хроматомембранной газовой экстракции при движении фаз в одном направлении // Вестн. СПбГУ. Сер. 4. 2001. Вып. 3 (№ 20). С. 68.
Родинков О.В., Москвин Л.Н. Непрерывная двухмерная хроматомембранная газовая экстракция. Тарелочная модель и практические следствия // Журн. аналит. химии. 2000. Т. 55. № 9. С. 950. (Rodinkov O.V., Moskvin L.N. Continuous two-dimensional chromatomembrane gas extraction: A plate model and its practical consequences // J. Anal. Chem. 2000. V. 55. № 9. P. 854.)
Родинков О.В., Москвин Л.Н. Закономерности противоточной хроматомембранной газовой экстракции // Журн. аналит. химии. 2003. Т. 58. № 6. С. 611. (Rodinkov O.V., Moskvin L.N. Regularities in counterflow chromatomembrane gas extraction // J. Anal. Chem. 2003. V. 58. № 6. P. 530.)
Гиндин Л.М. Экстракционные процессы и их применение. М.: Наука, 1984. 144 с.
Родинков О.В., Москвин Л.Н., Майорова Н.А. Быстродействие различных схем непрерывной хроматомембранной газовой экстракции // Журн. аналит. химии. 2005. Т. 60. № 8. С. 727. (Rodinkov O.V., Moskvin L.N., Maiorova N.A. Operation rates of different schemes of continuous chromatomembrane gas extraction // J. Anal. Chem. 2005. V. 60. № 8. P. 820.)
Витенберг А.Г., Новикайте Н.В. Газохроматографическое определение летучих примесей в воде методом проточного парофазного анализа // Журн. аналит. химии. 1999. Т. 54. № 3. С. 300. (Vitenberg A.G., Novikaite N.V. Gas-chromatographic determination of volatile impurities in water by flow headspace analysis // J. Anal. Chem. 1999. V. 54. № 3. P. 264.)
Москвин Л.Н., Родинков О.В. От жидкостно-газовой хроматографии к хроматомембранному массообменному процессу // Журн. аналит. химии. 2019. Т. 74. № 10. С. 729. (Moskvin L.N., Rodinkov O.V. From liquid–gas chromatography to a chromatomembrane mass-exchange process // J. Anal. Chem. 2019. V. 74. № 10. P. 955.)
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии