

Журнал аналитической химии, 2022, T. 77, № 3, стр. 248-254
Определение микроэлементов в биологических жидкостях методом дуговой атомно-эмиссионной спектрометрии
С. С. Савинов a, *, А. И. Дробышев a
a Санкт-Петербургский государственный университет, Институт химии
199034 Санкт-Петербург, Университетская наб., 7/9, Россия
* E-mail: s.s.savinov@spbu.ru
Поступила в редакцию 11.06.2021
После доработки 05.07.2021
Принята к публикации 04.08.2021
- EDN: GGDOBR
- DOI: 10.31857/S0044450222010121
Аннотация
Определение макро- и микроэлементов в различных биологических жидкостях, в том числе в слюне и сыворотке крови, широко используется для медицинской диагностики и оценки воздействия антропогенных факторов на состояние здоровья человека. Наиболее популярные методы спектрального анализа имеют ряд недостатков, в том числе необходимость пробоподготовки. В качестве альтернативы апробирована схема прямого (без минерализации) анализа биожидкостей методом дуговой атомно-эмиссионной спектрометрии по способу сухого остатка. Показано, что экспериментально найденные оптимальное количество спектрального буфера (0.15 мг NaCl) и силы тока (20 А) обеспечивают высокоинтенсивный аналитический сигнал и снижение предела обнаружения. Выбранные условия также нивелируют влияние матрицы слюны и сыворотки на параметры дуговой плазмы (температуру и концентрацию электронов), что позволяет использовать водные стандартные растворы солей элементов для построения градуировочных зависимостей. При нанесении на торец электрода 150 мкл пробы цельной слюны пределы обнаружения Ag, Al, Cd, Cu, Fe, Mn, Zn находятся на уровне не более мкг/л, Cr, Pb, Ti – нескольких мкг/л. В случае сыворотки крови удается наносить на торец не более 50 мкл пробы, пределы обнаружения в этом случае соответственно в три раза выше. Проведена оценка правильности определения микроэлементов в реальных образцах слюны и сыворотки. Дополнительно показана возможность определения макроэлементов (Ca, Mg, P, Si) в слюне.
Микроэлементы, присутствующие в организме человека, в зависимости от их роли принято классифицировать на эссенциальные и токсичные [1]. Однако любой элемент, в том числе эссенциальный, при избыточном содержании в организме может привести к негативным последствиям [2]. Для диагностики микроэлементного дисбаланса чаще всего используют такие биожидкости, как кровь и ее компоненты, а также мочу. Примерами биомаркеров экспозиции могут служить концентрации Pb в крови, Ni в моче и плазме [3], Cr в моче и сыворотке [4], Cd в моче и крови [3, 5], Mn в крови [6], слюне и волосах [7]. Слюна также рассматривается как проба, подходящая для биомониторинга. Анализ слюны считается одним из перспективных подходов к неинвазивному мониторингу состояния здоровья и диагностике заболеваний [8].
Для определения концентраций элементов в биожидкостях используют целый ряд методов, включая атомно-эмиссионную спектрометрию с индуктивно связанной плазмой (АЭС-ИСП), масс-спектрометрию с ИСП, атомно-абсорбционную спектрометрию с электротермической атомизацией (ЭТААС), рентгенофлуоресцентный анализ с полным внешним отражением (РФА-ПВО) [1, 9]. Каждый из указанных методов имеет определенные преимущества, однако при анализе биожидкостей неизбежны матричные влияния, для устранения которых используют пробоподготовку. Обычно пробу подвергают кислотной минерализации [6, 10–13], однако данный этап увеличивает сложность, стоимость и продолжительность анализа [14]. При анализе слюны некоторые исследователи прибегают к упрощенной пробоподготовке, заключающейся в центрифугировании пробы и последующем разбавлении супернатанта [15–18]. Однако содержания веществ, в том числе микроэлементов, в супернатанте и цельной слюне отличаются, что приводит к некорректности сопоставления результатов различных исследований [19–21].
Метод дуговой АЭС также используют для анализа жидких проб, при этом для концентрирования элементов применяют как выпаривание пробы на графитовом коллекторе с последующим анализом порошковой пробы, так и последовательное нанесение на электрод капель жидкой пробы и их выпаривание с анализом сухого остатка [22, 23]. Последний подход не требует использования угольного порошка, что минимизирует возможные загрязнения пробы. Дуговая АЭС по способу сухого остатка позволяет анализировать малые объемы проб от 10 мкл (аналогично ЭТА-АС) с пределами обнаружения на уровне мкг/л (близкими к АЭС-ИСП) [22]. Однако методика анализа жидких проб, оптимизированная для регистрации с помощью фотоэлектронных умножителей, не позволяет проводить прямой анализ биожидкостей из-за матричного влияния и недостаточно низких пределов обнаружения.
Цель данной работы – оптимизация схемы анализа методом дуговой АЭС по способу сухого остатка для определения микроэлементов в биожидкостях без стадии пробоподготовки (кислотной минерализации).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
За основу взяли разработанную ранее методику определения микроэлементов в природных водах по способу сухого остатка [22, 24]. Каплю жидкой пробы объемом 10 мкл с помощью микрошприца наносили на торец подготовленного угольного электрода (класс F, Карботек, Россия) и высушивали под ИК-лампой. Затем аналогично наносили и высушивали каплю объемом 10 мкл раствора спектрального буфера. Электрод с пробой вместе с противоэлектродом, заточенным на конус, устанавливали в разрядной камере, для создания переменнотоковой дуговой плазмы использовали генератор ИВС-28 (АОМЗ, Россия). При горении разряда сухой остаток пробы испарялся с торца электрода и поступал в плазму, где происходили атомизация и возбуждение аналитов. Для разложения света в спектр использовали спектральный прибор МФС-8 (ЛОМО, Россия) с шириной входной щели 50 мкм для увеличения светосилы [25, 26]. Спектр регистрировали с помощью детектора МАЭС (ВМК-Оптоэлектроника, Россия), представляющего собой сборку из восьми фотодиодных линеек, каждая из которых содержит 2580 ячеек-фотодиодов шириной 12.5 мкм, динамический диапазон регистрации 104 [27, 28]. Детектор позволяет регистрировать спектр в цифровом виде в диапазоне 197–343 нм. Для управления работой системы и обработки данных применяли программу “Атом” [29]. В качестве аналитического сигнала использовали интенсивность по всему контуру спектральной линии с вычетом фонового излучения и холостого опыта [30].
Градуировочные зависимости строили с использованием растворов солей элементов, приготовленных из головного раствора CertiPUR IV (Merck, Германия) и одноэлементных стандартных растворов (ЦСОВВ, Россия) разбавлением 1%-ной HNO3. Градуировочные растворы наносили на электроды и анализировали по аналогичному алгоритму. Для оптимизации схемы анализа использовали как модельные водные растворы, так и реальные образцы биожидкостей.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Оптимизация условий возбуждения спектра. Ранее на примере водных растворов и образцов слюны показано, что увеличение силы тока дугового разряда до 20 А приводит к увеличению как амплитуды, так и суммарной по времени экспозиции интенсивности спектральных линий [31]. При указанной силе тока происходит “импульсное” поступление элементов в зону плазмы (рис. 1), т.е. уменьшается время испарения сухого остатка пробы с торца электрода и ее поступления в плазму, что позволяет уменьшить время регистрации спектра. Данное значение силы тока было выбрано в качестве рабочего; оно является максимально возможным для используемого генератора и позволяет увеличить отношение сигнал/шум в 1.5–3 раза в зависимости от элемента (по сравнению с силой тока 8 А).
Рис. 1.
Гистограмма выгорания (зависимость интенсивности I от порядкового номера N спектра) аналитической линии Zn 213.86 нм при силе тока 8 А (а) и 20 А (б) при времени полной экспозиции 40 с и времени базовой экспозиции 2 с.
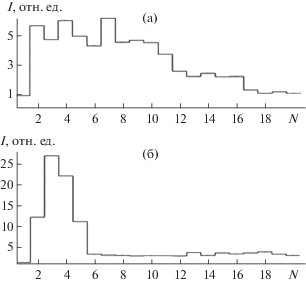
Интенсивность излучения спектральных линий во многом определяется температурой плазмы дугового разряда, влияющей на процессы атомизации и возбуждения. На степень ионизации, а значит и на интенсивность атомных и ионных линий, оказывает влияние концентрация электронов в дуговой плазме. При этом основным фактором, определяющим параметры плазмы, является состав атомного газа в дуговом промежутке, зависящий от состава исследуемых образцов [23, 32]. Анализируемые пробы, особенно биологические, содержат неопределяемые вещества, качественный и количественный состав которых может варьироваться в широком диапазоне. Для стабилизации параметров плазмы широко используют введение спектрального буфера, в частности различных легкоионизируемых элементов [32]. При введении достаточного количества спектрального буфера именно он будет задавать параметры плазмы, т.е. становиться основой пробы, нивелируя матричное влияние. В настоящей работе изучали влияние на интенсивность спектральных линий добавок NaCl и NaF, которые наносили на торец электрода после пробы в виде капли объемом 10 мкл водного раствора, приготовленного на деионизированной воде из чистых солей (Merc-k, Германия). Оба галогенида одинаково влияют на аналитический сигнал, однако при использовании NaCl интенсивности спектральных линий несколько выше. Поскольку уровень фонового излучения для обоих спектральных буферов одинаков, NaCl выбрали для дальнейшего исследования. Как видно из рис. 2, при увеличении массы спектрального буфера на торце электрода с 0 до 0.15 мг аналитический сигнал в холостом опыте практически не изменяется. При этом увеличивается интенсивность спектральных линий при нанесении на электрод исследуемых растворов, что приводит к увеличению отношения сигнал/шум (в 1.5–4 раза для различных элементов). Отмечено, что в присутствии NaCl на зависимостях интенсивностей спектральных линий от времени горения дугового разряда раньше появляется максимум, который при этом выше по амплитуде по сравнению со случаем отсутствия спектрального буфера. Добавка NaCl, вероятно, влияет на условия испарения элементов из сухого остатка с торца угольного электрода [24]. Кроме того, при введении спектрального буфера увеличивается концентрация электронов в плазме, что приводит к уменьшению степени ионизации и росту интенсивности атомных спектральных линий. В качестве оптимального параметра выбрали 0.15 мг спектрального буфера (что соответствует нанесению на электрод 10 мкл раствора NaCl с концентрацией 15 г/л).
Рис. 2.
Зависимость интенсивности I аналитической линии Pb 283.31 нм (◇) и фонового сигнала (◻), отношения сигнал/шум I/S (Δ) от массы m NaCl на торце электрода.
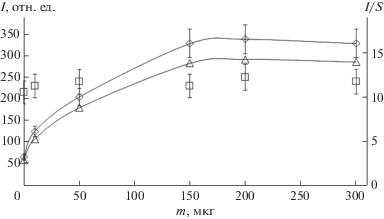
Макросостав исследуемых биожидкостей и водных растворов солей элементов, используемых для построения градуировочных зависимостей, отличается по содержанию как органических веществ, так и неорганических, в частности солей натрия (среднее содержание для слюны составляет 200 мг/л [33], для сывороки – 3 г/л [34]). Естественно, натрий, содержащийся в биопробах, также может выступать в качестве легкоионизируемого элемента. Тем не менее при нанесении на электрод 15 капель (объем каждой капли 10 мкл) пробы слюны с концентрацией натрия 200 мг/л в сухом остатке будет содержаться ≈0.03 мг элемента, при нанесении пяти капель пробы сыворотки с концентрацией натрия 3 г/л в сухом остатке будет содержаться ≈0.15 мг. Как видно из рис. 2, изменение массы спектрального буфера в диапазоне 0.15–0.30 мг не сказывается на интенсивности спектральных линий. Таким образом, нет необходимости учитывать натрий из биопроб.
Построение градуировочных зависимостей. Градуировочные растворы готовились в диапазоне концентраций элементов 10–6–1 г/л. В качестве аналитических использовались преимущественно самые интенсивные линии в рабочем диапазоне длин волн, свободные от спектральных наложений и позволяющие получить наиболее широкий диапазон линейности градуировочных зависимостей в координатах lgI–lgc. Для Ca, Mg, P, Si на самых интенсивных линиях происходило “зашкаливание” аналитического сигнала для растворов с большими концентрациями. Следует отметить, что перечисленные элементы являются макроэлементами в исследуемых биожидкостях, и в их спектрах имеются менее интенсивные линии, которые и использовали в качестве аналитических.
Каждую точку на градуировочной зависимости получали как среднее из восьми параллельных измерений интенсивности аналитического сигнала. Диапазон линейности для всех определяемых элементов составлял не менее трех порядков величины концентрации с коэффициентом корреляции 0.99 и более. Для некоторых элементов (Cd, Mn) линейность градуировочной зависимости сохранялась в диапазоне до пяти порядков величины концентрации. Концентрационные пределы обнаружения, рассчитанные по 3s-критерию (s – стандартное отклонение аналитического сигнала в контрольном опыте при n = 8) при нанесении на торец электрода 150 мкл пробы, составили (мкг/л): Ag 0.5, Al 0.07, Ca 0.7, Cd 0.2, Cr 2, Cu 0.2, Fe 0.7, Mg 0.7, Mn 0.07, P 70, Pb 4, Si 200, Ti 13, Zn 0.3. Полученные пределы обнаружения, естественно, зависят от наносимого объема пробы, который определяется, в том числе, возможностью получения тонкого равномерного слоя сухого остатка. В случае слюны нанесение на электрод 150 мкл пробы не составляет проблем, но для сыворотки крови это условие трудновыполнимо при объеме более 50 мкл, из-за чего пределы обнаружения элементов в ней в три раза выше. Оптимизированная схема анализа по пределам не уступает исходной методике с регистрацией с помощью ФЭУ [24], но выгодно отличается от нее возможностью прямого (без минерализации) анализа биопроб. Пределы обнаружения данной схемы также близки к пределам, достигаемым при анализе слюны методами АЭС-ИСП и ЭТААС с предварительной пробоподготовкой. Для большинства элементов при анализе проб слюны значение sr не превышает 0.2, что удовлетворительно для дугового разряда. Кроме того, для некоторых элементов, содержащихся в больших концентрациях в биожидкостях (P, Ca, Mg, Zn), sr < 0.1.
Оценка матричного влияния. При анализе биожидкостей по предлагаемой схеме, когда для построения градуировочной зависимости используют водные растворы, необходимо убедиться в отсутствии влияния матрицы пробы. Поскольку матричное влияние в дуге переменного тока проявляется прежде всего в изменении параметров плазмы, экспериментально определили их для следующих случаев: водный раствор (10 мкл), пробы слюны (150 мкл) и сыворотки крови (50 мкл). Для расчета температуры плазмы использовали графический метод Орнштейна [35] по линиям железа, концентрацию электронов рассчитывали из соотношения интенсивностей атомной и ионной линий магния [36]. Как видно из табл. 1, для всех проб параметры плазмы в оптимизированных условиях отличаются незначимо, что свидетельствует об отсутствии влияния на них макросостава биожидкостей.
Таблица 1.
Параметры плазмы (температура T и концентрация электронов Ne) дугового разряда при возбуждении спектров проб различной природы
| Проба | T, К | Ne, см–3 |
|---|---|---|
| Водный раствор | 4800 ± 400 | (11 ± 6) × 1013 |
| Слюна | 4700 ± 400 | (10 ± 5) × 1013 |
| Сыворотка крови | 4800 ± 400 | (13 ± 6)× 1013 |
Оценили правильность анализа реальных образцов с использованием различных подходов. В случае цельной слюны проводили сравнительный анализ методом разбавления и добавки, который показал удовлетворительные результаты [31]. Кроме того, анализировали образец слюны при нанесении на торец электрода различных объемов пробы (100, 150 и 200 мкл); при этом получили удовлетворительно сходящиeся результаты, что также подтвердило отсутствие влияния макросостава на результаты анализа [21]. Дополнительно провели сравнительный анализ одного образца по предлагаемой схеме с нанесением на торец электрода 150 мкл пробы и методом АЭС-ИСП после кислотной минерализации (2.5 мл пробы, 1.5 мл конц. HNO3 ос. ч. (Вектон, Россия) и 0.5 мл конц. H2O2 ос. ч. (НеваРактив, Россия) с последующим разбавлением до 25 мл) в микроволновой печи. Полученные результаты удовлетворительно согласуются как для микроэлементов, так и для ряда макроэлементов (табл. 2).
Таблица 2.
Результаты (мг/л) определения содержания ряда элементов в цельной слюне различными методами (n = 8, Р = 0.95)
| Элемент | Предлагаемая схема | АЭС-ИСП |
|---|---|---|
| Ag | 0.009 ± 0.001 | 0.010 ± 0.001 |
| Al | 0.011 ± 0.002 | <0.2 |
| Ca | 168 ± 8 | 161 ± 5 |
| Cd | <0.0002 | <0.004 |
| Cr | 0.038 ± 0.004 | 0.041 ± 0.004 |
| Cu | 0.026 ± 0.004 | 0.028 ± 0.003 |
| Fe | 0.16 ± 0.02 | 0.14 ± 0.01 |
| Mg | 14 ± 1 | 13 ± 1 |
| Mn | 0.012 ± 0.001 | 0.011 ± 0.001 |
| P | 380 ± 20 | 400 ± 20 |
| Pb | 0.012 ± 0.002 | <0.04 |
| Si | 0.69 ± 0.09 | 0.73 ± 0.09 |
| Ti | 0.074 ± 0.008 | 0.079 ± 0.007 |
| Zn | 0.24 ± 0.03 | 0.25 ± 0.03 |
В случае сыворотки крови правильность определения микроэлементов контролировали путем сравнительного анализа одного образца несколькими способами. Образец венозной крови отбирали в полимерные медицинские пробирки с гелем-разделителем и активатором свертывания и центрифугировали при 3600 об/мин в течение 10 мин для отделения сыворотки. Затем полученную пробу анализировали по предлагаемой схеме с нанесением на торец электрода 10 и 50 мкл. При попытке нанесения большего объема после высушивания под ИК-лампой наблюдали образование пузырей и твердой корки, которая выносится из зоны плазмы при пробое дугового разряда. Исследуемый образец сыворотки подвергали также кислотной минерализации (6 мл пробы, 7.5 мл конц. HNO3 ос. ч. (Вектон, Россия) и 2.5 мл конц. H2O2 ос. ч. (НеваРактив, Россия)) в микроволновой печи. Полученный минерализат разбавляли в мерной колбе емк. 25 мл до метки деионизованной водой с последующим анализом по предлагаемой схеме, а также методами АЭС-ИСП, ЭТААС, РФА-ПВО. Результаты определения ряда микроэлементов представлены в табл. 3, концентрации Ag, Al, Cr, Mn в исследуемом образце достоверно не удалось определить. Как видно, данные, полученные при прямом анализе сыворотки, удовлетворительно согласуются с результатами анализа минерализата различными методами, что также подтверждает отсутствие матричного влияния.
Таблица 3.
Результаты (мг/л) определения ряда элементов в сыворотке крови различными методами (n = 8, Р = 0.95)
| Метод | Проба | Cd | Cu | Fe | Pb | Zn |
|---|---|---|---|---|---|---|
| Предлагаемая схема | Исходная, 10 мкл | 0.005 ± 0.002 | 0.8 ± 0.2 | 1.5 ± 0.3 | <0.06 | 0.11 ± 0.03 |
| Исходная, 50 мкл | 0.0045 ± 0.0009 | 0.6 ± 0.1 | 1.2 ± 0.2 | 0.04 ± 0.01 | 0.12 ± 0.02 | |
| Минерализат | 0.004 ± 0.001 | 0.7 ± 0.2 | 1.1 ± 0.2 | <0.06 | 0.16 ± 0.03 | |
| АЭС-ИСП | Та же | <0.005 | 0.60 ± 0.06 | 1.5 ± 0.1 | <0.04 | 0.14 ± 0.01 |
| ЭТААС | $\gg $ | 0.0038 ± 0.0002 | 0.65 ± 0.04 | – | 0.035 ± 0.002 | 0.12 ± 0.01 |
| РФА-ПВО | $\gg $ | <0.010 | 0.8 ± 0.1 | 1.0 ± 0.2 | <0.06 | – |
* * *
Таким образом, показано, что использование оптимизированных условий атомизации и возбуждения, а именно большой силы тока дугового разряда (20 А) и значительного количества спектрального буфера на торце электрода (0.15 мг NaCl) позволяет не только увеличить отношение сигнал/шум, но и нивелировать влияние матрицы биопроб на параметры дуговой плазмы (ее температуру и концентрацию электронов). Это дает возможность определять элементы в биожидкостях без предварительной минерализации методом дуговой атомно-эмиссионной спектрометрии по способу сухого остатка с использованием водных стандартных растворов солей элементов для построения градуировочных зависимостей.
Исследование выполнено при финансовой поддержке гранта Президента РФ в рамках научного проекта № МК-2476.2021.1.3. Авторы выражают благодарность ресурсному центру Научного парка СПбГУ “Методы анализа состава вещества” и ООО “ВМК-Оптоэлектроника”, чье оборудование было использовано при выполнении исследования.
Список литературы
Parsons P.J., Barbosa F.Jr. Atomic spectrometry and trends in clinical laboratory medicine // Spectrochim. Acta B. 2007. V. 62. № 9. P. 992. .https://doi.org/10.1016/j.sab.2007.03.007
Savory J., Wills M.R. Trace-metals – essential nutrients or toxins // Clin. Chem. 1992. V. 38. № 8. P. 1565.
Kakkar P., Jaffery F.N. Biological markers for metal toxicity // Environ. Toxicol. Pharmacol. 2005. V. 19. № 2. P. 335. https://doi.org/10.1016/j.etap.2004.09.003
Bonde J.P., Christensen J.M. Chromium in biological samples from low-level exposed stainless-steel and mild-steel welders // Arch. Environ. Health. 1991. V. 46. № 4. P. 225. https://doi.org/10.1080/00039896.1991.9937453
Jarup L., Elinder C.G., Spang G. Cumulative blood-cadmium and tubular proteinuria – A dose-response relationship // Int. Arch. Occup. Environ. Health. 1988. V. 60. № 3. P. 223. https://doi.org/10.1007/BF00378700
Zheng W., Jiang Y.-M., Zhang Y., Jiang W., Wang X., Cowan D.M. Chelation therapy of manganese intoxication with para-aminosalicylic acid (PAS) in Sprague-Dawley rats // Neurotoxicology. 2009. V. 30. № 2. P. 240. https://doi.org/10.1016/j.neuro.2008.12.007
Zheng W., Fu S.X., Dydak U., Cowan D.M. Biomarkers of manganese intoxication // Neurotoxicology. 2011. V. 32. № 1. P. 1. https://doi.org/10.1016/j.neuro.2010.10.002
Lee J.M., Garon E., Wong D.T. Analyzing saliva to diagnose and monitor health status // Dental Abstracts. 2011. V. 56. № 1. P. 53.
Taylor A., Day M.P., Hill S., Marshall J., Patriarca M., White M. Atomic spectrometry update: review of advances in the analysis of clinical and biological materials, foods and beverages // J. Anal. At. Spectrom. 2015. V. 30. № 3. P. 542. https://doi.org/10.1039/c5ja90001h
Lopez-Jornet P., Juan H., Alvaro P.F. Mineral and trace element analysis of saliva from patients with BMS: a cross-sectional prospective controlled clinical study // J. Oral Pathol. Med. 2014. V. 43. № 2. P. 111. https://doi.org/10.1111/jop.12105
Menegario A.A., Gine M.F. Micro-scale flow system for on-line multielement preconcentration from saliva digests and determination by inductively coupled plasma optical emission spectrometry // Spectrochim. Acta B. 2001. V. 56. № 10. P. 1917. https://doi.org/10.1016/S0584-8547(01)00321-4
Monaci F., Bargagli E., Bravi F., Rottoli P. Concentrations of major elements and mercury in unstimulated human saliva // Biol. Trace Elem. Res. 2002. V. 89. № 3. P. 193. https://doi.org/10.1385/BTER:89:3:193
МУК 4.1.1482-03. Определение содержания химических элементов в диагностируемых биосубстратах, поливитаминных препаратах с микроэлементами, в биологически активных добавках к пище и в сырье для их изготовления методом атомно-эмиссионной спектрометрии с индуктивно связанной аргоновой плазмой.
Marco L.M., Greaves E.D., Alvarado J. Analysis of human blood serum and human brain samples by total reflection X-ray fluorescence spectrometry applying Compton peak standardization // Spectrochim. Acta B. 1999. V. 54. № 10. P. 1469. https://doi.org/10.1016/S0584-8547(99)00085-3
Guo W., Dong S., Jin Y., Pan Z., Pearce E.N., Wu W., Zhang Y., Chen W., Zhang W. Evaluation of variation of saliva iodine and recommendations for sample size and sampling time: Implications for assessing iodine nutritional status // Clin. Nutr. 2021. V. 40. № 5. P. 3559. https://doi.org/10.1016/j.clnu.2020.12.010
de Almeida G.R.C., Tavares C.F.D., de Souza A.M., de Sousa T.S., Funayama C.A.R., Barbosa F., Tanus-Santos J.E., Gerlach R.F. Whole blood, serum, and saliva lead concentrations in 6-to 8-year-old children // Sci. Total Environ. 2010. V. 408. № 7. P. 1551. https://doi.org/10.1016/j.scitotenv.2009.12.034
Wang D., Du X., Zheng W. Alteration of saliva and serum concentrations of manganese, copper, zinc, cadmium and lead among career welders // Toxicol. Lett. 2008. V. 176. № 1. P. 40. https://doi.org/10.1016/j.toxlet.2007.10.003
Olmedo P., Pla A., Hernandez A.F., Lopez-Guarnido O., Rodrigo L., Gil F. Validation of a method to quantify chromium, cadmium, manganese, nickel and lead in human whole blood, urine, saliva and hair samples by electrothermal atomic absorption spectrometry // Anal. Chim. Acta. 2010. V. 659. № 1–2. P. 60. https://doi.org/10.1016/j.aca.2009.11.056
Novo D.L., Mello J.E., Rondan F.S., Henn A.S., Mello P.A., Mesko M.F. Bromine and iodine determination in human saliva: Challenges in the development of an accurate method // Talanta. 2019. V. 191. P. 415. https://doi.org/10.1016/j.talanta.2018.08.081
Савинов С.С., Анисимов А.А., Дробышев А.И. Проблемы и оптимизация отбора образцов, их хранения и пробоподготовки при определении микроэлементного состава слюны человека // Журн. аналит. химии. 2016. Т. 71. № 10. С. 1063. (Savinov S.S., Anisimov A.A., Drobyshev A.I. Problems and optimization of sampling, storage, and sample preparation in the determination of the trace element composition of human saliva // J. Anal. Chem. 2016. V. 71. № 10. P. 1016. https://doi.org/10.1134/S106193481608012810.1134/S1061934816080128)https://doi.org/10.7868/S0044450216080120
Савинов С.С., Анисимов А.А. Влияние условий отбора образцов слюны человека на результаты определения макро- и микроэлементов // Журн. аналит. химии. 2020. Т. 75. № 4. С. 327. (Savinov S.S., Anisimov A.A. Effect of conditions for sampling of human saliva on the results of determination of macro- and micronutrients // J. Anal. Chem. 2020. V. 75. № 4. P. 453. https://doi.org/10.1134/S106193482004013910.1134/S1061934820040139)https://doi.org/10.31857/S0044450220040143
Дробышев А.И., Емелина О.И. Анализ вод и растворов атомно-эмиссионным методом на квантометре МФС-8 // Журн. аналит. химии. 1999. Т. 54. № 12. С. 1300. (Drobyshev A.I., Emelina O.I. Analysis of water samples and aqueous solutions by atomic emission spectrometry using an MFS-8 spectrometer // J. Anal. Chem. 1999. V. 54. № 12. P. 1152.)
Соломенцева Н.С., Шуваева О.В. Определение микроэлементов в природных водах методом атомно-эмиссионной спектрометрии сухих остатков на торцах графитовых электродов // Журн. аналит. химии. 2007. Т. 62. № 7. С. 719. (Solomentseva N.S., Shuvaeva O.V. Determination of trace elements in natural waters by atomic emission spectrometry of solution dry residues at the ends of graphite electrodes // J. Anal. Chem. 2007. V. 62. № 7. P. 645. )https://doi.org/10.1134/S1061934807070064
Дробышев А.И., Емелина О.И. Спектроаналитические возможности униполярной дуги переменного тока при фотоэлектрической регистрации спектра // Журн. аналит. химии. 1996. Т. 51. № 10. С. 1047. (Drobyshev A.I., Emelina O.I. Spectrochemical analytical possibilities of a unipolar alternating-current arc in the photoelectric recording of spectra // J. Anal. Chem. 1996. V. 51. № 10. P. 958.)
Дробышев А.И., Савинов С.С. Экспериментальное исследование светосилы цифрового спектрографа на базе МФС−МАЭС // Оптика и спектроскопия. 2016. Т. 120. № 2. С. 349. (Drobyshev A.I., Savinov S.S. An experimental study of the luminosity of an MFC–MAES-based digital spectrograph // Opt. Spectrosс. 2016. V. 120. № 2. P. 335. https://doi.org/10.1134/S0030400X1602007710.1134/S0030400X16020077)https://doi.org/10.7868/S0030403416020070
Дробышев А.И., Савинов С.С. Экспериментальное исследование аппаратной функции и разрешающей способности оптического цифрового спектрографа на базе полихроматора МФС // Оптический журн. 2014. Т. 81. № 1. С. 44. (Drobyshev A.I., Savinov S.S. Experimental study of the spread function and resolving power of an optical digital spectrograph based on an MFS polychromator // J. Opt. Technol. 2014. V. 81. № 1. P. 33. )
Labusov V.A. Setup complexes for atomic-emission spectral analysis based on grand spectrometer // Inorg. Mater. 2009. V. 45. № 14. P. 1529. https://doi.org/10.1134/S0020168509140039
Лабусов В.А., Гаранин В.Г., Шелпакова И.Р. Многоканальные анализаторы атомно-эмиссионных спектров. Современное состояние и аналитические возможности // Журн. аналит. химии. 2012. Т. 67. № 7. С. 697. (Labusov V.A., Garanin V.G., Shelpakova I.R. Multichannel analyzers of atomic emission spectra: Current state and analytical potentials // J. Anal. Chem. 2012. V. 67. № 7. P. 632. )https://doi.org/10.1134/S1061934812070040
Гаранин В.Г., Неклюдов О.А., Петроченко Д.В., Семёнов З.В., Шаталов И.Г., Панкратов С.В. Программное обеспечение атомно-эмиссионного спектрального анализа (программа “Атом”) // Заводск. лаборатория. Диагностика материалов. 2012. V. 78. № 1(2). P. 69.
Дробышев А.И., Савинов С.С. О некоторых особенностях регистрации спектра и фотометрирования спектральных линий с помощью цифрового спектрографа на базе МФС–МАЭС // Приборы и техника эксперимента. 2013. № 6. С. 56. (Drobyshev A.I., Savinov S.S. On certain features of spectrum recording and photometric measurements of spectral lines using a MFS–MAES-based digital spectrograph // Instrum. Exp. Tech. 2013. V. 56. № 6. P. 693. https://doi.org/10.1134/S002044121305013810.1134/S0020441213050138)https://doi.org/10.7868/S0032816213050133
Дробышев А.И., Савинов С.С. Дуговой атомно-эмиссионный цифровой спектрографический анализ жидких биопроб с использованием МАЭС // Заводск. лаборатория. Диагностика материалов. 2015. № 1(II). С. 142.
Torok T., Mika J., Gegus E. Emission Spectrochemical Analysis. Budapest: Akademiai Kiado, 1978. 692 c.
Нотова С.В., Орджоникидзе Г.З., Нигматуллина Ю.Ф. Содержание химических элементов в слюне и волосах детей, проживающих в районном центре Саракташ Оренбургской области // Вестник ОГУ. 2003. № 6. С. 146.
Bardarov K.V., Buchvarov I., Yordanova T., Georgiev P.A. Laser-induced break down spectroscopy for quantitative analysis of electrolytes (Na, K, Ca, Mg) in human blood serum // Proc. SPIE. 2019. V. 11332. P. 113320A. https://doi.org/10.1117/12.2552209
Вуколов А.В. Спектральный метод измерения температуры плазмы. Лабораторный практикум. Томск: ТПУ, 2008. 14 с.
Спектральный анализ чистых веществ / Под ред. Зильберштейна Х.И. СПб: Химия, 1994. 336 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии