

Журнал аналитической химии, 2022, T. 77, № 5, стр. 471-477
Внутрикапиллярная хиральная дериватизация аминокислот
Л. А. Карцова a, *, Д. О. Москвичев a
a Санкт-Петербургский государственный университет, Институт химии
198504 Санкт-Петербург, Петродворец, Университетский просп., 26, Россия
* E-mail: kartsova@gmail.com
Поступила в редакцию 06.09.2021
После доработки 15.09.2021
Принята к публикации 18.09.2021
- EDN: TUFDMK
- DOI: 10.31857/S004445022205005X
Аннотация
Обсуждаются возможности проведения внутрикапиллярной хиральной дериватизации аминокислот с использованием орто-фталевого альдегида и N-ацетил-L-цистеина. Выявлено влияние концентрации боратного буферного раствора и добавок в фоновый электролит 2-гидроксипропил-β-циклодекстрина и α-циклодекстрина на селективность разделения образующихся диастереомерных производных. Найденные условия адаптированы к электрофоретическому анализу культуральной жидкости бактерий Escherichia coli для обнаружения продуцируемых ими аминокислот. Идентифицированы тирозин, аланин, серин и глутаминовая кислота. Важным результатом является обнаружение в культуральной жидкости D-серина и D-аланина.
Аминокислоты (АК) являются важной группой аналитов, определение которых востребовано при диагностике различных заболеваний, оценке качества пищевых продуктов и кормов, при изучении клеточного метаболизма и др. В настоящее время отмечен возрастающий интерес к обнаружению D-аминокислот [1], играющих важную роль в жизни бактерий, которые с помощью D-аминокислот управляют процессом “сборки” клеточной стенки, защищая ее таким образом от неблагоприятных факторов окружающей среды. Эндогенный D-серин выполняет в мозгу млекопитающих функцию нейротрансмиттера, а D-аспарагиновая кислота, обнаруженная в нейроэндокринных тканях млекопитающих, модулирует гормональную секрецию. Одним из интересных объектов анализа для выявления продуцируемых аминокислот являются культуральные жидкости (КЖ) бактерий [2].
Для исследования энантиомерного состава смеси АК или их производных, получаемых в процессах дериватизации, применяют различные физико-химические методы анализа: газовую [3] и жидкостную хроматографию (ТСХ, ВЭЖХ) [4], капиллярный зонный электрофорез (КЗЭ) и мицеллярную электрокинетическую хроматографию [5, 6], лигандообменный капиллярный электрофорез [7], спектрометрию ионной подвижности [8]. Необходимость получения производных при определении АК обусловлена отсутствием хромофорных групп в составе большинства их молекул.
Проведение дериватизации возможно как в автономном режиме, так и в режиме онлайн, т.е. непосредственно в кварцевом капилляре. При этом дериватизирующие реагенты могут находиться в составе фонового электролита (ФЭ), матрицы пробы или же вводиться в кварцевый капилляр последовательно с анализируемой пробой. Весьма заманчивым является сочетание в одном аналитическом цикле внутрикапиллярной дериватизации [9] и онлайн концентрирования [10–13]. В работе [12] обсуждается вариант определения аминокислот методом капиллярного электрофореза с внутрикапиллярной дериватизацией орто-фталевым альдегидом (ОФА) совместно с тиольной компонентой в качестве нуклеофила. Реагенты добавляли непосредственно в фоновый электролит с последующим детектированием образующихся производных флуоресцентным детектором. Для регулирования селективности разделения в ФЭ вводили различные концентрации β-циклодекстрина (β-ЦД). Авторы показали возможность разделения энантиомеров 15 аминокислот и адаптировали полученные результаты к электрофоретическому анализу белкового гидролизата.
Установлено, что на скорость реакции и стабильность образующегося продукта дериватизации сильно влияет природа нуклеофила. Роль тиольной компоненты в работе [12] выполнял хиральный реагент N-ацетил-L-цистеин (N-АЦ). Такой выбор обусловлен высокой скоростью реакции (~1 мин) с первичными аминогруппами с образованием соответствующих диастереомеров [14, 15]. Продукты этой реакции (схема 1 ) относительно нестабильны, однако время, затрачиваемое на внутрикапиллярную дериватизацию, разделение аналитов и детектирование, оказалось достаточным для проведения полного анализа.
Позднее те же авторы использовали аналогичную дериватизирующую систему для разделения биогеных аминов, но в режиме мицеллярной электрокинетической хроматографии (в фоновый электролит добавляли мицеллообразующий агент – додецилсульфат натрия в концентрации 20 мМ) [11].
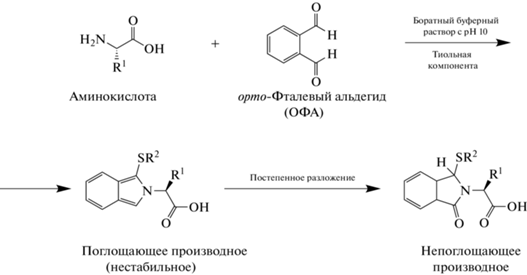
Схема 1 . Схема дериватизации аминокислот орто-фталевым альдегидом и тиольной компонентой [13].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Аппаратура. Для электрофоретического разделения применяли систему капиллярного электрофореза КАПЕЛЬ-105M со спектрофотометрическим детектором (Люмэкс, Санкт-Петербург). Использовали кварцевые капилляры с внешним полиимидным покрытием и общей длиной 60 или 75 см и эффективной длиной 50 или 65 см соответственно (внутренний диаметр 50 мкм, внешний диаметр 360 мкм). Обработку результатов проводили с помощью программного обеспечения “Эльфоран” для Windows.
Для контроля pH при приготовлении буферных растворов использовали pH-метр HI 2210-2216 (Hanna, Германия).
Реагенты. Гидроксид натрия ч. д. а. (Химреактив, Россия); соляная кислота х. ч., диметилформамид ч., диметилсульфоксид, ацетон (Реахим, Россия); дигидрофосфат натрия двухводный х. ч.; ацетон х. ч. (Вектон, Россия); метанол (J.T.Baker, Нидерланды); борная кислота, (2-гидроксипропил)-β-циклодекстрин (2-ГП-β-ЦД), N-ацетил-L-цистеин, о-фталевый альдегид (Sigma-Aldrich, Индия); D,L-протеиногенные аминокислоты: аланин (Ala), аспарагиновая кислота (Asp), валин (Val), глутаминовая кислота (Glu), метионин (Met), серин (Ser), тирозин (Tyr), треонин (Thr), триптофан (Trp); L-аминокислоты (Sigma-Aldrich, США): аланин (Ala), аспарагиновая кислота (Asp), валин (Val), глутаминовая кислота (Glu), метионин (Met), серин (Ser), тирозин (Tyr), треонин (Thr), триптофан (Trp); деионизованная вода.
Подготовка растворов реагентов и анализируемых объектов. Для приготовления 1 мл 400 мМ раствора о-фталевого альдегида в ацетонитриле брали его навеску массой 0.05365 г. Разбавленные растворы готовили путем добавления боратного буферного раствора cоответствующей концентрации (50–200 мМ) с pH 9.5. Для приготовления 1 мл раствора N-ацетилцистеина с концентрацией 600 мМ навеску массой 0.0979 г растворяли в боратном буферном растворе. Смесь 50 мкг/мл D,L-аминокислот готовили из концентрированных растворов и разбавляли 15 мМ фосфатным буферным раствором (pH 5.6).
Образцы культуральной жидкости бактерий E. coli, выращенных на глюкозо-минеральной среде, предоставленные коллегами из Государственного научно-исследовательского института особо чистых биопрепаратов (С.-Петербург), разбавляли 30 мМ фосфатным буферным раствором (pH 5.6) в соотношении 1 : 1 (по объему).
Внутрикапиллярная хиральная дериватизация аминокислот о-фталевым альдегидом и N-ацетил-L-цистеином. Перед проведением электрофоретических анализов кварцевый капилляр промывали метанолом (5 мин), затем 0.1 М раствором HCl (5 мин) и 0.1 М раствором NaOH (5 мин), после чего контролировали электроосмотический поток (ЭОП) в 5 мМ боратном буферном растворе. В качестве маркера ЭОПа использовали диметилсульфоксид. Перед каждым анализом капилляр предварительно промывали 0.1 М раствором NaOH (5 мин) и фоновым электролитом (5 мин), затем вводили 150 мМ раствора N-ацетил-L-цистеина (2 с, 10 мбар), раствор пробы (10–60 с, 30 мбар) и, наконец, 100 мМ раствор о-фталевого альдегида (2 с, 10 мбар). Анализ проводили в боратном буферном растворе при концентрации в диапазоне 50–200 мМ и pH 9.5. Напряжение составляло 25 кВ, длина волны детектирования – 340 нм. В результате выбрали следующие условия: 160 мМ боратный буфер, pH 9.5.
Анализ культуральных жидкостей E. coli. Для анализа КЖ использовали фоновый электролит следующего состава: 160 мМ боратный буферный раствор с pH 9.5, 0.3 мМ 2-ГП-β-ЦД. Режим ввода: 150 мМ раствор N-ацетил-L-цистеина (2 с, 10 мбар), раствор пробы (80 с, 30 мбар) и 100 мМ раствор о-фталевого альдегида (2 с, 10 мбар).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В последние годы возможности внутрикапиллярной дериватизации аминокислот рассматривались в целом ряде работ [13, 14, 16]. В работе [16] используемые для этой цели реагенты ОФА и N‑АЦ не входили в состав фонового электролита, а вводились отдельными зонами в кварцевый капилляр до и после ввода зоны пробы (так называемый метод “сэндвича”): сначала раствор N-АЦ, затем – разбавленный раствор анализируемой пробы и, наконец, раствор ОФА (5 с).
За счет разбавления пробы 60 мМ фосфатным буферным раствором с pH 6 достигалось онлайн концентрирование. По данным авторов работы [16], в этом случае реализуется смешанный вариант концентрирования – pH-скачок и изотахофорез: при вводе пробы в течение 100 с достигалось увеличение чувствительности в 40 раз по сравнению с вводом в течение 3 с. Добавление 1 мМ β-ЦД в ФЭ обеспечило увеличение селективности разделения дериватов [16]. Подобный принцип использован при выявлении энантиомерной чистоты метионина, триптофана, фениланалина и аспарагиновой кислоты [13]. В работе [13] смесь реагентов (30 мМ ОФА и 30 мМ N-АЦ в 20 мМ боратном буферном растворе) вводили в кварцевый капилляр до и после зоны пробы (5 с, 0.5 атм.), а в качестве дополнительного хирального селектора в фоновом электролите использовали γ-циклодекстрин. Сопоставлено влияние β-ЦД и γ-ЦД на разделение диастереомеров. Показано, что при использовании γ-ЦД для большинства АК значения факторов разрешения выше. Однако применение γ-ЦД не позволяет разделить энатиомеры серина, которые при использовании β-ЦД разделяются с разрешением 1.5.
Нами предпринята попытка внутрикапиллярной хиральной дериватизации с применением реагентов ОФА и N-АЦ в сочетании с онлайн концентрированием, а также исследовано влияние 2‑ГП-β-ЦД и α-ЦД на селективность разделения образующихся диастереомеров. Для проведения внутрикапиллярной дериватизации за основу взяли условия из работы [16]. Варьировали концентрацию буферного электролита (50–200 мМ), время гидродинамического ввода пробы (2–100 с, 30 мбар) и время ввода реагентов дериватизации (2–5 с, 10–30 мбар). Данный вариант предполагал последовательное введение N-АЦ, раствора пробы и затем – раствора ОФА. Электрофоретическое разделение образующихся производных происходило в боратном буферном растворе с pH 9.5, в котором растворялись и реагенты дериватизации. В отличие от последних, анализируемую пробу аминокислот растворяли в фосфатном буферном растворе с pH 5.4, что обеспечило не только внутрикапиллярную хиральную дериватизацию, но и онлайн концентрирование (pH-скачок).
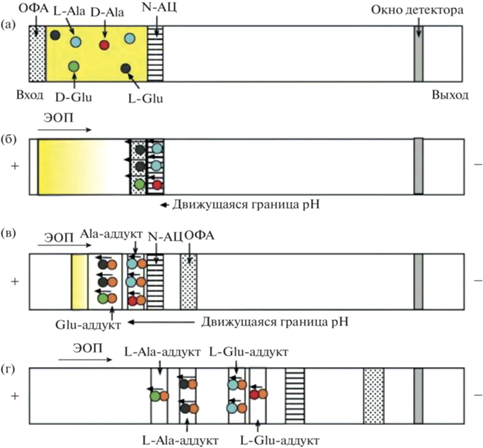
Суть механизма описана в работе [16]. В капилляр последовательно вводят раствор хирального реагента N-АЦ, далее – анализируемую пробу в фосфатном буферном растворе и, наконец, раствор ОФА (рис. 1а). После приложения напряжения положительно заряженные молекулы аминокислот мигрируют к катоду и концентрируются на границе зон пробы и N-ацетилцистеина. Навстречу им движутся отрицательно заряженные молекулы N-ацетил-L-цистеина. В зону пробы начинают проникать гидроксильные ионы, поскольку концентрация боратного буферного раствора в фоновом электролите выше, чем фосфатного в пробе (рис. 1б). Это приводит к изменению значения pH и аналитической формы аминокислот из катионной в анионную, после чего сконцентрированные аминокислоты совместно с N-АЦ начинают мигрировать к аноду, т.е. в противоположную сторону относительно электроосмотического потока, и в определенный момент достигают зоны о-фталевого альдегида. Здесь и происходит реакция образования соответствующих изоиндольных производных (рис. 1в). Молекулы о-фталевого альдегида в боратном буферном растворе с pH 9.5 не имеют собственной электрофоретической подвижности и мигрируют совместно с ЭОП. Образующиеся и сконцентрированные диастереомеры аминокислот далее разделяются в условиях КЗЭ (рис. 1г).
Рис. 1.
Схема внутрикапиллярной хиральной дериватизации аминокислот. (а): Последовательное гидродинамическое введение растворов N-ацетил-L-цистеина (N-АЦ), пробы и о-фталевого альдегида (ОФА); (б): онлайн концентрирование аминокислот; (в): внутрикапиллярная дериватизация за счет смешения с зоной ОФА/N-АЦ; (г) хиральное разделение аддуктов диастереомерных аминокислот. Концентрирование аминокислот происходит на разных стадиях электромиграции – как до, так и после внутрикапиллярной дериватизации [16].
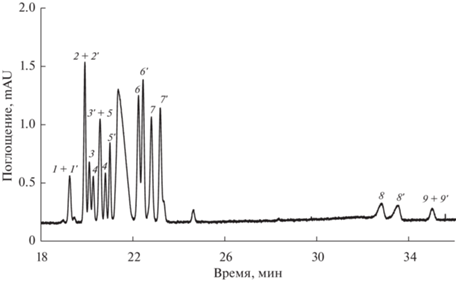
Описанные выше эксперименты по хиральной внутрикапиллярной дериватизации проводили как для модельных систем аминокислот (рис. 2), так и для каждой АК в отдельности. Для увеличения эффективности концентрацию фосфатного буферного раствора в пробе снизили с 60 мМ (данные работы [16]) до 15 мМ. По нашим данным, D-энантиомеры имеют большую электрофоретическую подвижность и мигрируют первыми.
Рис. 2.
Электрофореграмма, полученная при внутрикапиллярной дериватизации и онлайн концентрировании модельной смеси аминокислот. Условия: 160 мМ боратный буферный раствор с pH 9.5; режим ввода: 1 – 150 мМ раствор N-ацетил-L-цистеина (2 с, 10 мбар); 2 – раствор анализируемой пробы (50 с, 30 мбар); 3 – 100 мМ раствора о-фталевого альдегида (2 с, 10 мбар); 25 кВ; 340 нм. Обозначения производных аминокислот: 1, 1' – Trp; 2, 2' – Tyr; 3, 3' – Met; 4, 4' – Val; 5, 5' – Thr; 6, 6' – Ser; 7, 7' – Ala; 8, 8' – Glu; 9, 9' – Asp; D-изомеры обозначены цифрой, а L-изомеры – цифрой со штрихом.
Исследовано влияние концентрации боратного буферного раствора на энантиоселективное разделение диастереомеров аминокислот. Приемлемые времена миграции и требуемую селективность разделения наблюдали при концентрации фонового электролита 150 мМ.
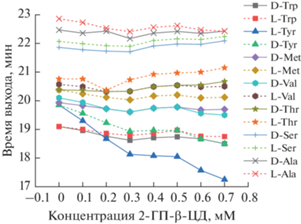
В найденных условиях достигнуто разделение энантиомеров производных Ser, Val, Met, Thr и Ala, однако D- и L-формы Tyr и Thr не разделяются. Выполнили серию специальных экспериментов по установлению влияния макроциклического агента 2-ГП-β-ЦД в диапазоне концентраций 0.1–0.7 мМ в составе фонового электролита на миграционные характеристики хиральных производных аминокислот (рис. 3). При увеличении концентрации 2-ГП-β-ЦД в фоновом электролите энантиоселективность возрастает для Trp, Tyr, Val, Met и Thr, но при этом снижается для Ala и Val. Наиболее заметное влияние увеличения концентрации макроцикла на время миграции энантиомеров обнаружено в случае тирозина. При этом только для тирозина в этих условиях время миграции производного L-изомера меньше, чем D-изомера.
Рис. 3.
Влияние концентрации (2-гидроксипропил)-β-циклодекстрина на селективность разделения диастереомерных производных аминокислот. Условия разделения см. в подписи к рис. 2.
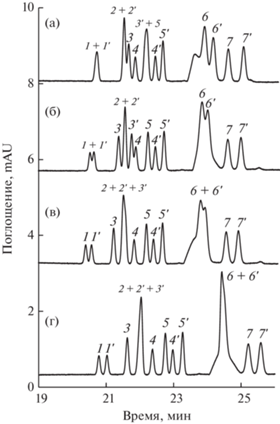
Анализ результатов, полученных на модельной системе аминокислот, позволил выбрать наиболее подходящую концентрацию используемого макроциклического агента, которая составила 0.1 мМ (рис. 4). Факторы энантиоселективности (Rs) производных D- и L-аминокислот при данной концентрации представлены ниже:
Рис. 4.
Электрофореграмма диастереомеров производных аминокислот (Trp, Tyr, Ala, Ser, Val, Met, Thr) c добавкой (2-гидроксипропил)-β-циклодекстрина (2-ГП-β-ЦД) в фоновый электролит, полученная в условиях внутрикапиллярной дериватизации и онлайн концентрирования. Условия: 160 мМ боратный буферный раствор с pH 9.5, 0.1 мМ 2-ГП-β-ЦД; режим ввода: 1) – 150 мМ раствора N-ацетил-L-цистеина, 5 с, 15 мбар; 2) – раствор анализируемой пробы, 10 с, 30 мбар; 3) – 100 мМ раствор о-фталевого альдегида, 5 с, 15 мбар; 25 кВ; 340 нм. Обозначения производных аминокислот см. в подписи к рис. 2.
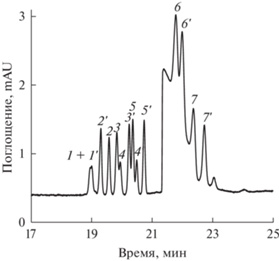
Наличие в составе фонового электролита макроцикла с меньшим размером полости – α-цикодекстрина – влияет в первую очередь на времена миграции диастереомеров метионина, улучшая разделение с энантиомерами других АК, а также обеспечивает разделение энантиомеров производных триптофана. При этом D- и L-формы тирозина в этих условиях не разделяются (рис. 5). Для сокращения продолжительности анализа, начиная с 28-й минуты, подавали дополнительное давление 30 мбар. Введение добавок сульфо-β-циклодекстрина не привело к изменению селективности разделения энантиомеров.
Рис. 5.
Влияние концентрации α-циклодекстрина (α‑ЦД) на селективность разделения диастереомерных производных аминокислот. Начиная с 28-й минуты, подается давление 30 мбар. Концентрация α‑ЦД в фоновом электролите, мМ: 0 (а), 1.5 (б), 2 (в), 2.5 (г). Остальные условия разделения см. в подписи к рис. 2.
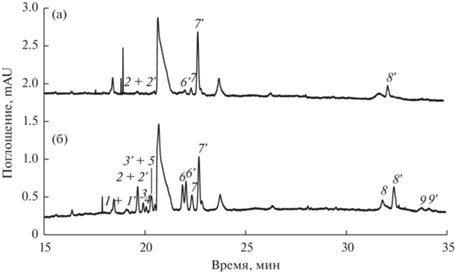
Выбранные условия (см. рис. 3) применили при электрофоретическом исследовании аминокислотного состава культуральной жидкости бактерии E. coli (рис. 6а). Для увеличения чувствительности анализируемую пробу вводили в течение 50 с при давлении 30 мбар. С целью идентификации компонентов КЖ добавляли смесь всех исследуемых АК (5 мкг/мл) (рис. 6б). Путем варьирования времени ввода пробы установлено, что при 80 с достигается максимальная чувствительность при сохранении селективности разделения. Пределы обнаружения составили 0.01 мМ для Glu и Asp, для других АК – в диапазоне 3–7 мкМ.
Рис. 6.
Электрофореграмма культуральной жидкости E. coli в условиях хиральной дериватизации (а); с добавкой аминокислот (5 мкг/мл) (б). Ввод пробы – 50 с, 30 мбар. Остальные условия разделения см. в подписи к рис. 2.
* * *
Таким образом, достигнуто электрофоретическое разделение энантиомеров аминокислот за счет внутрикапиллярной хиральной дериватизации с применением реагентов ОФА и N-АЦ в сочетании с онлайн концентрированием. Выбранные условия использованы для определения продуцируемых аминокислот в культуральной жидкости бактерий E. coli. Электрофоретический анализ показал присутствие Tyr, Ala, Ser и Glu. Важным результатом явилось электрофоретическое обнаружение в культуральной жидкости D-Ser и D-Ala.
Работа выполнена при финансовой поддержке проекта РНФ № 19-13-00370. Выражаем благодарность Ресурсному центру СПбГУ “Методы анализа состава вещества” за предоставленное оборудование.
Список литературы
Tanwar S., Bhushan R. Enantioresolution of amino acids: A decade’s perspective, prospects and challenges // Chromatographia. 2015. V. 78. № 17–18. P. 1113. https://doi.org/10.1007/s10337-015-2933-8
Kartsova L.A., Makeeva D.V., Kravchenko A.V., Moskvichev D.O., Polikarpova D.A. Capillary electrophoresis as a powerful tool for the analyses of bacterial samples // Trends Anal. Chem. 2021. V. 134. Article 116110. https://doi.org/10.1016/j.trac.2020.116110
Schurig V. Gas chromatographic enantioseparation of derivatized α-amino acids on chiral stationary phases-Past and present // J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2011. V. 879. № 29. P. 3122. https://doi.org/10.1016/j.jchromb.2011.04.005
Ilisz I., Aranyi A., Pataj Z., Péter A. Recent advances in the direct and indirect liquid chromatographic enantioseparation of amino acids and related compounds: A review // J. Pharm. Biomed. Anal. 2012. V. 69. P. 28. https://doi.org/10.1016/j.jpba.2012.01.020
Poinsot V., Carpéné M.A., Bouajila J., Gavard P., Feurer B., Couderc F. Recent advances in amino acid analysis by capillary electrophoresis // Electrophoresis. 2012. V. 33. № 1. P. 14. https://doi.org/10.1002/elps.201100360
Giuffrida A., Maccarrone G., Cucinotta V., Orlandini S., Contino A. Recent advances in chiral separation of amino acids using capillary electromigration techniques // J. Chromatogr. A. 2014. V. 1363. P. 41. https://doi.org/10.1016/j.chroma.2014.08.041
Zhao L., Qiao J., Zhang K., Li D., Zhang H., Li Q. Construction of chiral ligand exchange capillary electrochromatography for D,L-amino acids enantioseparation and its application in glutaminase kinetics study // J. Chromatogr. A. 2018. V. 1548. P. 104. https://doi.org/10.1016/j.chroma.2018.03.031
Pérez-Míguez R., Bruyneel B., Castro-Puyana M., Marina M.L.,Somsen G.W., Domínguez-Vega E. Chiral discrimination of DL-amino acids by trapped ion mobility spectrometry after derivatization with (+)-1-(9-fluorenyl)ethyl chloroformate // Anal. Chem. 2019. V. 91. № 5. P. 3277. https://doi.org/10.1021/acs.analchem.8b03661
Marina M.L., Castro-Puyana M. Derivatization in capillary electrophoresis / Capillary Electrophoresis. Methods in Molecular Biology / Schmitt-Kopplin P. (eds). N.Y.: Humana Press, 2016. P. 37. https://doi.org/10.1007/978-1-4939-6403-1_3
Cancelliere G., D’Acquarica I., Gasparrini F., Misiti D., Villani C. Synthesis and applications of novel, highly efficient HPLC chiral stationary phases: A chiral dimension in drug research analysis // Pharm. Sci. Technol. Today. 1999. V. 2. № 12. P. 484. https://doi.org/10.1016/S1461-5347(99)00218-7
Oguri S., Watanabe S., Abe S. Determination of histamine and some other amines by high-performance capillary electrophoresis with on-line mode in-capillary derivatization // J. Chromatogr. A. 1997. V. 790. № 1–2. P. 177. https://doi.org/10.1016/S0021-9673(97)00719-X
Oguri S., Yokoi K., Motohase Y. Determination of amino acids by high-performance capillary electrophoresis with on-line mode in-capillary derivatization // J. Chromatogr. A. 1997. V. 787. № 1–2. P. 253. https://doi.org/10.1016/S0021-9673(97)00664-X
Kühnreich R., Holzgrabe U. Indirect enantioseparation of amino acids by CE using automated in-capillary derivatization with ortho-phthalaldehyde and N-acetyl-l-cysteine // Chromatographia. 2016. V. 79. № 15–16. P. 1013. https://doi.org/10.1007/s10337-016-3122-0
Celá A., Glatz Z. Homocyclic o-dicarboxaldehydes: Derivatization reagents for sensitive analysis of amino acids and related compounds by capillary and microchip electrophoresis // Electrophoresis. 2020. V. 41. № 21–22. P. 1851. https://doi.org/10.1002/elps.202000041
García Alvarez-Coque M.C., Medina Hernández M.J., Villanueva Camañas R.M., Mongay Fernández C. Formation and instability of o-phthalaldehyde derivatives of amino acids // Anal. Biochem. 1989. V. 178. № 1. P. 1. https://doi.org/10.1016/0003-2697(89)90346-1
Ptolemy A.S., Tran L., Britz-McKibbin P. Single-step enantioselective amino acid flux analysis by capillary electrophoresis using on-line sample preconcentration with chemical derivatization // Anal. Biochem. 2006. V. 354. № 2. P. 192. https://doi.org/10.1016/j.ab.2006.04.016
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии