

Журнал аналитической химии, 2022, T. 77, № 5, стр. 412-426
Современная практика нецелевого химического анализа
Б. Л. Мильман a, b, *, И. К. Журкович b
a Институт экспериментальной медицины
197376 Санкт-Петербург, ул. Академика Павлова, 12, Россия
b Научно-клинический центр токсикологии имени академика С.Н. Голикова ФМБА России
192019 Санкт-Петербург, ул. Бехтерева, 1, Россия
* E-mail: bormilman@yandex.ru
Поступила в редакцию 19.06.2021
После доработки 08.08.2021
Принята к публикации 09.08.2021
- EDN: PNQSPF
- DOI: 10.31857/S0044450222050085
Аннотация
Представлен обзор основных методов, процедур и информационных продуктов, применяемых в нецелевом анализе (НЦА) при установлении неизвестного состава веществ. Предпочтительны методы пробоотбора и пробоподготовки, обеспечивающие извлечение определяемых соединений из анализируемых образцов в широком диапазоне свойств аналитов с наименьшими их потерями. Необходимые методы анализа представляют собой различные варианты хроматографии–тандемной масс-спектрометрии высокого разрешения (ХМС), которые обеспечивают получение индивидуальных характеристик аналитов (масс-спектров, характеристик удерживания) с целью их правильной идентификации. Приоритизация аналитической стратегии позволяет отбросить ненужные определения и тем самым повысить эффективность НЦА. Химические базы данных, массивы справочных масс-спектров и характеристик удерживания, алгоритмы и программы обработки данных ХМС незаменимы в НЦА.
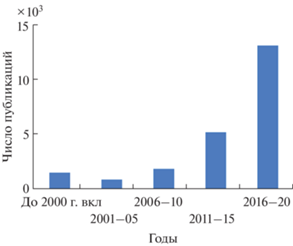
Нецелевой химический анализ (НЦА) представляет собой установление неизвестных аналитику компонентов анализируемых образцов (“известных неизвестных”, а также “неизвестных неизвестных”, табл. 1). В общем случае наиболее вероятно обнаружение тех или иных аналитов, относящихся к массиву нескольких миллионов распространенных индивидуальных соединений [1, 2]. Нецелевой химический анализ занимает все более значительное место в современных научных исследованиях химиков и биохимиков и практической деятельности технологов и инженеров. Это определяется тремя факторами. Первый из них отражает возрастающие потребности в таких аналитических определениях, которые связаны с появлением новых (emerging) загрязнителей окружающей среды, более полным контролем качества продуктов питания, последовательным усилением внимания к здоровью человека и т.д. Два других фактора позволяют реализовать возможности таких анализов. Здесь следует указать на современный высокий уровень развития аналитической методологии как следствие разработки новых типов и моделей хроматографов и масс-спектрометров, новых вариантов извлечения соединений из анализируемых сред. Наконец, важно отметить бурное развитие информатики, сопровождающееся наращиванием характеристик компьютеров и их сетей; появлением новых баз данных (БД); созданием алгоритмов и соответствующих программ, позволяющих эффективно манипулировать большими объемами получаемых данных и справочной информации. Названные факторы обусловливают резкий рост числа публикаций в области НЦА: более половины научных статей приходится на последние пять лет (рис. 1).
Таблица 1.
Основные термины нецелевого химического анализа
| Английский термин | Русский эквивалент | Комментарии |
|---|---|---|
| Untargeted/non-target/ non-targeted/nontargeted/nontarget analysis/screening | Нецелевой анализ/скрининг | Определение неизвестных аналитику компонентов анализируемых образцов; глагольные формы термина (окончание -ed) типичны прежде всего для описаний анализа биообъектов (метаболомика, протеомика) |
| (General) unknown analysis/screening | Анализ пробы/образца/ вещества неизвестного состава | Старый, стабильный термин (эквивалентный нецелевому анализу) в английском языке; используется в криминалистике, судебной медицине, токсикологии |
| Screening | Скрининг | Быстрое обнаружение аналитов с предварительным заключением об их природе. Наблюдаются отчетливые аналитические сигналы (хроматографические и массовые пики), надежная идентификация проблематична |
| Suspect analysis | Определение ожидаемых соединений (ООС) | Промежуточен между целевым анализом (ЦА), реализуемым по стандартным методикам, и НЦА |
| Effect-directed analysis | Анализ, направляемый эффектом | Анализ части пробы, которая содержит биологически активные соединения, проявляя ту или иную биологическую активность |
| Analytical strategy | Аналитическая стратегия | Совокупность и последовательность основных используемых методов, методик и операций |
| Prioritiz(s)ation | Приоритизация | Предпочтение в определении тех или иных аналитов, имеющих те или иные свойства |
| Аnnotation | Аннотация | Формальная процедура приписания характеристик определяемого соединения аналитическому сигналу. Термин достаточно часто используется вместо “идентификации” |
| Identification level | Уровень идентификации | Степень детализации в заключении о формуле и структуре аналита |
| Chemical space | Химическое пространство | Совокупность всех известных и/или возможных химических соединений |
| Known unknown | “Известное неизвестное” соединение | Известное соединение, его состав и строение установлены ранее, но факт присутствия в пробе аналитику неизвестен |
| Unknown unknown | “Неизвестное неизвестное” соединение | Новое соединение; его строение и свойства еще не установлены в результате экспериментальных исследований |
Рис. 1.
Динамика числа публикаций в области нецелевого химического анализа. Оценка проведена в начале 2021 г. суммированием числа статей и других документов, которые найдены в результате поисков в системе Google Академия по различным англоязычным терминам, обозначающих этот вид анализа (см. табл. 1).
Методология НЦА, используемая практически во всех областях химической аналитики, рассмотрена в многочисленных обзорах, часто связанных с отдельными объектами исследований (табл. 2). Методы и способы НЦА непрерывно совершенствуются, и имеет смысл зафиксировать современный уровень его общего развития, типичный для большинства объектов анализа. Такого рода общие характеристики НЦА будут рассмотрены в данной статье в сжатом обзорном виде. Наряду с методами анализа, подготовки проб и обработки информации, будет обсуждаться вопрос об эффективности НЦА, уровне его ошибок. В большинстве случаев, однако, не существует способов достоверной оценки таких ошибок. О правильности результатов НЦА часто можно судить лишь по тому, что реализована современная надлежащая практика его проведения. Это означает, что соответствующая работа включает основные необходимые стадии анализа, приборы, программы и базы данных, кратко рассмотренные в этой обзорной статье.
Таблица 2.
Основные области нецелевого химического анализа и соотвествующие объекты анализа
| Область | Объект/матрица | |||
|---|---|---|---|---|
| воздух, вода, почва, донные отложения, отходы, мусор | пищевые продукты и напитки | моча, кровь, ткани и др. биоматрицы | образцы растительного и животного происхождения | |
| Объекты окружающей среды [3–9] | ++ | + | + | |
| Пища, продовольственное сырье, напитки [10–12] | + | ++ | + | + |
| Лекарства, наркотики, допинг [13–15]* | ++ | |||
| Токсикология [13–15]* | + | + | ++ | + |
| Метаболомика [16–18] | + | ++ | + | |
| Природные соединения [19, 20] | + | ++ | ||
В обзоре затрагиваются низкомолекулярные соединения; преимущественно цитируется наиболее значимая литература последних лет, содержащая ссылки на предыдущие исследования. Следует отметить новые руководства по проведению НЦА в отдельных научных областях [16, 18, 21]; эти публикации имеют особое значение при начале работы в рассматриваемой области аналитики.
КРАТКАЯ ХАРАКТЕРИСТИКА НЕЦЕЛЕВОГО АНАЛИЗА
В наиболее широком смысле НЦА включает определение всех компонентов образца, состав которого неизвестен аналитику до эксперимента. Выбор приоритетов (см. ниже) может внести поправки в количество и природу определяемых соединений. Стадии НЦА формально совпадают для большинства объектов анализа (рис. 2). Результат анализа – соединения, обнаруженные и идентифицированные в пробе. Количественные определения обычно не ассоциируются с НЦА, но могут быть проведены вслед за идентификацией аналитов. Полноценное количественное определение требует подходящих методик анализа и аналитических стандартов. Полуколичественное определение возможно при использовании в качестве стандартов соединений со сходной структурой или при оценке относительной чувствительности методов к разным соединениям [12].
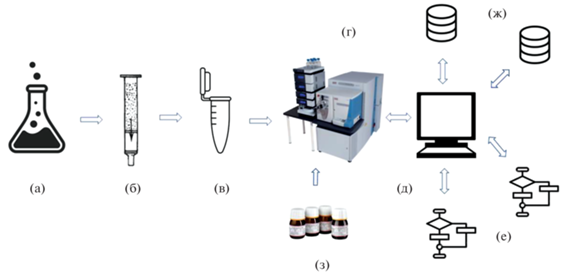
Рис. 2.
Схематичное изображение НЦА образца (а). Аналиты (в) выделяют методом экстракции (б) и вводят в хромато-масс-спектрометр (г). Работа прибора управляется компьютером (д), снабженным многочисленными программами (е) и связанным с разнообразными базами данных (ж). Предусмотрено обеспечение аналитическими стандартами (з) для окончательной идентификации аналитов.
Основные методы НЦА – это различные варианты хроматографии-масс-спектрометрии (ХМС). В случае летучих соединений – это газовая хроматография (ГХ)-масс-спектрометрия (МС) с электронной ионизацией (ЭИ) и одним квадрупольным масс-анализатором. При определении нелетучих аналитов, к которым относится большинство биологически важных соединений, применяют высокоэффективную или ультраэффективную жидкостную хроматографию (В/УЭЖХ) в сочетании с тандемной масс-спектрометрией высокого разрешения (МС2ВР); в качестве устройства ионизации используют электрораспыление (ИЭР). Другие варианты масс-спектрометрии небесполезны, но, как правило, менее эффективны. В последние годы развивается весьма ценное дополнение к ХМС – спектрометрия ионной подвижности [22]. При обнаружении и определении структуры новых, неописанных в научной литературе соединений (“неизвестных неизвестных”) целесообразно их выделение из анализируемых образцов и дальнейшее применение спектроскопии ЯМР (в дополнение к МС).
Нецелевой химический анализ преимущественно касается органических (биоорганических) соединений. В обзоре рассмотрен именно этот предмет НЦА. Тем не менее к нему может относиться и определение элементов, если рассматривается молекулярная форма их присутствия (speciation analysis); при этом применяется сочетание ВЭЖХ и МС с индуктивно связанной плазмой [23]). Другие виды “неорганического” НЦА важны, скорее, в историческом отношении.
ЭФФЕКТИВНОСТЬ НЕЦЕЛЕВОГО АНАЛИЗА
Показатель, вынесенный в заголовок раздела, мы предлагаем определить как долю соединений, обнаруженных и/или правильно идентифицированных в результате НЦА. В случае скрининга эффективность НЦА можно выразить величиной Pс и формулой (1):
(1)
${{P}_{{\text{с}}}}\left( \% \right) = {\text{ }}100{\text{ }} \times {\text{ }}\frac{{\left( {{\text{Количество обнаруженных соединений}}} \right)}}{{\left( {{\text{Количество компонентов пробы}}} \right)}}.~$Результат скрининга – обнаруженные компоненты анализируемой пробы и предварительное заключение об их природе. “Полноценный” же НЦА, где требуется максимально полно и надежно идентифицировать компоненты образца, характеризуется величиной PНЦА:
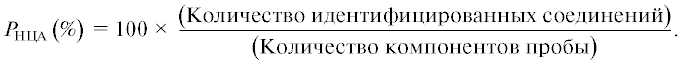
В общем случае величины Pс и PНЦА оценить очень сложно, поскольку состав анализируемой пробы и количество компонентов (знаменатель дроби) неизвестны. Числитель этих дробей вычислить легче, хотя не всегда понятно, насколько надежна соответствующая идентификация (см. ниже). Значения PНЦА можно оценить в случае модельной ситуации, например, используя специальные искусственные смеси соединений, представляющие интерес для рассматриваемой области анализа [24, 25], и оценивая количество идентифицированных и неидентифицированных соединений. Величина PНЦА в этом случае представляет собой показатель правильных положительных результатов (ПП) [26, 27], который в тест-экспериментах со смесями, выпускаемыми американским агентством EPA, не превышал 65% для методов, основанных на ВЭЖХ-МС2ВР [24].
ПРИОРИТИЗАЦИЯ
Если приоритеты НЦА (табл. 1) сформулированы, проводится поиск соответствующих соединений, которые наиболее важны и обладают определенными свойствами/признаками. При этом число определяемых соединений сокращается, что уменьшает знаменатель формул (1) и (2) и, следовательно, увеличивает величину Р.
Наиболее простой вариант НЦА реализуется в том случае, если ожидается присутствие в пробе тех или иных соединений (ООС, табл. 1) [4, 5, 7]. Такой анализ может быть близок к целевому, тем более что некоторые современные методики анализа включают определение многих сотен аналитов [28]. К ожидаемым относятся: (а) соединения, присутствующие в списках приоритетных загрязнителей окружающей среды, например в БД NORMAN [29]; (б) другие токсичные соединения, в том числе такие, опасные характеристики которых предсказаны; (в) вещества, часто встречающиеся в тех или иных матрицах; (г) соединения, родственные приоритетным, похожие на них по физико-химическим свойствам, в том числе продукты их трансформации (например, метаболиты).
Различие между ООС и “настоящим” НЦА заключается в том, что в первом случае число кандидатов на идентификацию (возможных аналитов) сравнительно невелико (соответствует выбранным приоритетам). Часто к ним относятся распространенные соединения, в отношении которых имеется представительная справочная хроматографическая и масс-спектрометрическая информация, что облегчает их идентификацию. В остальных случаях НЦА, которые охватывают менее популярные или менее ожидаемые соединения, в целом объем справочных данных не так велик и носит фрагментарный характер, поэтому поиск информации требует обращения к самым большим БД.
Если указывать другие варианты приоритизации, то к ним следует отнести “анализ, направляемый эффектом” (табл. 1). В этом случае контролируют (в том числе выделяют фракционированием) и анализируют ту часть пробы, которая проявляет биологическую активность. Еще один пример предпочтительного определения – хлор- и/или броморганические соединения, многие из которых относятся к опасным веществам. Их присутствие легко выявляется по характерной изотопной картине в масс-спектрах. Наконец, следует отметить тривиальный приоритет – основные компоненты проб, определяемые по наиболее интенсивным сигналам на хроматограммах и в масс-спектрах [8]. Родственная методология при поиске новых биологически активных соединений носит название “дерепликации”: здесь в первую очередь идентифицируют основные компоненты проб (как правило, известные соединения) и далее исключают их из рассмотрения [19].
Приоритизация может распространяться не только на аналиты, но и на сами анализируемые образцы. Часть образцов общего происхождения можно пропускать, а детально анализировать только те из них, которые демонстрируют (а) интенсивные аналитические сигналы в ожидаемых участках хроматограмм и масс-спектров и, более того, (б) рост интенсивностей таких сигналов в серии проб.
ПРЕАНАЛИТИЧЕСКИЕ ПРОЦЕДУРЫ
Качество этих процедур – отбора, транспортировки и хранения образцов – должно быть таким, чтобы не потерять искомые аналиты (приводит к ложным отрицательным результатам, ЛО; табл. 3) и не загрязнить пробу посторонними веществами (ложные положительные результаты, ЛП). Необходимые стандартные требования к таким процедурам сформулированы [16, 18, 21, 30], хотя трудно утверждать, что они выполняются во всех текущих исследованиях.
Таблица 3.
Источники и причины ложных результатов нецелевого химического анализа
| Процедура, метод | ЛП* | ЛО |
|---|---|---|
| Преаналитические процедуры | Плохая сорбция при пробоотборе. Трансформация аналитов |
|
| Пробоподготовка | Потери аналитов, неэффективные процедуры экстракции, неполные целевые химические превращения (дериватизация, деконъюгация) | |
| Хроматография | Совпадение характеристик удерживания разных соединений | Плохое разделение, слабые сигналы, вариация характеристик удерживания, испарение летучих аналитов в В/УЭЖХ, невозможность прямого определения нелетучих аналитов в ГХ, отсутствие справочных данных |
| Масс-спектрометрия | Совпадение масс-спектров разных соединений | Плохая ионизация, матричные эффекты, множественные формы ионов, содержащих молекулы аналита, слабые сигналы, ошибки в значениях ионных масс, плохая воспроизводимость масс-спектров, наложение спектров разных аналитов, отсутствие справочных данных |
Техника отбора проб влияет на представительство различных аналитов в отобранном веществе и появление ложных результатов (табл. 3). При пассивном пробоотборе проб воздуха или воды соотношение между аналитами может зависеть от типа сорбента. В разовых пробах воды, отобранных простым зачерпыванием (grab), такая дискриминация компонентов смесей отсутствует [7].
Посторонние соединения (ЛП) в анализируемой пробе сравнительно легко выявляются при анализе холостых проб (подготовленных из матриц, материалов, растворителей, реактивов и др. [16]). Труднее выявить ЛО. Для этого следует установить факт потери/разложения/превращения (в том числе биотрансформации) определяемых соединений до начала или в ходе анализа с использованием внутренних стандартов, что непросто сделать в НЦА, поскольку аналиты неизвестны. Тем не менее рекомендуют использовать добавки в анализируемые пробы – по одному соединению из каждого класса (группы) ожидаемых веществ [21] или из группы, представляющей тот или иной интервал физико-химических характеристик аналитов, например коэффициента распределения н-октанол–вода KOW [8]. Аналогичным образом формируют тест-смеси для обеспечения качества анализа [18].
ПРОБОПОДГОТОВКА
Эта необходимая процедура представляет собой выделение аналитов из отобранных образцов, сопровождающееся концентрированием первых. В большинстве проводимых анализов жидких (водных) образцов используется жидкостная или твердофазная (ТФЭ) экстракция. Разные аналиты обладают неидентичными физико-химическими свойствами, поэтому селективность рассматриваемых процедур и различная потеря аналитов неизбежны. Химические реакции (разложение, дегидратация, окисление, полимеризация), невольно сопровождающие процедуры пробоподготовки, приводят в ряде случаев к ложным результатам. К ним же приводит неполнота химических процедур, входящих в процесс пробоподготовки (табл. 3) [16].
В качестве экстрагентов для крови, плазмы крови и некоторых других биологических матриц используют водно-метанольные и водно-ацетонитрильные смеси, обеспечивающие близкую степень извлечения многих метаболитов. Кроме того, изменяя состав экстрагентов, можно поочередно извлекать и затем определять компоненты биологических матриц с разной полярностью [18]. Перспективен выбор тройных систем в качестве экстрагентов для плазмы крови, например использование смеси ацетонитрил–изопропанол–вода для извлечения полярных и умеренно полярных аналитов в диапазоне 25 порядков их величин KOW [17]. Для биологических матриц неизбежны быстрые превращения (энзимные реакции), так или иначе влияющие на степень извлечения аналита [18]. Эти процессы останавливают замораживаем проб или добавлением холодных растворителей.
Еще одна процедура, реализуемая в начале подготовки твердых образцов, а именно гомогенизация, может повлиять на результаты анализа, если не будет сформирована представительная проба [21]. В этих случаях проводят измельчение в специальных мельницах, где также необходимо подавить активность ферментов по отношению к компонентам образцов растительного и животного происхождения. Аналиты из таких измельченных проб извлекают методами экстракции, например с использованием популярной процедуры QuEChERS [11, 12].
В некоторых случаях (сравнительно высокие концентрации определяемых веществ) возможен прямой анализ (прямая инжекция в хроматограф) жидких образцов, таких как сточные воды и моча, без существенной потери многих аналитов; при этом часто практикуют предварительное разбавление исходных проб (методы dilute-and-shoot) [8]. Тем не менее и здесь очень вероятны ЛО. При сравнении таких процедур и ТФЭ продемонстрировано, что во втором случае можно обнаружить в воде больше полярных соединений, нежели при прямом вводе сравнительно больших объемов (0.5–1 мл) воды в жидкостный хроматограф [9].
ХРОМАТОГРАФИЯ
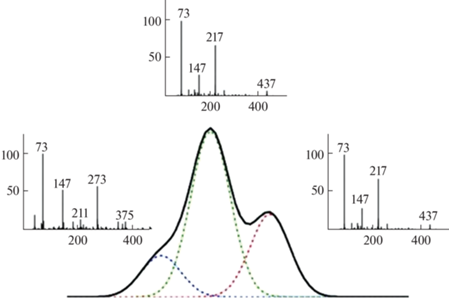
Как и в ЦА, хроматография является основным методом разделения в НЦА, хотя применение капиллярного электрофореза [31] и спектрометрии ионной подвижности (в дополнение к жидкостной хроматографии) [32] в каких-то ситуациях может привести к успеху. Неполное хроматографическое разделение аналитов искажает параметры удерживания и масс-спектры, что приводит к ошибкам в идентификации мажорных аналитов и потере минорных компонентов анализируемых смесей (табл. 3). Наиболее трудны для анализа сложные образцы, содержащие десятки, сотни, тысячи компонентов, поскольку соответствующие хроматограммы содержат очень много пиков, значительная часть которых в той или иной степени перекрывается (рис. 3). По этой причине в хромато-масс-спектрометрии реализуют процедуру деконволюции – разделение на индивидуальные хроматограммы, соответствующие отдельным компонентам смесей, с использованием их масс-спектрометрических сигналов (рис. 4, программы, см. [16]).
Рис. 3.
Пример сложной хроматограммы, адаптирован из работы [33]. Образец ткани одного из участков головного мозга. Метод УЭЖХ-МС2ВР. Горизонтальная ось – время удерживания, мин.
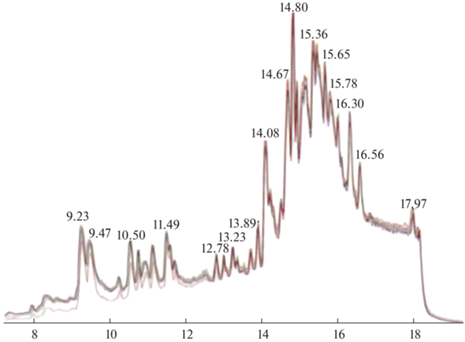
Рис. 4.
Пример деконволюции хроматографического пика в ГХ-МС. Адаптирован из работы [34]. Масс спектры, горизонтальная ось – значения m/z, вертикальная ось – относительная интенсивность, %. Сложный хроматографический пик разделен на три гауссовых сигнала. Второй и третий масс-спектры очень похожи. Для различения этих аналитов необходимы справочные параметры удерживания и, в конечном счете, аналитические стандарты. Еще один путь анализа – применение МС2 и МСВР.
Два основных вида хроматографии – газовая и жидкостная – применяются при определении летучих и нелетучих соединений соответственно.
В случае ГХ нелетучие соединения так или иначе теряются. Дериватизация (силилирование и другие реакции) может обеспечить их определение, хотя неполная дериватизация или испарение наиболее летучих производных могут привести к ЛО [16]. Вариант двумерной ГХ (ГХ-ГХ) приводит к более эффективному разделению смесей и решению некоторых важных аналитических задач качественного анализа II [27] – идентификации (характеризации, аутентификации и т.п.) самих анализируемых образцов [35, 36]. Традиционное сочетание ГХ-МС1 дополняется в последние годы комбинациями ГХ (ГХ-ГХ) с более сложными вариантами масс-спектрометрии: МСВР и МС2 [35, 36].
Для определения нелетучих соединений традиционно применяют обращенно-фазовую (ОФ) жидкостную хроматографию, в том числе в варианте, обеспечивающем лучшее разделение (ОФ-УЭЖХ). В последние годы набрала популярность гидрофильная хроматография (HILIC) демонстрирующая в случае очень полярных аналитов лучшие характеристики разделения по сравнению с ОФ-В/УЭЖХ (где эти аналиты не удерживаются колонкой) [17, 18]. Тем не менее в случае HILIC наблюдаются более существенные вариации времен удерживания и более частое присутствие матричных эффектов (типичное для ХМС подавление ионизации отдельных аналитов в масс-спектрометре) [18]. Это может приводить к ЛО вследствие плохого прогнозирования времен удерживания (порядка элюирования аналитов) и незначительных масс-спектрометрических сигналов соответственно.
МАСС-СПЕКТРОМЕТРИЯ
Масс-спектрометр – основной инструмент идентификации аналитов в НЦА [26, 27]. Определение “известных неизвестных” летучих соединений – сравнительно легко решаемая задача, обеспеченная сопоставлениями экспериментальных масс-спектров ЭИ с соответствующими справочными/библиотечными спектрами. Спектры этого вида достаточно хорошо воспроизводятся и зарегистрированы для подавляющего большинства распространенных летучих соединений (см. ниже).
В случае нелетучих соединений наибольшие возможности НЦА обеспечиваются масс-спектрометрией высокого разрешения и тандемной масс-спектрометрией, прежде всего в сочетании В/УЭЖХ-ИЭР-МС2ВР. В этом тандеме первый масс-спектрометр представляет собой квадрупольный масс-анализатор или ионную ловушку, второй – времяпролетный масс-анализатор или специальную ионную ловушку Orbitrap. Между двумя масс-анализаторами располагается та или иная ячейка столкновений ионов (ионов-предшественников) с газовой мишенью, формирующая фрагментные ионы, которые необходимы для идентификации аналитов.
Отметим, что наибольшее массовое разрешение – у приборов ионно-циклотронного резонанса [37], недоступных для подавляющего большинства химических лабораторий из-за своей очень высокой стоимости. Технику и методологию с использованием Orbitrap в последнее время могут называть “масс-спектрометрией высокого разрешения с определением точной массы” (high-resolution accurate-mass mass spectrometry) [38]. Применение масс-спектрометров с обычным “единичным” разрешением в какой-то степени также может приводить к успеху [39] при решении менее сложных задач НЦА. Различные аналитические методы основаны на комбинации разных вариантов хроматографии (ОФ или HILIC, ВЭЖХ или УЭЖХ) и масс-спектрометрии (различные масс-анализаторы, положительные или отрицательные ионы и др.)
Указанные приборы высокого разрешения обеспечивают точность измерения масс (значений m/z) ионов-предшественников на уровне ppm, ионов-фрагментов – несколько хуже. Ионы-предшественники отбираются во всей совокупности их пиков в МС1-спектрах (сбор данных, независимых от исходной информации, data-independent acquisition) или могут задаваться заранее для ожидаемых соединений (сбор данных, зависимых от исходной информации, data-dependent acquisition). Наложение некоторых массовых пиков ионов-предшественников может потребовать их деконволюции и разделения МС2-спектров [40]. Важно иметь в виду, что молекулы аналита образуют в процессе ИЭР не только протонированные молекулы основных изотопных форм, но и ионы-аддукты, включая катионированные молекулы, заряженные частицы других изотопных форм (“изотопологи”). Исключение последних ионов из рассмотрения (процедура “компонентизации” [8]) существенно упрощает обработку хромато-масс-спектрометрических данных. При идентификации чаще всего прибегают к сравнению экспериментальных спектров со справочными. Приборы, включающие обычные ионные ловушки, приводят к спектрам МС2, которые хуже сопоставимы со спектрами, полученными с применением других типов тандемных масс-спектрометров [27, 41].
Природа ложных результатов, которые могут быть следствием применения МС, отражена в табл. 3 (см. также [18]).
ИДЕНТИФИКАЦИЯ
Правильность/надежность идентификации определяется соответствием полученных данных определенным критериям. В случае ЦА эти критерии, основанные на хроматографических и масс-спектрометрических данных, хорошо очерчены [26, 27, 42]. В НЦА ситуация оказывается гораздо менее определенной.
Наиболее надежная идентификация с получением ПП достигается (а) совпадением хроматографических и массовых пиков при совместном анализе пробы, содержащей определяемое соединение, и соответствующего аналитического стандарта [26, 27, 42, 43]. На втором месте по надежности – (б) способ идентификации, заключающийся в сходстве экспериментальных и справочных масс-спектров и параметров хроматографического удерживания. Сходство может быть выражено его общим показателем (см. ниже) или степенью близости интенсивностей нескольких основных пиков одних и тех же (различающихся только в пределах погрешности) масс. Правильность идентификации летучих соединений по библиотекам справочных масс-спектров ЭИ составляет в общем случае ~80%; у нелетучих аналитов и масс-спектров ИЭР-МС2 ситуация более неопределенная, доля ПП варьируется в широких пределах [26, 27, 41].
Второй способ идентификации (б) может быть столь же надежным как первый вариант (а), но при выполнении ряда условий: значения указанных величин должны быть получены в сходных экспериментальных условиях (одинаковые типы и модели приборов, близкие режимы получения данных), и эти значения должны быть уникальны для идентифицируемого соединения. Другие способы идентификации (интерпретация данных, сравнение с предсказанными спектрами и хроматографическими характеристиками [27, 43, 44]) менее надежны, но, по-видимому, применимы для отбора кандидатов на идентификацию.
Если ряд таких “кандидатских” соединений имеет общие элементы структуры, то на этом этапе идентификацию называют групповой [26, 27]. Такую группу соединений позволяют выявить “молекулярные сети” – графы структурно сходных соединений, построенные по похожим масс-спектрам [45, 46].
В англоязычной научной литературе весьма популярен термин “уровень идентификации” (табл. 1). Его смысл в подробной трактовке одной из публикаций [3] и с нашими комментариями прояснен в табл. 4. Добавим, что высота уровня идентификации коррелирует с ее надежностью (долей ПП).
Таблица 4.
Уровни и детали идентификации методом (ВЭЖХ)-МС2ВР [3]
| Уровень | Надежность (confidence) | Результат идентификации | Наши комментарии |
|---|---|---|---|
| 1 |  |
Структура, подтвержденная аналитическим стандартом | Наиболее надежная идентификация индивидуального соединения, очень высокая вероятность ПП |
| 2 | Структура, подтвержденная библиотечным поиском и/или характеристичными ионами (diagnostic evidence), совпадающими параметрами удерживания (retention behavior), присутствием родственных веществ | Результат, по надежности сопоставимый с уровнем 1 при выполнении ряда условий (см. текст) | |
| 3 | Условный кандидат на идентификацию; входит в список предполагаемых аналитов. Установлены элементы структуры (substructure) и класс соединений. ООС начинается с этого уровня | В общем случае — это уровень неоднозначно идентифицированных соединений большинством методов МС | |
| 4 | Однозначное определение молекулярной формулы, недостаток структурной информации | Может считаться кандидатом на идентификацию при учете метаинформации (см. текст) | |
| 5 | Недостаток полученной информации, несовпадение со справочными данными или свойствами аналитического стандарта. Старт НЦА | Идентификация отсутствует |
Существуют независимые количественные показатели надежности [26, 27, 43]. К ним относятся α- и β-критерии принятия статистических гипотез при рассмотрении идентификации как процедуры их проверки. Весьма популярна концепция точек идентификации – совпадающих значений измеряемых масс-спектрометрических и хроматографических величин с учетом их различной значимости. Показатели сходства масс-спектров, такие как точечная функция или вероятность их совпадения, можно также рассматривать как частную меру надежности. Недавно предложена общая шкала надежности, учитывающая уровни идентификации, степень совпадения характеристик удерживания и число точек идентификации [47].
ИНФОРМАТИКА
Несколько видов информационных продуктов незаменимы при проведении НЦА.
Библиотеки масс-спектров – основной ресурс справочной информации. Наиболее крупные из них представлены в табл. 5. Сравнительно хорошо обстоит дело со спектрами ЭИ-МС1 летучих соединений: в библиотеках представлено большинство известных и наиболее важных соединений этого класса. Библиотеки спектров МС2 и тем более МС2-ВР, преимущественно относящихся к нелетучим аналитам, т.е. большинству биологически важных соединений, стали создаваться значительно позже. Масс-спектры многих соединений в библиотеках отсутствуют, и спектры МС2 в целом недостаточно хорошо воспроизводятся, зависят от типа тандемного масс-спектрометра и энергии столкновений, приводящих к распаду ионов-предшественников [26, 27, 41]. Необходимость пополнения и повышения качества библиотек тандемных масс-спектров широко осознается [41, 43].
Таблица 5.
Основные библиотеки масс-спектров низкомолекулярных соединений
| Название | Количество данных | Комментарии | |
|---|---|---|---|
| спектры | соединения | ||
| Wiley Registry 12th, ЭИ-МС1 [48]* | 817 290 | 668 435 | Летучие соединения |
| NIST 20, ЭИ-MС1 [49]** | 350 643 | 306 869 | Летучие соединения |
| NIST 20, MС2 [49]*** | 1 320 389 | 31 000 | |
| METLIN [50] | >500 000 | MС2, преобладают метаболиты | |
| MassBank of North America (MONA) [51] |
691 548 | 226 303 | Преобладают МС2 спектры биологически активных соединений. Присутствуют in silico масс-спектры |
| mzCloud [53] | 8 0651 35 | 19 109 | MСn, лекарственные соединения, метаболиты |
| The Global Natural Product Social Molecular Networking (GNPS) [45] | 221 083 | 18 163 | MС2, природные соединения. Данные 2016 г. |
| MassBank [52] | 86 576 | 16 537 | MC1, MCn, летучие и нелетучие соединения |
| Human Metabolome Database (HMDB) [54] –ИЭ-MС1 –MС2 |
1763 279 972 | 1220 1249 | Метаболиты, преобладают in silico масс-спектры. Данные 2018 г. |
Сводки параметров удерживания. База данных НИСТ включает 447285 значений газохроматографических индексов удерживания (ИУ) для 139693 соединений [49]. Для В/УЭЖХ концепция ИУ применима в меньшей степени, чем в ГХ, но соответствующие работы появляются, как и методы оценки значений этих величин [55].
Химические базы данных. Наиболее крупные БД, содержащие сведения о химических соединениях, указаны в табл. 6. Информация, содержащаяся в них и дополняющая экспериментальные данные ХМС, носит название априорной или метаинформации. Она полезна и даже необходима при отборе и ранжировании соединений – кандидатов на идентификацию.
Таблица 6.
Наиболее крупные химические базы данных
| Название | Массив соединений | Свободный доступ | Комментарии |
|---|---|---|---|
| Chemical Abstracts Service (CAS) [56] | 182 млн веществ, включая индивидуальные соединения* и смеси** и 68 млн биополимеров (пептиды, белки и др.) | – | Полная библиографическая информация по химии и смежным наукам и сопутствующие фактографические данные |
| PubChem [57] | 110 млн индивидуальных соединений*, 271 млн веществ*, ** | + | Биологически значимые соединения, свойства, биологическая активность, токсичность и др. Компиляция из 795 источников |
| ChemSpider [1, 58] |
≥ 80–90 млн структур (индивидуальных соединений*) | + | То же, более короткие справки о веществах, но с большим количеством опций поиска, в том числе по массам ионов. Компиляция из 279 источников |
При использовании этих массивов информации наиболее важны три обстоятельства. Во-первых, БД ChemSpider позволяет проводить поиск молекулярных формул по экспериментально установленным массам молекулярных ионов (МС1) или протонированных (катионированных) молекул (МС2). Во-вторых, эти БД дают возможность оценить популярность/распространенность химических соединений – по числу источников информации о них, по количеству такой метаинформации и др. [1, 2, 27]. При проверке идентификационных гипотез [26, 27] имеет смысл начинать с наиболее популярных соединений (при прочих равных условиях). В-третьих, имеющаяся в БД информация о получении и свойствах химических соединений, их присутствии в различных объектах также полезна при отборе кандидатов на идентификацию.
Предсказание масс-спектров и параметров удерживания. Существует несколько методов, алгоритмов и соответствующих программ предсказания масс-спектров (in silico масс-спектров). Они основаны на машинном обучении, эвристике (правилах фрагментации ионов), комбинаторике (перебору и оценке вероятностей появления различных комбинаций атомов исходного иона), квантово-химических расчетах и смешанных принципах [59]. В среднем результаты предсказания спектров оказываются умеренно правильными. Так, мы провели исследование возможностей различения структурных изомеров одним из методов машинного обучения. Доля ПП при сравнении выборок предсказанных и экспериментальных спектров составила ~50–60% [44], и это далеко не самый низкий результат в рассматриваемой области вычислений [59]. Тем не менее методы предсказания масс-спектров бурно развиваются, и можно ожидать повышения их эффективности. Даже сейчас эту методологию можно использовать в НЦА, (а) рассчитывая масс-спектры кандидатов на идентификацию, которые отобраны по массе ионов-предшественников и поискам в химических базах данных, и (б) отбрасывая наиболее непохожие in silico спектры.
Несмотря на существование большой коллекции экспериментальных ГХ-ИУ (см. выше), проводятся работы по предсказанию и этих показателей, хотя бы с целью проверки эффективности вычислительных методов. Применение методологии машинного обучения позволяет рассчитывать их значения с удовлетворительной точностью [60]. Подобные алгоритмы предсказания были использованы для расчёта ВЭЖХ-ИУ; полученные данные не имели существенного самостоятельного значения для идентификации, но все-таки улучшали ее результаты, основанные на предсказании масс-спектров [55].
В случае ЖХ чаще предсказывают не ИУ, а (относительные) времена удерживания (ВУ) [61–63]. Некоторые результаты предсказания оказываются удовлетворительными. Из прогнозов ВУ для 80 тыс. соединений, которые включены в БД METLIN (табл. 5), следует, что в 70% случаев соответствующие аналиты оказываются в числе трех наиболее вероятных кандидатов на идентификацию [62].
Различные программы. Программы обработки хромато-масс-спектрометрических данных незаменимы в анализе сложных образцов, который приводит к многочисленным хроматографическим пикам. Надлежащая практика НЦА подразумевает проведение в автоматическом режиме [4, 6, 8, 16, 40, 64, 65]:
• деконволюции хроматографических пиков с выделением сигналов отдельных компонентов и их масс-спектров,
• фильтрации массовых пиков с удалением фоновых и слабых пиков, выбросов,
• аннотации пиков в масс-спектрах: приписания пикам значений масс и даже формул соответствующих ионов, выведенных из точных масс (значений m/z) и изотопной картины,
• сравнения масс-спектров, а также характеристик удерживания с соответствующими справочными данными, оценивание их сходства,
• формирования домашних библиотек масс-спектров,
• взаимной подгонки разных хроматограмм по временам удерживания и/или массам ионов реперных соединений для сравнения между собой различных образцов.
Соответствующими программами обеспечено серийно выпускаемое оборудование, они поставляются и другими фирмами/организациями [16, 64].
Следует таже упомянуть алгоритмы и программы многомерного статистического анализа (хемометрии), позволяющего по результатам НЦА группировать и классифицировать изучаемые образцы, например образцы продовольствия [10, 12, 66].
* * *
Многочисленные инновации в аналитическом приборостроении и информатике сделали доступным одновременное определение многих десятков, сотен и даже тысяч органических (биоорганических) соединений, неизвестных аналитику до проведения эксперимента, в самых сложных матрицах (биологические и медицинские объекты, продукты питания, объекты окружающей среды и т.д.). Среди аналитических приборов первостепенное значение приобрели тандемные масс-спектрометры высокого разрешения, соединенные с хроматографами, в значительном количестве поступившие в аналитические лаборатории в последние 10–15 лет. Параллельно достигнут значительный прогресс в области информатики, который привел к появлению больших БД и новых программ обработки хромато-масс-спектрометрических данных. Результатом успехов приборостроения и информатики явился взрывной рост работ в области НЦА.
Следует выделить два аспекта этих публикаций. В их числе оказалось много соответствующих тематических обзоров, связанных с отдельными методами и/или объектами анализа. Совместное рассмотрение этих работ позволяет очертить общую практику НЦА, выявить его стандартные приемы, применение определенных методов пробоподготовки, анализа, извлечения и обработки информации. Вторая сторона обсуждаемых научных публикаций заключается в том, что они отражают новый, достаточно полный анализ самых разных объектов во всем их географическом, биологическом, природном, промышленном и т.п. многообразии. Такого рода исследования с использованием методологии НЦА продолжаются, ожидается появление новых фактических данных, касающихся неизвестного ранее состава веществ. Полученные в последние годы и ожидаемые новые данные, вероятно, также будут нуждаться в широком обобщении, которое будет интересно химикам многих специальностей.
Список литературы
Milman B.L., Zhurkovich I.K. Statistics of the popularity of chemical compounds in relation to the non-target analysis // Molecules. 2021. V. 26. № 8. P. 2394. https://doi.org/10.3390/molecules26082394
Мильман Б.Л., Журкович И.К. Популярность химических соединений. О чем это? // Аналитика. 2020. Т. 10. № 6. С. 464. https://doi.org/10.22184/2227-572X.2020.10.6.464. 469
Schymanski E.L., Singer H.P., Slobodnik J., Ipolyi I.M., Oswald P., Krauss M., Schulze T., Haglund P., Letzel T., Grosse S., Thomaidis N.S., Bletsou A., Zwiener C., Ibáñez M., Portolés T., De Boer R., Reid M.J., Onghena M., Kunkel U., Schulz W., Guillon A., Noyon N., Leroy G., Bados P., Bogialli S., Stipaničev D., Rostkowski P., Hollender J. Non-target screening with high-resolution mass spectrometry: Critical review using a collaborative trial on water analysis // Anal. Bioanal. Chem. 2015. V. 407. № 21. P. 6237. https://doi.org/10.1007/s00216-015-8681-7
Hollender J., Schymanski E.L., Singer H.P., Ferguson P.L. Nontarget screening with high resolution mass spectrometry in the environment: Ready to go? // Environ. Sci. Technol. 2017. V. 51. № 20. P. 11505. https://doi.org/10.1021/acs.est.7b02184
Ccanccapa-Cartagena A., Pico Y., Ortiz X., Reiner E.J. Suspect, non-target and target screening of emerging pollutants using data independent acquisition: Assessment of a Mediterranean River basin // Sci. Total Environ. 2019. V. 687. P. 355. https://doi.org/10.1016/j.scitotenv.2019.06.057
Ljoncheva M., Stepišnik T., Džeroski S., Kosjek T. Cheminformatics in MS-based environmental exposomics: Current achievements and future directions // Trends Environ. Anal. Chem. 2020. V. 28. Article e00099. https://doi.org/10.1016/j.teac.2020.e00099
Menger F., Gago-Ferrero P., Wiberg K., Ahrens L. Wide-scope screening of polar contaminants of concern in water: A critical review of liquid chromatography-high resolution mass spectrometry-based strategies // Trends Environ. Anal. Chem. 2020. V. 28. Article e00102. https://doi.org/10.1016/j.teac.2020.e00102
Schulze B., Jeon Y., Kaserzon S., Heffernan A.L., Dewapriya P., O’Brien J., Ramos M.J.G., Gorji S.G., Mueller J.F., Thomas K.V., Samanipour S. An assessment of quality assurance/quality control efforts in high resolution mass spectrometry non-target workflows for analysis of environmental samples // Trends Anal. Chem. 2020. V. 133. Article 116063. https://doi.org/ 10.1016/j.trac.2020.116063
Kutlucinar K.G., Hann S. Comparison of preconcentration methods for nontargeted analysis of natural waters using HPLC-HRMS: Large volume injection versus solid-phase extraction // Electrophoresis. 2021. V. 42. № 4. P. 490. https://doi.org/10.1002/elps.202000256
Riedl J., Esslinger S., Fauhl-Hassek C. Review of validation and reporting of non-targeted fingerprinting approaches for food authentication // Anal. Chim. Acta. 2015. V. 885. P. 17. https://doi.org/10.1002/elps.202000256.org/10.1016/ j.aca.2015.06.003
Knolhoff A.M., Croley T.R. Non-targeted screening approaches for contaminants and adulterants in food using liquid chromatography hyphenated to high resolution mass spectrometry // J. Chromatogr. A. 2016. V. 1428. P. 86. https://doi.org/10.1016/j.chroma.2015.08.059
Fisher C.M., Croley T.R., Knolhoff A.M. Data processing strategies for non-targeted analysis of foods using liquid chromatography/high-resolution mass spectrometry // Trends Anal. Chem. 2021. V. 136. Article 116188. https://doi.org/10.1016/j.trac.2021.116188
Chen C., Wohlfarth A., Xu H., Su D., Wang X., Jiang H., Feng Y., Zhu M. Untargeted screening of unknown xenobiotics and potential toxins in plasma of poisoned patients using high-resolution mass spectrometry: Generation of xenobiotic fingerprint using background subtraction // Anal. Chim. Acta. 2016. V. 944. P. 37. https://doi.org/10.1016/j.aca.2016.09.034
Oberacher H., Arnhard K. Current status of non-targeted liquid chromatography-tandem mass spectrometry in forensic toxicology // Trends Anal. Chem. 2016. V. 84. P. 94. https://doi.org/10.1016/j.trac.2015.12.019
Mollerup C.B., Dalsgaard P.W., Mardal M., Linnet K. Targeted and non-targeted drug screening in whole blood by UHPLC-TOF-MS with data-independent acquisition // Drug Test. Anal. 2017. V. 9. № 7. P. 1052. https://doi.org/10.1002/dta.2120
Mastrangelo A., Ferrarini A., Rey-Stolle F., Garcia A., Barbas C. From sample treatment to biomarker discovery: A tutorial for untargeted metabolomics based on GC-(EI)-Q-MS // Anal. Chim. Acta. 2015. V. 900. P. 21. https://doi.org/10.1016/j.aca.2015.10.001
Cajka T., Fiehn O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics // Anal. Chem. 2016. V. 88. № 1. P. 524. https://doi.org/10.1021/acs.analchem.5b04491
Pezzatti J., Boccard J., Codesido S., Gagnebin Y., Joshi A., Picard D., Gonzalez-Ruiz V., Rudaz S. Implementation of liquid chromatography–high resolution mass spectrometry methods for untargeted metabolomic analyses of biological samples: A tutorial // Anal. Chim. Acta. 2020. V. 1105. P. 28. https://doi.org/10.1016/ j.aca.2019.12.062
Hubert J., Nuzillard J.M., Renault J.H. Dereplication strategies in natural product research: how many tools and methodologies behind the same concept? // Phytochem. Rev. 2017. V. 16. № 1. P. 55. https://doi.org/10.1007/s11101-015-9448-7
Aydoğan C. Recent advances and applications in LC-HRMS for food and plant natural products: A critical review // Anal. Bioanal. Chem. 2020. V. 412. № 9. P. 1973. https://doi.org/10.1007/s00216-019-02328-6
Caballero-Casero N., Belova L., Vervliet P., Antignac J.P., Castaño A., Debrauwer L., López M.E., Huber C., Klanova J., Krauss M., Lommen A., Mol H.G.J., Oberacher H., Pardo O., Price E.J., Reinstadler V., Vitale C.M., Van Nuijs A.L.N., Covaci A. Towards harmonized criteria in quality assurance and quality control of suspect and non-target LC-HRMS analytical workflows for screening of emerging contaminants in human biomonitoring // Trends Anal. Chem. 2021. V. 136. Article 116201. https://doi.org/10.1016/j.trac.2021.116201
Mairinger T., Causon T.J., Hann S. The potential of ion mobility–mass spectrometry for non-targeted metabolomics // Curr. Opin. Chem. Biol. 2018. V. 42. P. 9. https://doi.org/10.1016/j.cbpa.2017.10.015
Lorenc W., Hanć A., Sajnóg A., Barałkiewicz D. LC/ICP-MS and complementary techniques in bespoke and nontargeted speciation analysis of elements in food samples // Mass Spectrom. Rev. 2020. P. 1. https://doi.org/10.1002/mas.21662
Sobus J.R., Grossman J.N., Chao A., Singh R., Williams A.J., Grulke C.M., Richard A.M., Newton S.R., McEachran A.D., Ulrich E.M. Using prepared mixtures of ToxCast chemicals to evaluate non-targeted analysis (NTA) method performance // Anal. Bioanal. Chem. 2019. V. 411. № 4. P. 835. https://doi.org/10.1007/s00216-018-1526-4
Knolhoff A.M., Premo J.H., Fisher C.M. A Proposed quality control standard mixture and its uses for evaluating nontargeted and suspect screening LC/HR-MS method performance // Anal. Chem. 2021. V. 93. № 3. P. 1596. https://doi.org/10.1021/acs.analchem.0c04036
Мильман Б.Л. Введение в химическую идентификацию. СПб: ВВМ, 2008. 180 с.
Milman B.L. Chemical Identification and Its Quality Assurance. Berlin: Springer, 2011. 281 p.
Мильман Б.Л., Журкович И.К. Большие данные в современном химическом анализе // Журн. анал. хим. 2020. Т. 75. №. 4. С. 316. https://doi.org/ 10.31857/S0044450220020139 (Milman B.L., Zhurkovich I.K. Big data in modern chemical analysis // J. Anal. Chem. 2020. V. 75. № 4. P. 443. https://doi.org/ 10.1134/S1061934820020124)
NORMAN Database System. https://www.norman-network.com/nds (05.06.2021).
Stevens V.L., Hoover E., Wang Y., Zanetti K.A. Pre-analytical factors that affect metabolite stability in human urine, plasma, and serum: A review // Metabolites. 2019. V. 9. № 8. P. 156. https://doi.org/10.3390/metabo9080156
García A., Godzien J., López-Gonzálvez Á., Barbas C. Capillary electrophoresis mass spectrometry as a tool for untargeted metabolomics // Bioanalysis. 2017. V. 9. № 1. P. 99. https://doi.org/10.4155/bio-2016-0216
Mairinger T., Causon T. J., Hann S. The potential of ion mobility–mass spectrometry for non-targeted metabolomics // Curr. Opin. Chem. Biol. 2018. V. 42. P. 9. https://doi.org/10.1016/j.cbpa.2017.10.015
Geng C., Guo Y., Qiao Y., Zhang J., Chen D., Han W., Yang M., Jiang P. UPLC-Q-TOF-MS profiling of the hippocampus reveals metabolite biomarkers for the impact of Dl-3-n-butylphthalide on the lipopolysaccharide-induced rat model of depression // Neuropsychiatr. Dis. Treat. 2019. V. 15. P. 1939. https://doi.org/10.2147/NDT.S203870
Koek M.M., Jellema R.H., Van der Greef J., Tas A.C., Hankemeier T. Quantitative metabolomics based on gas chromatography mass spectrometry: Status and perspectives // Metabolomics. 2011. V. 7. № 3. P. 307. https://doi.org/10.1007/s11306-010-0254-3
Aspromonte J., Wolfs K., Adams E. Current application and potential use of GC × GC in the pharmaceutical and biomedical field // J. Pharm. Biomed. Anal. 2019. V. 176. Article 112817. https://doi.org/10.1016/j.jpba. 2019.112817
Franchina F.A., Zanella D., Dubois L.M., Focant J.F. The role of sample preparation in multidimensional gas chromatographic separations for non-targeted analysis with the focus on recent biomedical, food, and plant applications // J. Sep. Sci. 2021. V. 44. № 1. P. 188. https://doi.org/10.1002/jssc.202000855
Ghaste M., Mistrik R., Shulaev V. Applications of Fourier transform ion cyclotron resonance (FT-ICR) and orbitrap based high resolution mass spectrometry in metabolomics and lipidomics // Int. J. Mol. Sci. 2016. V. 17. № 6. P. 816. https://doi.org/10.3390/ijms17060816
Strupat K., Scheibner O., Bromirski M. High-resolution, accurate-mass orbitrap mass spectrometry–definitions, opportunities, and advantages // Thermo Technical Note. 2013. № 64287. P. 1. https://assets.thermofisher.com/TFS-Assets/CMD/Technical-Notes/ tn-64287-hram-orbitrap-ms-tn64287-en.pdf (06.06. 2021).
Alon T., Amirav A. Comparison of isotope abundance analysis and accurate mass analysis in their ability to provide elemental formula information // J. Am. Soc. Mass Spectrom. // 2021. V. 32. № 4. P. 929. https://doi.org/10.1021/jasms.0c00419
Samanipour S., Reid M.J., Bæk K., Thomas K.V. Combining a deconvolution and a universal library search algorithm for the nontarget analysis of data-independent acquisition mode liquid chromatography–high-resolution mass spectrometry results // Environ. Sci. Technol. 2018. V. 52. № 8. P. 4694. https://doi.org/10.1021/acs.est.8b00259
Oberacher H., Sasse M., Antignac J.P., Guitton Y., Debrauwer L., Jamin E.L., Schulze T., Krauss M., Covaci A., Caballero-Casero N., Rousseau K., Damont A., Fenaille F., Lamoree M., Schymanski E.L. A European proposal for quality control and quality assurance of tandem mass spectral libraries // Environ. Sci. Eur. 2020. V. 32. № 1. P. 1. https://doi.org/10.1186/s12302-020-00314-9
Мильман Б.Л., Журкович И.К. Обобщенные критерии идентификации химических соединений методами хроматографии–масс-спектрометрии // Аналитика и контроль. 2020. Т. 24. №. 3. С. 164. https://doi.org/10.15826/analitika.2020.24.3.003
Milman B.L. General principles of identification by mass spectrometry // Trends Anal. Chem. 2015. V. 69. P. 24. https://doi.org/10.1016/j.trac.2014.12.009
Milman B.L., Ostrovidova E.V., Zhurkovich I.K. Isomer differentiation using in silico MS2 spectra. A case study for the CFM-ID mass spectrum predictor // Mass Spectrom. Lett. 2019. V. 10. № 3. P. 93. https://doi.org/10.5478/MSL.2019.10.3.93
Global Natural Products Social Molecular Networking. https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp (06.06.2021).
Vincenti F., Montesano C., Di Ottavio F., Gregori A., Compagnone D., Sergi M., Dorrestein P. Molecular networking: A useful tool for the identification of new psychoactive substances in seizures by LC–HRMS // Front. Chem. 2020. V. 8. Article 572952. https://doi.org/10.3389/fchem.2020.572952
Rochat B. Proposed confidence scale and ID score in the identification of known-unknown compounds using high resolution MS data // J. Am. Soc. Mass Spectrom. 2017. V. 28. № 4. P. 709. https://doi.org/10.1007/s13361-016-1556-0
Wiley Registry of Mass Spectral Data, 12th Ed. https://www.sisweb.com/software/wiley-registry. htm#1 (07.06.2021).
The NIST 20 Mass spectral library. https:// www.sisweb.com/software/ms/nist.htm#stats (07. 06.2021).
METLIN. https://metlin.scripps.edu/landing_page.php?pgcontent=mainPage (07.06.2021).
MONA – MassBank of North America. https://mona.fiehnlab.ucdavis.edu/spectra/statistics?tab=0 (07. 06.2021).
MassBank. https://massbank.eu/MassBank/Contents (07.06.2021).
mzCloud. https://www.mzcloud.org (07.06.2021).
The human metabolome database (HMDB). https://hmdb.ca (07.06.2021).
Samaraweera M.A., Hall L.M., Hill D.W., Grant D.F. Evaluation of an artificial neural network retention index model for chemical structure identification in nontargeted metabolomics // Anal. Chem. 2018. V. 90. № 21. P. 12752. https://doi.org/10.1021/acs.analchem.8b03118
CAS. https://www.cas.org/about/cas-content (07.06. 2021).
PubChem. https://pubchem.ncbi.nlm.nih.gov (07.06. 2021).
ChemSpider. http://www.chemspider.com (07.06. 2021).
Krettler C.A., Thallinger G.G. A map of mass spectrometry-based in silico fragmentation prediction and compound identification in metabolomics // Briefings Bioinf. 2021. P. 1. https://doi.org/10.1093/bib/bbab073
Matyushin D.D., Buryak A.K. Gas chromatographic retention index prediction using multimodal machine learning // IEEE Access. 2020. V. 8. P. 223140. https://doi.org/10.1109/ACCESS.2020.3045047
McEachran A.D., Mansouri K., Newton S.R., Beverly B.E., Sobus J.R., Williams A.J. A comparison of three liquid chromatography (LC) retention time prediction models // Talanta. 2018. V. 182. P. 371. https://doi.org/ 10.1016/j.talanta.2018.01.022
Domingo-Almenara X., Guijas C., Billings E., Montenegro-Burke J.R., Uritboonthai W., Aisporna A.E., Chen E., Benton H.P., Siuzdak G. The METLIN small molecule dataset for machine learning-based retention time prediction // Nat. Commun. 2019. V. 10. № 1. P. 1. https://doi.org/10.1038/s41467-019-13680-7
Witting M., Böcker S. Current status of retention time prediction in metabolite identification // J. Sep. Sci. 2020. V. 43. № 9–10. P. 1746. https://doi.org/ 10.1002/jssc.202000060
Kind T., Tsugawa H., Cajka T., Ma Y., Lai Z., Mehta S.S., Wohlgemuth G., Barupal D.K., Showalter M.R., Arita M., Fiehn O. Identification of small molecules using accurate mass MS/MS search // Mass Spectrom. Rev. 2018. V. 37. № 4. P. 513. https://doi.org/ 10.1002/mas.21535
Helmus R., Ter Laak T.L., Van Wezel A.P., De Voogt P., Schymanski E.L. patRoon: Open source software platform for environmental mass spectrometry based non-target screening // J. Cheminf. 2021. V. 13. № 1. P. 1. https://doi.org/10.1186/s13321-020-00477-w
Cavanna D., Righetti L., Elliott C., Suman M. The scientific challenges in moving from targeted to non-targeted mass spectrometric methods for food fraud analysis: A proposed validation workflow to bring about a harmonized approach // Trends Food Sci. Technol. 2018. V. 80. P. 223. https://doi.org/10.1016/j.tifs.2018.08.007
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии