

Журнал аналитической химии, 2022, T. 77, № 6, стр. 540-549
Спектрофотометрическое определение аминосодержащих лекарственных препаратов в мицеллярных матрицах при использовании планов бокса–бенкена
Р. Ф. Бакеева a, С. Ю. Гармонов a, *, О. Е. Вахитова a, В. Ф. Сопин a
a Казанский национальный исследовательский технологический университет
420015 Казань, ул. К. Маркса, 68, Россия
* E-mail: serggar@mail.ru
Поступила в редакцию 06.06.2021
После доработки 06.08.2021
Принята к публикации 06.08.2021
- EDN: KOANUR
- DOI: 10.31857/S0044450222060032
Аннотация
Представлены результаты использования методологии “поверхности отклика” и дизайна Бокса–Бенкена при спектрофотометрическом определении лекарственных веществ на примере дапсона и прокаина в мицеллярной матрице. В качестве реагента использовали 5,7-дихлор-4,6-динитробензофуроксан. Дизайн матрицы состава неонол–вода (80%)–диметилсульфоксид (20%) варьировали за счет факторов: концентрация реагента, рН, концентрация неонола. Оптимизацию проводили с использованием трехуровневого плана Бокса–Бенкена. Показано существенное увеличение интенсивности полосы поглощения продуктов реакции в мицеллярной псевдофазе. Найдены оптимальные условия, при которых возможно спектрофотометрическое определение лекарственных веществ в жидких и твердых лекарственных формах.
Прокаин (2-(диэтиламино)этил-4-аминобензоат) и дапсон (4,4'-сульфонилбис[бензоламин]) относятся к лекарственным веществам (ЛВ), содержащим в своем составе функциональные первичные аминогруппы. Прокаин применяют в медицине как эффективное местноанестизирующее средство, а дапсон обладает оригинальным антибактериальным и противолепрозным действием [1, 2].
При фармацевтическом анализе широко используют спектрофотометрию, что обусловлено ее проработанной методической базой, доступностью аппаратуры и универсальностью по отношению ко многим объектам фармации. Однако в случае производных ароматических аминов спектрофотометрическое определение часто ограничивается проблемой избирательности и чувствительности анализа, что приводит к необходимости применения селективных реагентов, как правило, в органической или смешанной среде. Все это значительно усложняет процедуру анализа, требует обеспечения устойчивости спектрохимических реагентов и образующихся производных, повышает стоимость анализа и его опасность за счет использования токсичных органических растворителей [3, 4].
Один из возможных путей решения этой проблемы – использование в фармацевтическом анализе организованных сред, в частности, содержащих мицеллярные системы. При этом в мицеллах поверхностно-активных веществ (ПАВ) увеличивается растворимость гидрофобных и гидрофильных реагентов, повышается агрегативная устойчивость фотометрируемых систем, изменяется скорость реакций реагентов с ЛВ. На практике чаще всего эти эффекты проявляются одновременно. Реализация такого подхода позволяет более широко использовать спектрофотометрию в водных средах в фармацевтическом анализе. Так, в обзоре [5] описано спектрофотометрическое определение ЛВ различных фармакологических групп на основе реакций получения их окрашенных производных (карбонильные соединения, диазотирование с последующим азосочетанием, образование хинониминов и др.) в организованных средах. Следует отметить, что из-за сложного состава анализируемой матрицы эти приемы могут быть малоизбирательными, не всегда обеспечивать достаточную чувствительность при анализе биологических жидкостей, а также быть многостадийными и длительными.
Формирование мицеллярных систем – сложный процесс, зависящий от большого количества независимых переменных. Если мицеллярные среды используются как матрица для проведения аналитических определений, то возможна ее оптимизация с помощью дизайна Бокса–Бенкена. Аналитическому применению этого подхода посвящена обзорная статья [6]. Дизайн Бокса–Бенкена успешно зарекомендовал себя как инструмент многофакторного анализа при оптимизации ионной ловушки масс-спектрометра при определении 15 поли- и перфторалкильных веществ [7], при определении пропранолола методом капиллярного электрофореза в сочетании с индуцированной лазером флуоресценцией в биологических жидкостях [8], при экстракции флавоноидов и определении целевых аналитов в пробах оливкового, миндального, кунжутного и коричного масел [9, 10], а также при оптимизации ряда спектроаналитических методов [11–13].
Ранее нами для спектрофотометрического определения ароматических аминов в мицеллярной среде использован 5,7-дихлор-4,6-динитробензофуроксан [14], который обеспечил высокую селективность и чувствительность анализа при контроле качества токсичной примеси 4-аминофенола в лекарственных препаратах на основе парацетамола.
В настоящей работе мицеллярные системы использовали при спектрофотометрическом определении прокаина и дапсона с реагентом 5,7-дихлор-4,6-динитробензофуроксаном для получения окрашенных производных в водных и водно-органических средах. Приведены результаты применения методологии поверхности отклика с дизайном Бокса–Бенкена для определения прокаина и дапсона при использовании в качестве матрицы растворов мицелл. Применение мицеллярных систем на основе оксиэтилированного нонилфенола позволяет избирательно и с высокой чувствительностью определять аналиты в лекарственных формах в виде таблеток и растворов для инъекций.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и аппаратура. 5,7-Дихлор-4,6-динитробензофуроксан был синтезирован по известной методике [15] и любезно предоставлен проф. Л.М. Юсуповой. Использовали субстанции прокаина и дапсона фармакопейной чистоты. Для создания мицеллярных матриц использовали неионное ПАВ – оксиэтилированный нонилфенол (APh 9-10, неонол) (Fluka Chemie AG, Швейцария). Для определения дапсона и прокаина применяли мицеллярную матрицу APh 9-10 в водном растворе диметилсульфоксида (ДМСО, 20 об. %) до и после формирования мицелл. Критическая концентрация мицеллобразования (ККМ) в системе APh 9-10 + Н2О (80%) + ДМСО (20%) составила 2.51 × 10–4 М. Остальные реактивы были марки х.ч. Применяли лекарственные формы прокаина и дапсона коммерческого изготовления.
Тензиометрические измерения проводили на тензиометре Дю-Нуи, оснащенном платиновым кольцом, в соответствии с ГОСТ 29232–91 (ИСО 4311–79).
Значения рН определяли на рН-метре (рН 150 М).
Для спектрофотометрических измерений применяли спектрофотометр Agilent 8453 в спектральном диапазоне от 400 до 700 нм. При измерении оптической плотности растворов использовали кварцевые кюветы с толщиной поглощающего слоя 0.1, 0.50, 1.00 см. Эксперименты проводили при 25°С.
Статистическую обработку результатов оптимального эксперимента проводили для пяти измерений при доверительной вероятности 0.95. Результаты представляли как X ± ΔX. где X – среднее значение и ΔX – доверительный интервал. Случайную погрешность определения оценивали по величине относительного стандартного отклонения (sr). Для расчета эффектов и построения поверхностей отклика использовали пакет программ Statistica 10.
Методика определения. Жидкие лекарственные формы (инъекционные растворы) разбавляют водой до содержания лекарственных веществ в интервале 0.01–0.05 мг/мл, далее аликвотную часть раствора помещают в мерную колбу емк. 50 мл, добавляют водный раствор ПАВ для создания концентрации АPh 9-10 5.3–10 мг/мл, 2 мл 0.05 М боратного буферного раствора (рН 8.76 для прокаина и 11 для дапсона), ацетонитрильный раствор 5,7-дихлор-4,6-динитробензофуроксана до концентрации (4.6–6.8) × 10–4 М и доводят смесью ДМСО–вода (20 : 80, по объему) до метки. Оптическую плотность раствора измеряют при длине волны 510 нм (дапсон) и 500 нм (прокаин) в кювете с длиной поглощающего слоя 1 см. Содержание лекарственных веществ находят по градуировочной зависимости или методом добавок.
При анализе таблеток их растирают до порошка, отбирают навеску 0.5 г, прибавляют 20 мл ДМСО и встряхивают 10 мин. Повторяют извлечение. Раствор переносят в мерную колбу емк. 50 мл, доводят до метки ДМСО и выполняют определение как описано выше.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В качестве аналитической реакции использовали нуклеофильное замещение аминогрупп при взаимодействии дапсона (ДПС) и прокаина (ПК) с 5,7-дихлор-4,6-динитробензофуроксаном (ДХДНБФ) в мицеллярной системе APh 9-10 + + Н2О (80%) + ДМСО (20%), в результате которой появляются продукты взаимодействия с полосами поглощения (ПП) при 510 и 500 нм соответственно. Интенсивности ПП А510 и А500 выбрали как целевые функции для количественного определения ДПС и ПК.
Особенностью систем, содержащих ПАВ, является формирование мицелл с определенной критической концентрацией мицеллообразования. Влияние мицеллярных агрегатов можно проследить, если исследовать действие системы до и после формирования мицелл. Формирование мицелл происходит по достижении ККМ, значение которой определяли из тензиометрической кривой по точке изменения наклона ветвей кривых зависимости σ = f(cAPh 9-10) (рис. 1). Самопроизвольное образование мицелл в системе APh 9-10 + Н2О (80%) + ДМСО (20%) наблюдается при ККМ 2.51 × 10–4 М APh 9-10. Присутствие ДМСО приводит к увеличению ККМ (в системе APh 9-10 + Н2О ККМ составляет 1.02 × 10–4 М). Это отмечалось и в работах [16, 17], поэтому следует учитывать влияние природы компонентов при дизайне мицеллярных матриц.
Рис. 1.
Тензиометрическая кривая для системы APh 9-10 + Н2О (80%) + ДМСО (20%), 25°С, ККМ 2.51 × × 10–4 М.

Значимыми независимыми факторами являлись: концентрация аналитического реагента с(ДХДНБФ), мМ; величина рН; с(APh 9-10), М, поскольку в результате однофакторных экспериментов показано их влияние на аналитический сигнал.
Для трех независимых факторов число испытаний N, в соответствии с данными [6], равно N = = 2K(K-1) + CP, где K – число независимых факторов, СР – центральные точки для оценки экспериментальной погрешности. В нашем случае СР = 3. Общее число опытов было равно 15.
Дальнейшая оптимизация включала планирование и выполнение факторного эксперимента; построение математической модели (y = f(Xi)); изучение влияния факторов (Xi) на целевую функцию (Y = А510 или А500) путем построения поверхности отклика; анализ поверхности отклика с целью поиска точки оптимума; выявление оптимальных условий.
В табл. 1 и 2 приведены кодированные и декодированные значения факторов и отклика (А510 и А500), которые использовали в дизайне Бокса–Бенкена. Диаграмма Парето основных эффектов показана на рис. 2. Для описания математической модели выбрали значения с уровнем значимости р < 0.05, которое соответствует доверительной вероятности 0.95. Адекватность моделей подтверждается коэффициентами детерминации (R2 = = 0.9598 для ДПС и R2 = 0.9903 для ПК), характеризующими меру отклонения среднего в модели. Таким образом, модель может описать 95.98% влияния на отклик для ДПС и 99.03% для ПК.
Таблица 1.
Факторы, действительные и кодированные уровни и матрица для дизайна Бокса–Бенкена при определении дапсона в системе APh 9-10 + Н2О (80%) + ДМСО (20%)
| Фактор | Уровень | ||||||
|---|---|---|---|---|---|---|---|
| нижний –1 |
центральный 0 |
верхний +1 |
|||||
| Х1 | с(ДХДНБФ), мМ | 0.100 | 0.400 | 0.700 | |||
| Х2 | pHрс(APh 9-10) | 2.00 | 6.00 | 10.00 | |||
| Х3 | 4.601 | 3.602 | 2.603 | ||||
| № опыта |
Реальные (декодированные) уровни факторов | Кодированные уровни факторов | Отклик А510 |
||||
| ${{X}_{{{{1}^{\centerdot }}}}}$ | Х2 | ${{X}_{{{{3}^{\centerdot }}}}}$ | $X_{1}^{*}$ | $X_{2}^{*}$ | $X_{3}^{*}$ | $Y_{{{\text{ср}}}}^{4}$ | |
| 1 | 0.700 | 10.00 | 3.60 | +1 | +1 | 0 | 0.618 |
| 2 | 0.700 | 2.00 | 3.60 | +1 | –1 | 0 | 0.625 |
| 3 | 0.100 | 10.00 | 3.60 | –1 | +1 | 0 | 0.261 |
| 4 | 0.100 | 2.00 | 3.60 | –1 | –1 | 0 | 0.242 |
| 5 | 0.700 | 6.00 | 2.60 | +1 | 0 | +1 | 0.949 |
| 6 | 0.700 | 6.00 | 4.60 | +1 | 0 | –1 | 0.053 |
| 7 | 0.100 | 6.00 | 2.60 | –1 | 0 | +1 | 0.377 |
| 8 | 0.100 | 6.00 | 4.60 | –1 | 0 | –1 | 0.027 |
| 9 | 0.400 | 10.00 | 2.60 | 0 | +1 | +1 | 1.131 |
| 10 | 0.400 | 10.00 | 4.60 | 0 | +1 | –1 | 0.039 |
| 11 | 0.400 | 2.00 | 2.60 | 0 | –1 | +1 | 0.385 |
| 12 | 0.400 | 2.00 | 4.60 | 0 | –1 | –1 | 0.040 |
| 13 | 0.400 | 6.00 | 3.60 | 0 | 0 | 0 | 0.779 |
| 14 | 0.400 | 6.00 | 3.60 | 0 | 0 | 0 | 0.771 |
| 15 | 0.400 | 6.00 | 3.60 | 0 | 0 | 0 | 0.782 |
| 0.682∙ | 11.04 | 1.51 | 0.94 | 1.26 | 1.51 | 1.158 ± 0.004 | |
Таблица 2.
Факторы, действительные и кодированные уровни и матрица для дизайна в Бокса–Бенкена для определения прокаина в системе APh 9-10 + Н2О (80%) + ДМСО (20%)
| Фактор | Уровень | ||||||
|---|---|---|---|---|---|---|---|
| нижний –1 |
центральный 0 |
верхний +1 |
|||||
| Х1 | с(DCDNBP), мМ | 0.050 | 0.200 | 0.350 | |||
| Х2 | pH | 2.00 | 6.00 | 10.00 | |||
| Х3 | рс(APh 9-10) | 4.60 | 3.60 | 2.60 | |||
| № опыта | Реальные (декодированные) уровни факторов | Кодированные уровни факторов | Отклик А500 | ||||
| ${{X}_{{{{1}^{\centerdot }}}}}$ | Х2 | ${{X}_{{{{3}^{\centerdot }}}}}$ | $X_{1}^{*}$ | $X_{2}^{*}$ | $X_{3}^{*}$ | Ycp | |
| 1 | 0.350 | 10.00 | 2.60 | +1 | +1 | 0 | 0.351 |
| 2 | 0.350 | 2.00 | 2.60 | +1 | –1 | 0 | 0.203 |
| 3 | 0.050 | 10.00 | 2.60 | –1 | +1 | 0 | 0.101 |
| 4 | 0.050 | 2.00 | 2.60 | –1 | –1 | 0 | 0.052 |
| 5 | 0.350 | 6.00 | 2.60 | +1 | 0 | +1 | 0.570 |
| 6 | 0.350 | 6.00 | 4.60 | +1 | 0 | –1 | 0.100 |
| 7 | 0.050 | 6.00 | 2.60 | –1 | 0 | +1 | 0.164 |
| 8 | 0.050 | 6.00 | 4.60 | –1 | 0 | –1 | 0.041 |
| 9 | 0.200 | 10.00 | 2.60 | 0 | +1 | +1 | 0.382 |
| 10 | 0.200 | 10.00 | 4.60 | 0 | +1 | –1 | 0.071 |
| 11 | 0.200 | 2.00 | 2.60 | 0 | –1 | +1 | 0.174 |
| 12 | 0.200 | 2.00 | 0.25 | 0 | –1 | –1 | 0.052 |
| 13 | 0.200 | 6.00 | 3.60 | 0 | 0 | 0 | 0.389 |
| 14 | 0.200 | 6.00 | 3.60 | 0 | 0 | 0 | 0.386 |
| 15 | 0.200 | 6.00 | 3.60 | 0 | 0 | 0 | 0.387 |
| 0.459 ∙ | 8.76 | 1.830 | 1.7253 | 0.6906 | 1.7687 | 0.611 ± 0.019 | |
Рис. 2.
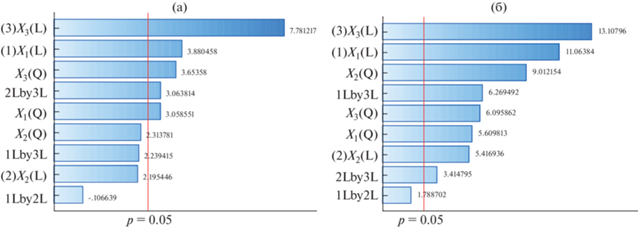
Диаграмма Парето стандартизированных эффектов на отклик. (а) – интенсивность полосы поглощения А510 для дапсона; (б) – интенсивность полосы поглощения А500 для прокаина в мицеллярной матрице APh 9-10 + Н2О (80%) + ДМСО (20%). L – линейные, Q – квадратичные эффекты, X1 – c(ДХДНБФ), мМ; X2 – рН; X3 – pc(APh 9-10).
Уровни факторов Х1с(DCDNBP), Х2 рН, Х3с(APh 9-10) кодировали в соответствии с рекомендациями [6] по формуле $X_{i}^{*} = \frac{{{{X}_{i}} - M}}{H},$ где $X_{i}^{*}$ – кодированный уровень i-го фактора, Хi – декодированный (реальный) уровень i-го фактора, М – среднее значение, Н – полуширина интервала.
Декодирование уровней факторов выполняли по формуле Хi = $X_{i}^{*}H$ + M.
Полученное нами соотношение между откликом (А510) и тремя значимыми факторами для ДПС является полиномом второй степени и выглядит следующим образом:
На рис. 3–5 представлены 3D графики поверхности отклика А510 в зависимости от значимых факторов при определении ДПС в мицеллярной матрице APh 9-10 + Н2О (80%) + ДМСО (20%). Можно отметить, что во всех случаях форма поверхности отклика неплоская, что свидетельствует о наличии эффектов взаимодействия между факторами. Из рис. 3 следует, что интенсивность ПП А510 существенно возрастает при формировании мицеллярной матрицы. Это можно объяснить солюбилизацией продукта реакции с ДПС за счет концентрирования реагента на или в мицелле, что использовали для определения ДПС.
Рис. 3.
Поверхность отклика зависимости А510 от концентрации реагента ДХДНБФ (X1) и pH (X2). (a) – при pc(APh 9‑10) $\left( {X_{{\text{3}}}^{*}} \right)$ = –1, до ККМ; (б) – при $X_{{\text{3}}}^{*}$ = +1, после ККМ.

Рис. 4.
Поверхность отклика зависимости А510 от концентрации реагента ДХДНБФ (X1) и pc(APh 9-10) (X3). (а) – при $X_{{\text{2}}}^{*}$ = –1, pH 2.00; (б)– при $X_{{\text{2}}}^{*}$ = 1, pH 10.00.

Рис. 5.
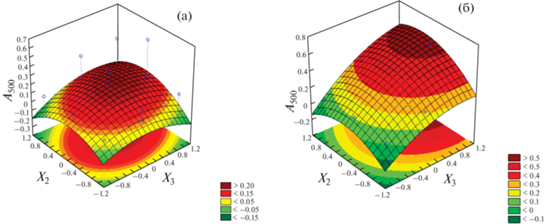
Поверхность отклика зависимости А510 от pc(APh 9-10) (Х3) и рН (Х2). (а) – при $X_{{\text{1}}}^{*}$ = –1, с(ДХДНБФ) = = 0.100 мМ, (б) – при $X_{{\text{1}}}^{*}$ = 1, с(ДХДНБФ) = 0.700 мМ.

Диаграмма Парето, представленная на рис. 2, показывает значимость каждого из факторов и свидетельствует о взаимодействиях этих факторов. Рассматривали те эффекты, для которых уровень значимости р < 0.05, (что соответствует доверительной вероятности 0.95). Наиболее существенный вклад в отклик при определении ДПС вносят концентрация неионного ПАВ APh 9-10, концентрация реагента ДХДНБФ, величина же рН влияет опосредованно. Максимально возможная величина А510 найдена нами с помощью функции “Critical values” и равна 1.1685. При этом состав мицеллярной матрицы в кодированных значениях следующий: Х1 0.9403; Х2 1.2609; Х3 1.5083. Декодированные значения составляют: с(DCDNBP) = 6.820 × 10–4 М, pH 11.04 и с(АPh 9-10) = = 8.097 × 10–3 М.
Для проверки модели провели пять параллельных экспериментов по определению значения отклика А510 в оптимальных условиях. Полученное значение (1.158 ± 0.004) вполне согласуется с расчетным. В этих условиях величина ελmax составляет 23200 л/(моль ⋅ см), что позволяет говорить о значительном увеличении чувствительности спектрофотометрического определения ДПС в результате проведенной оптимизации (ранее значение ελmax составляло 11200 л/(моль ⋅ см)).
Результаты дизайна поверхности отклика для ПК приведены на рис. 6–8. Полученные значения координат оптимума (максимума) в кодированных значениях факторов составили: фактор X1 1.7253; фактор X2 0.6906; фактор X3 1.7687. Декодированные значения составили: с(ДХДНБФ) = = 4.59 × 10–4 М, pH 8.76 и с(АPh 9-10) = 1.48 × 10–2 М. При этом величина отклика в области максимума составляет: Y = A500 = 0.612. Полученные экспериментальные значения отклика в оптимальных условиях (0.61 ± 0.02) согласуются с расчетными.
Рис. 6.
Поверхность отклика зависимости А500 от концентрации реагента ДХДНБФ (Х1) и pH (Х2). (a) – при $X_{{\text{3}}}^{*}$ = –1, c(APh 9-10) до ККМ, (б) – при $X_{{\text{3}}}^{*}$ = 1, c(APh 9-10) после ККМ.

Рис. 7.
Поверхность отклика зависимости А500 от pc(APh 9-10) (Х1) и ДХДНБФ (Х3). (a) – при $X_{{\text{2}}}^{*}$ = –1, рН 2.00; (б) – при $X_{{\text{2}}}^{*}$ = 1, рН 10.00.

Рис. 8.
Поверхность отклика зависимости А500 от pc(APh 9-10) (Х1) и рН (Х2). (a) – при $X_{{\text{3}}}^{*}$ = –1, c(ДХДНБФ) = = 0.100 мМ, и (б) – при $X_{{\text{3}}}^{*}$ = 1, c(ДХДНБФ) = 0.700 мМ.
В установленных оптимальных условиях оценили возможностьспектрофотометрического определения ДПС и ПК. Результаты демонстрируют линейность градуировочных зависимостей в широком интервале определяемых концентраций 0.003–0.05 мг/мл:
Значения пределов обнаружения, рассчитанные по 3s-критерию, составили 0.75 и 1.5 мкг/мл для дапсона и прокаина соответственно.
Изучили влияние различных органических и неорганических веществ, являющихся потенциальными компонентами лекарственных форм, на результаты определения ЛВ. Присутствие неорганических солей, других действующих и вспомогательных веществ, входящих в состав лекарственных форм, определению не мешает. Правильность методик спектрофотометрического определения проверяли сопоставлением результатов анализа лекарственных форм по Фармакопее РФ (табл. 3, 4). Такая избирательность определения возможна ввиду того, что полосы поглощения производных вспомогательных компонентов сдвинуты гипсохромно по сравнению с бензофуроксановыми производными лекарственных веществ.
Таблица 3.
Мешающее влияние компонентов на результаты определения лекарственных веществ (n = 5, P = = 0.95)
| Компонент (А) | Содержание компонента, с(А), мкг/мл | Введено аналита, мкг/мл | Найдено аналита, мкг/мл |
sr |
|---|---|---|---|---|
| Дапсон | ||||
| Фенол | 38.0 | 6.21 | 6.25 ± 0.19 | 0.03 |
| 1.90 | 12.40 | 12.20 ± 0.30 | 0.02 | |
| 9.50 | 24.80 | 25.30 ± 0.56 | 0.02 | |
| Крахмал | 40.0 | 12.40 | 12.55 ± 0.17 | 0.01 |
| Магния стеарат | 10.0 | 12.40 | 12.25 ± 0.22 | 0.02 |
| Лактозы моногидрат | 40.0 | 12.40 | 12.55 ± 0.35 | 0.02 |
| Натрия бензоат | 2.00 | 12.40 | 12.65 ± 0.30 | 0.02 |
| Глюкоза | 50.0 | 12.40 | 12.15 ± 0.31 | 0.02 |
| СаHPO4 | 40.0 | 12.40 | 12.63 ± 0.28 | 0.02 |
| Ацетилсалициловая кислота | 7.50 | 12.40 | 11.80 ± 0.48 | 0.03 |
| Сульфосалициловая кислота | 11.0 | 12.40 | 11.70 ± 0.51 | 0.03 |
| Прокаин | ||||
| Фенол | 50.0 | 27.28 | 28.00 ± 0.95 | 0.03 |
| Хлорид натрия | 145 | 27.28 | 27.02 ± 0.34 | 0.04 |
| Хлорид кальция | 27.0 | 27.28 | 27.45 ± 0.50 | 0.03 |
| Оксид цинка | 100 | 27.28 | 26.9 ± 0.60 | 0.03 |
Таблица 4.
Результаты определения лекарственных веществ в лекарственных формах (n = 4, Р = 0.95)
| Состав лекарственной формы | Определяемое вещество | Найдено, г | |
|---|---|---|---|
| по нормативной документации | по разработанной методике | ||
| Прокаина гидрохлорид 0.25 г; HCl (0.1 н) 0.3 мл; NaCl 0.85 г; вода до 100 мл | Прокаин | 0.24 ± 0.01 | 0.25 ± 0.01 |
| Раствора СаСl2 (6.0 г) 200,0 мл; NaBr 4.0 г; прокаина гидрохлорида 1.0 г | Прокаин | 0.98 ± 0.03 | 1.01 ± 0.03 |
| Эфедрина гидрохлорид 6.0 г; прокаина гидрохлорид (1%-ный) 200 мл; димедрол 2.0 г | Прокаин | 1.98 ± 0.06 | 1.99 ± 0.06 |
| Атропина сульфат 0.02 г; эфедрина гидрохлорид 0.05 г; прокаина гидрохлорид 0.04 г; вода до 10.0 мл | Прокаин | 0.043 ± 0.002 | 0.042 ± 0.002 |
| Димедрол 0.025 г; эфедрин 0.025 г; прокаина гидрохлорид 0.2 г; NaCl 0.06 г | Прокаин | 0.18 ± 0.01 | 0.19 ± 0.01 |
| Сульфокамфокаин | Прокаин | 50.4 ± 0.8 мг/мл | 50.3 ± 0.9 мг/мл |
| Бициллин-5 | Прокаиновая соль бензилпенициллина | 0.89 ± 0.01 | 0.90 ± 0.01 |
| Таблетки дапсона 0.05 г | Дапсон | 0.049 ± 0.002 | 0.050 ± 0.001 |
* * *
Таким образом, предложено использование методологии поверхности отклика и дизайна Бокса–Бенкена для спектрофотометрического определения в лекарственных препаратах ЛВ, содержащих ароматическую аминогруппу, на примере дапсона и прокаина. Существенным достижением является сокращение времени поиска оптимальной матрицы для проведения определений и экологичность предложенных мицеллярных матриц за счет существенного снижения доли органического растворителя.
Список литературы
Becker D.E., Reed K.L. Essentials of local anesthetic pharmacology // Anesth. Prog. Fall 2006. V. 53. № 3. P. 98. https://doi.org/10.2344/0003-3006(2006)53[98:EOLAP]2.0.CO
Zhu Y.I., Stiller M.J. Dapsone and sulfones in dermatology: Overview and update // J. Am. Ac. Dermatol. 2001. V. 45. P. 420. https://doi.org/10.1067/mjd.2001.114733
Полюдек-Фабини Р., Бейрих Т. Органический анализ. Л.: Химия, 1981. 624 с.
Фармацевтическая химия / Под ред. Плетеневой Т.В. М.: ГЭОТАР-Медиа, 2017. С. 547.
Чернова Р.К., Доронин С.Ю., Гусакова Н.Н., Гаврилова Ю.Ю. Проблема избирательности и чувствительности спектрофотометрического определения компонентов лекарственных средств в водных, неводных и организованных средах / Проблемы аналитической химии. Т. 16. Фармацевтический анализ / Под ред. Будникова Г.К., Гармонова С.Ю. М.: Аргамак-Медиа, 2013. С. 187.
Ferreira S.L.C., Bruns R.E., Ferreira H.S., Matos G.D., David J.M., Brandao G.C., Silva E.G.P., Portugal L.A., Reisc P.S., Souza A.S., Santos W. Box-Behnken design: An alternative for the optimization of analytical methods // Anal. Chim. Acta. 2007. V. 597. P. 179. https://doi.org/10.1016/j.aca.2007.07.011
Munoz G., Duy S., Budzinski H., Labadie P., Liu J., Sauve S. Quantitative analysis of poly- and perfluoroalkyl compounds using high resolution mass spectrometry: Optimization for a laser diode thermal dersorbtion method // Anal. Chim. Acta. 2015. V. 881. P. 98.https://doi.org/10.1016/j.aca.2015.04.015
Schappler J., Staub A., Veuthey J.-L., Serge Rudaz S. Highly sensitive detection of pharmaceutical compounds in biological fluids using capillary electrophoresis coupled with laser-induced fluorescence // J. Chomatogr. A. 2008. V. 1204. P. 183 https://doi.org/10.1016/j.chroma.2008.04.044
Bi W., Tian M., Row K.H. Evalution of alcohol-based deep eutectic solvent in extraction and determination of flavonoids with response surface methodology optimization // J. Chromatogr. A. 2013. V. 1285. P. 22. https://doi.org/10.1016/j.chroma.2013.02.041
Tahere K., Ali D., Reza S. A green ultrasonic-assisted liquid-liquid microextracion based on deep eutectic solvent for the HPLC-UV determination of ferulic, caffeic, and cinnamic acid from olive, almond, sesame and cinnamon oil // Talanta. 2016. V. 150. P. 577. https://doi.org/10.1016/j.talanta.2015.12.077
Oliveira F.S., Korn M. Spectrophotometric determination of sulphate in automotive fuel ethanol by sequential injection analysis using dimethylsulphonazo(III) reaction // Talanta. 2005. V. 68. P. 992. https://doi.org/10.1016/j.talanta.2005.06.055
Brasil J.L., Ev R.R., Milcharek C.D., Martins L.C., Pavan F.A., Santos A.A.Jr., Dias S.L.P., Dupont J., C.P.Z. Norena C.P.Z., Lima E.C. Statistical design of experiments as a tool for optimizing the batch conditions to Cr(VI) biosorption on Araucaria angustifolia wastes // J. Hazard. Mater. 2006. V. 133. № 1–3. P. 143. https://doi.org/10.1016/j.jhazmat.2005.10.002
Bosque-Sendra J.M., Pescarolo S., Cuadros-Rodriguez L., Garcia-Campana A.M., Almansa-Lopez E.M. Optimizing analytical methods using sequential response surface methodology: Application to the pararosaniline determination of formaldehyde // Fresenius J. Anal. Chem. 2001. V. 369. P. 715. https://doi.org/10.1007/s002160100751
Бакеева Р.Ф., Горбунова Т.С., Вахитова О.Е., Гайсина А.И., Юсупова Л.М., Гармонов С.Ю., Сопин В.Ф. Cпектрофотометрическое определение п-аминофенола в лекарственных препаратах при использовании 5,7-дихлор-4,6-динитробензофуроксана как реагента в мицеллярной среде // Химико-фармацевтический журнал. 2010. Т. 44. № 4. С. 51. (Bakeeva R.F., Gorbunova T.S., Vakhitova O.E., Gaisina A.I., Yusupova L.M., Garmonov S.Y., Sopin V.F. Spectrophotometric determination of p-aminophenol in drugs using 5,7-dichloro-4,6-dinitrobenzofuroxan reagent in micellar medium // Pharm. Chem. J. 2010. V. 44. № 5. P. 282. https://doi.org/10.1007/s11094-010-0450-5)
Юсупова Л.М., Гармонов С.Ю., Горелова Е.Г., Васютина Е.А., Мухаметшина А.М. Способ синтеза 4,6-динитро-5,7-дихлорбензофуроксана. Патент РФ 2013132641. Заявка 2013132641/04 от 16.07.2013, опубл. 27.01.2015.
Арутюнян Л.Р., Маркарян Ш.А Изучение влияния диметилсульфоксида и диэтилсульфоксида на мицеллообразование додецилсульфата натрия в водных растворах методом флуоресценции // Коллоид. журн. 2007. Т. 69. № 4. С. 444. (Harutyunyan L., Markarian S.A. Investigation of the effect of dimethyl sulfoxide and diethyl sulfoxide on sodium dodecyl sulfate micellization in aqueous solutions by fluorescence method // Colloid. J. 2007. V. 69. № 4. P. 444. )https://doi.org/10.1134/S1061933X07040023
Бакеева Р.Ф., Каримуллин Р.А., Курабашева Р.Ф., Горбунова Т.С., Вахитова О.Е., Сопин В.Ф. Мицеллобразование в бинарном растворителе диметилсульфоксид–вода в присутствии NaCl. Планирование и оптимизация эксперимента // Жидкие кристаллы и их практическое использование. 2017. Т. 17. № 2. С. 51 DOI 10.18083/LCAppl.2017.2.51 (Bakeeva R.F., Karimullin R.A., Kurabasheva R.F., Gorbunova T.S., Vakhitova O.E., Sopin V.F. Micelle formation in dimethyl sulfoxide–water binary solvent in presence of NaCl. Planning and optimization of experiment // Liq. Cryst. Their Appl., 2017. V. 17. № 2. P. 51.)
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии