

Журнал аналитической химии, 2022, T. 77, № 6, стр. 533-539
Селективное вольтамперометрическое и проточно-инжекционное амперометрическое определение ацикловира и валацикловира на электроде с композитом восстановленный оксид графена‑полиглициновая пленка
Л. Г. Шайдарова a, *, А. В. Гедмина a, А. А. Поздняк a, И. А. Челнокова a, Г. К. Будников a
a Казанский федеральный университет, Химический институт им. А.М. Бутлерова
420008 Казань, ул. Кремлевская, 18, Россия
* E-mail: LarisaShaidarova@mail.ru
Поступила в редакцию 21.06.2021
После доработки 29.09.2021
Принята к публикации 29.09.2021
- EDN: NOKEBT
- DOI: 10.31857/S0044450222060172
Аннотация
Ацикловир и валацикловир окисляются на электроде из стеклоуглерода (СУ) с иммобилизованным восстановленным оксидом графена, покрытым полиглициновой пленкой. Подобраны условия получения полимерной пленки и восстановления оксида графена на СУ, а также регистрации максимального каталитического тока на композитной поверхности. Разработан способ селективного вольтамперометрического определения ацикловира и валацикловира на электроде с композитом восстановленный оксид графена–полиглициновая пленка. Предложен способ амперометрического детектирования противовирусных препаратов на основе гуанина на этом электроде в условиях проточно-инжекционного анализа. Зависимость аналитического сигнала от концентрации ацикловира и валацикловира линейна до 5 × 10–6 М в стационарном режиме и до 1 × 10–7 M в проточной системе. Разработанный вольтамперометрический способ определения ацикловира и валацикловира апробирован при анализе лекарственных средств.
Композитные электроды на основе полимерных пленок, полученных электрополимеризацией, привлекают к себе внимание благодаря высокой селективности и чувствительности их отклика при определении широкого круга низкомолекулярных органических соединений. Такие полимерные пленки отличаются гомогенностью, сильной адгезией к поверхности СУ и химической устойчивостью.
Полимерные пленки из полиглицина (поли-Гли), как и большинство электропроводящих полимерных пленок, наносят на поверхность электрода осаждением из раствора соответствующего мономера с последующей его полимеризацией. При электрополимеризации глицина на поверхности углеродной подложки формируется полиглицин типа II со спиральной конформацией [1]. Анодное окисление первичного амина происходит по радикальному катионному механизму, который объясняет однородность полученного тонкого покрытия. Полиглицин нерастворим в воде и в обычных органических растворителях.
В настоящее время получение композитов из проводящих полимерных пленок с восстановленным оксидом графена особенно привлекательно благодаря аддитивности, а в некоторых случаях синергизму электрокаталитических эффектов обоих компонентов [2–6].
Один из способов получения окида графена (ГО) с хорошим выходом – химическое окисление графита по методу Хаммера [7]. Для уменьшения количества оксидных форм графена обычно проводят химическое восстановление, однако для этого требуется высокотемпературная обработка, многократное повторение процесса восстановления и значительное время. Кроме того, используемый в качестве восстановителя моногидрат гидразина является токсичным реагентом и приводит к появлению в структуре графена гетероатомных примесей азота [8, 9]. По сравнению с химическим и термическим способами электрохимическое восстановление ГО является более экспрессным, технологичным и безопасным [10, 11].
Электропроводность электрохимически восстановленного оксида графена (ГОвос) выше, чем химически полученного ГОвос. Это возможно из-за остаточных дефектов в химически полученном ГОвос, которые могут быть сведены к минимуму в случае электрохимического восстановления ГО [12]. Восстановленный ГО обычно является неполной приведенной формой оксида графена и находится в промежуточном состоянии между оксидом графена и самим графеном. В этом состоянии число кислородсодержащих функциональных групп в материале мало, но имеются дефекты, которые могут удерживать химически активные участки для использования в каталитических реакциях и взаимодействовать со слоями полимеров [8].
В настоящей работе рассмотрена возможность вольтамперометрического определения в стационарных условиях и амперометрического детектирования в проточно-инжекционной системе ацикловира и валацикловира на химически модифицированном электроде (ХМЭ) на основе стеклоуглеродного электрода с иммобилизованным композитом восстановленный оксид графена–полиглициновая пленка.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Циклические вольтамперограммы (ЦВА) регистрировали с помощью вольтамперометрического анализатора Экотест-ВА (ООО “Эконикс-Эксперт”, Россия). Использовали трехэлектродную ячейку, в которой в качестве электрода сравнения использовали хлоридсеребряный электрод, в качестве вспомогательного – платиновую проволоку, в качестве рабочего применяли электроды из СУ с видимой поверхностью 0.10 см2, покрытые полимерной пленкой полиглицина (поли-Гли-СУ) и композитом из ГОвос и полимерной пленки полиглицина (поли-Гли-ГОвос-СУ).
Иммобилизацию пленки из поли-Гли проводили в условиях потенциодинамического осаждения. ЦВА регистрировали при скорости наложения потенциала 20 мВ/с. При получении полимерной пленки скорость циклирования потенциала составляла 100 мВ/с, а при восстановлении иммобилизованного на поверхность СУ оксида графена – 50 мВ/с. Поверхностную концентрацию модификатора на СУ определяли по площади катодного пика на ЦВА ферроцианид-иона [13].
Композитный электрод получали в две стадии: сначала на поверхность СУ наносили слой оксида графена, который получали методом капельного испарения под ИК-лампой из суспензии оксида графена с хитозаном, после чего проводили электрохимическое восстановление ГО до ГОвос на поверхности полученного электрода. Затем на поверхность СУ с ГОвос (ГОвос-СУ) наносили полимерную пленку, электрохимически осажденную из 5 мМ раствора глицина в фосфатном буферном растворе с рН 6.86.
Для измерений в условиях проточно-инжекционного анализа (ПИА) применяли установку, включающую перистальтический насос, инжектор, проточную электрохимическую ячейку и регистрирующее устройство [14]. Подачу и слив растворов осуществляли по проточным коммуникациям, изготовленным из силиконовых трубок внутренним диаметром 2.0 мм. Инжекцию осуществляли микрошприцем через уплотнительную мембрану.
В качестве аналитов в модельных системах использовали ацикловир и валацикловир (Aldrich, США). Стандартные растворы исследуемого объекта с концентрацией 5 × 10–3 М готовили путем растворения их точных навесок. Для построения градуировочной зависимости готовили растворы с меньшими концентрациями аналитов последовательным разбавлением исходных растворов непосредственно перед измерениями. В качестве фонового электролита использовали раствор серной кислоты с рН 2.0, ацетатный буферный раствор с pH 4.0 и фосфатный буферный раствор с рН 6.86. Контроль pH растворов осуществляли с помощью pH-метра типа рН-150.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Электроосаждение полиглициновой пленки на поверхности стеклоуглеродного электрода и восстановленном оксиде графена. Полиглициновую пленку осаждали на поверхности СУ с помощью электрополимеризации при многократном циклировании потенциала (Е) в выбранной области его значений от –0.8 до +1.8 В. Предварительно рассматривали медиаторные свойства пленки из полиглицина по отношению к окислению иона [Fe(CN)6]4–. ЦВА ферроцианид-иона, регистрируемые на немодифицированном СУ и на СУ с иммобилизованной пленкой из полиглицина, представлены на рис. 1. На ЦВА ферроцианид-иона, полученной на поли-Гли-СУ (кривая 2), по сравнению с СУ (кривая 1) отношение величин катодного тока (Iк) к анодному (Iа) близко к единице, а разность потенциалов катодного (Ек) и анодного (Еа) пиков составляет 60 мВ. Эти факты указывают на медиаторные свойства электронопроводящей полимерной пленки поли-Гли в обратимой редокс-системе.
Рис. 1.
Электрохимическое поведение 2.5 мМ К4[Fe(CN)6] в 0.1 М серной кислоте на СУ (1) и на поли-Гли-СУ (2).

Изучено влияние условий электрополимеризации пленки, таких как область циклического изменения потенциала и рН фонового электролита, на электрохимические характеристики ферроцианид-иона. Наилучшие вольт-амперные характеристики окисления ферроцианид-иона, такие как отношение величин катодного тока к анодному (Iк/Iа) (рис. 2а) и разность потенциалов катодного и анодного пиков (ΔЕ = Ек – Еа) (рис. 2б), наблюдаются при получении полиглициновой пленки в потенциодинамическом режиме в области потенциалов от –0.8 до 1.8 В из раствора глицина в фосфатном буферном растворе с рН 6.86. Этот режим близок к условиям обратимого окисления ферроцианид-иона (Iк/Iа = 1 и ΔЕ = 60 мВ).
Рис. 2.
Диаграммы зависимости электрохимических характеристик (отношение величин катодного к анодного токов (а) и разность потенциалов катодного и анодного пиков (б)) окисления ферроцианид-иона от области потенциалов электрополимеризации глицина и рН фонового электролита.

Для улучшения свойств пленки поли-Гли в качестве подложки для ее иммобилизации использовали восстановленный оксид графена, нанесенный на СУ методом капельного испарения из суспензии оксида графена с хитозаном.
Поверхностную концентрацию медиатора (Г) на поверхности СУ определяли по площади пика на катодной ветви ЦВА ферроцианид-иона, регистрируемой со скоростью наложения потенциала, равной 10 мВ/с. Размерность поверхностной концентрации представляли как отношение количества молей редокс-центров металлокомплекса, относящееся к площади поверхности электрода. Величину Г рассчитывали по формулам [15]:
где Q – заряд, Кл или А ⋅ с; n – число электронов; А – площадь поверхности рабочего электрода, см2; F – число Фарадея, равное 96 500 Кл/моль; Г – поверхностная концентрация медиатора, моль/см2. или Q = ${1 \mathord{\left/ {\vphantom {1 {v\int {I\partial \varphi } }}} \right. \kern-0em} {v\int {I\partial \varphi } }}$, где S – площадь под катодным пиком, мкА ⋅ мВ; v – скорость наложения потенциала, мВ/с; I –значение тока, мкА; φ – потенциал, мВ.Значение поверхностной концентрации модификатора увеличивается при переходе от пленки поли-Гли, нанесенной на СУ, к пленке, нанесенной на ГОвос-СУ, от 2.2 × 10–8 до 2.5 × 10–8 моль/см2, что связано с увеличением поверхности матрицы из ГОвос, на поверхность которого проводили электрополимеризацию пленки поли-Гли.
Электроокисление ацикловира и валацикловира на стеклоуглероде, модифицированном композитом из поли-Гли-ГОвос. На немодифицированном СУ окисление ацикловира протекает необратимо и с большим перенапряжением. При окислении ацикловира на поли-Гли-ГОвос-СУ на анодной ветви ЦВА при Е = 1.10 В регистрируется пик, многократно превышающий величины токов окисления модификатора на фоновой кривой (рис. 3, кривые 1, 2) и окисления ацикловира на немодифицированном СУ (табл. 1). Ток, регистрируемый на этом электроде, линейно зависит от концентрации ацикловира. Окисление валацикловира происходит при тех же потенциалах, что и окисление ацикловира. Многократный прирост тока, линейная зависимость величины тока пика от концентрации субстрата позволяют отнести ток электродной реакции к каталитическому. Наибольшее значение величины электрокаталитического эффекта, выраженного отношением величин каталитического тока окисления субстрата на ХМЭ (Iкат) и тока окисления модификатора (Iмод) – Iкат/Iмод, наблюдается при окислении ацикловира и валацикловира на поли-Гли-ГОвос-СУ (табл. 1).
Для выяснения особенностей электроокисления ацикловира на композитном электроде изучена зависимость тока (I) и потенциала пика (E) от скорости наложения потенциала (v), которую варьировали в диапазоне от 10 до 100 мВ/с. Полученное из линейной зависимости (lg I = 0.77 + + 0.81lg v) значение критерия Семерано, равное 0.81, свидетельствует об адсорбционной природе тока. Из зависимости величины потенциала пика от логарифма скорости наложения потенциала (Е = = 0.89 + 0.11lg v) рассчитаны кинетические параметры окисления ацикловира – величина наклона Тафеля (b), коэффициент переноса электрона (α) и константа скорости электрокаталитической реакции (ks) [16, 17]. Для электрокаталитического окисления ацикловира на немодифицированном стеклоуглеродном электроде с композитом поли-Гли-ГОвос найдены следующие кинетические характеристики: b = 110.2 мВ, α = 0.732 и ks = 0.53 × 102 с–1. Полученное значение ks характеризует высокую скорость электронного переноса.
Установлено, что продолжительность циклирования потенциала (N –количество циклов) и величина рН фонового электрода влияют на каталитический ток электроокисления ацикловира и валацикловира. Так, максимальный каталитический эффект наблюдается при восстановлении ГО путем циклического изменения потенциала в области от 0.60 до –1.50 В в течение 20 циклов (рис. 4а) и полимеризации глицина в области потенциалов от –0.80 до 1.80 В в течение 40 циклов (рис. 4б), а также при регистрации ЦВА ацикловира и валацикловира на фоне фосфатного буферного раствора с рН 6.86 (рис. 4в).
Рис. 3.
Циклические вольтамперограммы, полученные на поли-Гли-ГОвос-СУ в отсутствие (1) и в присутствии ацикловира (5 × 10–3 М) (2) на фоне фосфатного буферного раствора с рН 6.86.
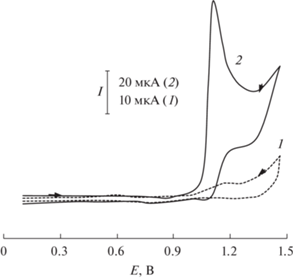
Рис. 4.
Зависимость величины каталитического эффекта при окислении ацикловира и валацикловира от количества циклов при восстановлении ГО (а), от количества циклов при электрополимеризации глицина (б) и от рН фонового электролита (в).

Вольтамперометрическое определение в стационарном режиме ацикловира и валацикловира на электроде, модифицированном композитом из поли-Гли-ГОвос. В мерную колбу емк. 10 мл вводили фиксированный объем раствора аналита (ацикловира или валанцикловира), в который добавляли фосфатный буферный раствор с рН 6.86 до метки. Полученный раствор количественно переносили в электрохимическую ячейку, в которую погружали электрод из СУ с композитом поли-Гли-ГОвос, хлоридсеребряный и вспомогательный электроды. Регистрировали ЦВА в интервале от –0.0 до +1.30 В и измеряли величину Iп окисления ацикловира при Еп 1.10 В.
Аналитические характеристики, демонстрирующие высокую чувствительность отклика, представлены в табл. 2. Нижняя граница определяемых содержаний составляет 5 × 10–6 М, правильность предлагаемых способов определения оценена методом введено–найдено. Относительное стандартное отклонение (Sr) не превышает 0.05 во всем рабочем диапазоне концентраций (табл. 3).
Таблица 1.
Вольт-амперные характеристики окисления ацикловира и валацикловирана на модифицированных электродах
| Субстрат | $E_{{\text{s}}}^{*}$, В | $I_{{\text{s}}}^{*}$, мкА | Модификатор | Емод, В | Екат, В | Iкат, мкА | Iкат/Iмод |
|---|---|---|---|---|---|---|---|
| Ацикловир | 1.15 | 18.0 | поли-Гли | 1.10 | 1.10 | 58.0 | 18.2 |
| поли-Гли-ГОвос | 1.10 | 1.10 | 104.3 | 26.1 | |||
| Валацикловир | 1.15 | 15.0 | поли-Гли | 1.10 | 1.10 | 53.2 | 16.6 |
| поли-Гли-ГОвос | 1.10 | 1.10 | 96.5 | 24.1 |
Таблица 2.
Аналитические характеристики определения ацикловира и валацикловира на электроде поли-Гли-ГОвос-СУ (n = 6, P = 0.95, tтабл = 2.57)
| Субстрат | Способ регистрации | Диапазон концентраций, М | Уравнение регрессии lg(I) = a + blg (c) (Iп, мкА; с, М) |
R | |
|---|---|---|---|---|---|
| а ± Δa | b ± Δb | ||||
| Ацикловир | Циклическая вольтамперометрия | 5 × 10–3–5 × 10–6 | 2.8 ± 0.3 | 0.41 ± 0.02 | 0.996 |
| Валацикловир | 5 × 10–3–5 × 10–6 | 2.7 ± 0.4 | 0.40 ± 0.03 | 0.996 | |
| Ацикловир | Амперометрия в ПИА | 1 × 10–3–1 × 10–7 | 2.3 ± 0.5 | 0.44 ± 0.03 | 0.998 |
| Валацикловир | 1 × 10–3–1 × 10–7 | 2.3 ± 0.4 | 0.42 ± 0.02 | 0.999 | |
Таблица 3.
Метрологические характеристики определения ацикловира и валацикловира на электроде поли-Гли-ГОвос-СУ (n = 6, P = 0.95, tтабл = 2.57)
| Аналит | Введено, мкМ | Найдено, (х ± Δх), мкМ |
sr |
|---|---|---|---|
| Циклическая вольтамперометрия | |||
| Ацикловир | 5.0 | 4.8 ± 0.3 | 0.05 |
| 10.0 | 9.9 ± 0.4 | 0.04 | |
| 100 | 101 ± 2 | 0.02 | |
| Валацикловир | 5.0 | 4.6 ± 0.3 | 0.07 |
| 10.0 | 9.6 ± 0.8 | 0.08 | |
| 100 | 101 ± 8 | 0.07 | |
| Амперометрия в ПИА | |||
| Ацикловир | 4.5 | 4.3 ± 0.1 | 0.03 |
| 45 | 46 ± 1 | 0.02 | |
| 90 | 88 ± 1 | 0.01 | |
| Валацикловир | 4.0 | 4.1 ± 0.1 | 0.03 |
| 40 | 39 ± 1 | 0.02 | |
| 80 | 77 ± 2 | 0.02 | |
Проведен анализ лекарственных средств (в таблетках) на содержание ацикловира и валанцикловира. Для этих целей использовали разработанные способы вольтамперометрического определения этих органических соединений на электроде поли-Гли-ГОвос-СУ. Результаты определения представлены в табл. 4. Статистическая оценка результатов с использованием величины t-критерия свидетельствует об отсутствии значимой систематической погрешности (tрасч < tтабл) (табл. 4).
Таблица 4.
Результаты определения ацикловира и валанцикловира в лекарственных средствах методом вольтамперометрии на поли-Гли-ГОвос-СУ (n = 6, P = 0.95, tтабл = 2.57, Fтабл = 5.79)
| Объект анализа | Аналит | Заявленное содержание в препарате, г | Найдено, г | sr | tрасч |
|---|---|---|---|---|---|
| “Ацикловир-Акри” | Ацикловир | 0.40 | 0.38 ± 0.02 | 0.05 | 2.45 |
| “Валвир” | Валацикловир | 0.50 | 0.49 ± 0.02 | 0.04 | 2.44 |
Показана возможность использования электрода поли-Гли-ГОвос-СУ для определения рассматриваемых биологически активных соединений в условиях ПИА. В качестве потока-носителя использовали фосфатный буферный раствор с рН 6.86.
Графические зависимости аналитического сигнала от электрохимических (накладываемый потенциал (Еп)) и гидродинамических (объем инжектируемой пробы (V) и скорость потока жидкости (u)) параметров проточной системы приведены для ацикловира (рис. 5). Максимум на зависимостях величины ПИА-сигнала ацикловира от накладываемого потенциала (рис. 5а) наблюдается при Еп 1.15 В, от объема инжектируемой пробы – при V 0.90 мл (рис. 5б), от скорости потока – при u 27.5 мл/мин (рис. 5в).
Рис. 5.
Зависимость ПИА-сигнала при окислении ацикловира (5 × 10–3 М) на поли-Гли-ГОвос-СУ от накладываемого потенциала (а), объема инжектируемой пробы (б) и скорости подачи потока-носителя (в).
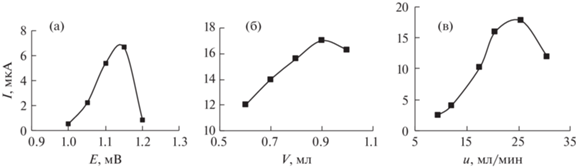
На основе полученных зависимостей выбрали рабочие условия регистрации ПИА-сигнала ацикловира и валацикловира на электроде поли-Гли-ГОвос-СУ. Для ацикловира наилучшими условиями регистрации тока являются: Еп +1.15 В, V 0.90 мл и u = 27.5 мл/мин. Аналогичные характеристики амперометрического детектирования в проточных условиях получены и для валоцикловира.
Для каждого аналита разработана методика амперометрического детектирования в условиях ПИА. Аналитические характеристики определения ацикловира и валацикловира представлены в табл. 2. Использование амперометрического сигнала в ПИА по сравнению со стационарными вольтамперометрическими условиями регистрации аналитического сигнала приводит к снижению нижней границы мопределяемых содержаний аналита до 1 × 10–7 М.
Метрологические характеристики определения ацикловира и валацикловира на электроде с композитом поли-Гли-ГОвос в условиях ПИА представлены в табл. 3. Нижняя граница определяемых содержаний составляет 1 × 10–7 М для проточных условий регистрации аналитического сигнала. При оценке этих характеристик сопоставили сходимость результатов анализа в стационарных и проточных системах. В условиях ПИА достигнута лучшая воспроизводимость результатов определения исследуемых аналитов по сравнению со стационарными условиями (табл. 3). Во всех случаях регистрации аналитического сигнала в проточных условиях относительное стандартное отклонение не превышало 3%.
Повышение воспроизводимости и стабильности каталитического отклика ХМЭ в ПИА-условиях связано с обновлением поверхности электрода потоком фонового электролита, с ограничением по времени сорбции исследуемых соединений или продуктов электрохимических реакций на поверхности электрода в потоке, что, в свою очередь, уменьшает риски отравления модификатора.
* * *
Таким образом, использование ПИА способствует автоматизации анализа, сочетание ПИА с амперометрическим способом определения ацикловира и валацикловира на ХМЭ позволяет улучшить воспроизводимость электрокаталитического отклика ХМЭ, повысить чувствительность и производительность метода по сравнению со статическим вольтамперометрическим способом определения.
Список литературы
Herlem G., Zeggari R., Rauch J.Y., Monney S., Anzola F.T., Guillaume Y., Andre C., Gharbi T. One-pot electrosynthesis of polyglycine-like thin film on platinum electrodes as transducer for solid state pH measurements // Talanta. 2010. V. 82. № 1. P. 417.
Han L. Zhao Y., Chang C., Li F. A novel electrochemical sensor based on poly(p-aminobenzene sulfonic acid)-reduced graphene oxide composite film for the sensitive and selective detection of levofloxacin in human urine // J. Electroanal. Chem. 2018. V. 817. P. 141.
Das T.K., Prusty S. Graphene-based polymer composites and their applications // Polymer-Plastics Technol. Eng. 2013. V. 52. № 4. P. 319.
Eda G., Chhowalla M. Graphene-based composite thin films for electronics // Nano Letters. 2009. V. 9. № 2. P. 814.
Terrones M., Martín O., González M., Pozuelo J., Serrano B., Cabanelas J.C., Vega-Díaz S.M., Baselga J. Interphases in graphene polymer-based nanocomposites: achievements and challenges // Adv. Mater. 2011. V. 23. № 44. P. 5302.
Liu H., Zhang G., Zhou Y., Gao M., Yang F. One-step potentiodynamic synthesis of poly(1,5-diaminoanthraquinone)/reduced graphene oxide nanohybrid with improved electrocatalytic activity // J. Mater. Chem A. 2013. V. 1. № 44. P. 13902.
William S., Hummers J.R., Richard E.O. Preparation of graphitic oxide // J. Am. Chem. Soc. 1958. V. 80. №. 6. P. 1339.
Palakollu V.N., Thapliyal N., Chiwunze T.E., Karpoormath R., Karunanidhi S., Cherukupalli S. Electrochemically reduced graphene oxide/poly-glycine composite modified electrode for sensitive determination of L-dopa // Mater. Sci. Eng. C. 2017. V. 77. P. 394.
Shin H.-J., Kim K.K., Benayad A., Yoon S.-M., Park H.K., Jung I.-S., Jin M.H., Jeong H.-K., Kim J.M., Choi J.-Y., Lee Y.H. Efficient reduction of graphite oxide by sodium borohydride and its effect on electrical conductance // Adv. Funct. Mater. 2009. V. 19. № 12. P. 1987.
Kumar D.R., Kesavan S., Baynosa M.L., Shim J.J. 3,5-Diamino-1,2,4-triazole electrochemically reduced graphene oxide film modified electrode for the electrochemical determination of 4-nitrophenol // Electrochim. Acta. 2017. V. 246. P. 1131.
Guo H.-L. Wang X.-F., Qian Q.-Y., Wang F.-B., Xia X.-H. A green approach to the synthesis of graphene nanosheets // ACS Nano. 2009. V. 3. №. 9. P. 2653.
Kuila T., Mishra A.K., Khanra P., Kim N.H., Lee J.H. Recent advances in the efficient reduction of graphene oxide and its application as energy storage electrode materials // Nanoscale. 2013. V. 5. № 1. P. 52.
Каплун М.М., Смирнов Ю.Е., Микли В., Малеев В.В. Структурное исследование пленок гексацианоферрата кобальта, синтезированных из комплексного электролита // Электрохимия. 2001. Т. 37. № 9. С. 1065.
Шайдарова Л.Г., Зиганшина С.А., Тихонова Л.Н., Будников Г.К. Электро-каталитическое окисление и проточно-инжекционное определение серосодержащих аминокислот на графитовых электродах, модифицированных пленкой из гексацианоферрата рутения // Журн. аналит. химии. 2003. Т. 58. № 12. С. 1277.
Дамаскин Б.Б. Практикум по электрохимии: учеб. пособие для хим. спец. вузов / Под ред. Дамаскина Б.Б. М.: Высшая школа, 1991. 288 с.
Будников Г.К., Улахович Н.А., Медянцева Э.П. Основы электроаналитической химии. Казань: Изд-во КГУ, 1986. 288 с.
Будников Г.К. Принципы и применение вольтамперной осциллографической полярографии. Казань: Изд-во КГУ, 1975. 197 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии