

Журнал аналитической химии, 2022, T. 77, № 7, стр. 645-652
Факторы, влияющие на результаты определения фтора в горных породах рентгенофлуоресцентным методом с использованием прессованных таблеток
Т. Г. Кузьмина a, *, М. А. Тронева a, Т. В. Ромашова a
a Институт геохимии и аналитической химии им. В.И. Вернадского Российской академии наук
119991 Москва, ул. Косыгина, 19, Россия
* E-mail: kuzminatg@inbox.ru
Поступила в редакцию 05.07.2021
После доработки 24.11.2021
Принята к публикации 24.11.2021
- EDN: FXNLEV
- DOI: 10.31857/S0044450222060093
Аннотация
Изучена возможность определения фтора в горных породах рентгенофлуоресцентным методом с использованием в качестве излучателей прессованных таблеток. Рассмотрены наиболее важные факторы, влияющие на результаты определения фтора: эффект обволакивания; систематические и случайные погрешности, связанные с измерением интенсивности линии фтора и пробоподготовкой; зависимость интенсивности FKα-линии от матричного состава пробы и от соединения, в состав которого входит этот элемент. Полученные результаты оценили с точки зрения допустимых погрешностей. Показано, что для прессованных таблеток невозможно использование единого градуировочного уравнения для горных пород с различными фторсодержащими минералами.
Фтор является носителем важной информации о процессах, происходящих в магматических системах. Летучие компоненты, в состав которых входит фтор, находятся в подчиненных количествах по отношению к петрогенным элементам. Однако “их химическая активность вызывает целый ряд существенных изменений в физико-химических свойствах магматического расплава, например, рост кристаллизационной способности минералов, понижение вязкости, снижение температуры кристаллизации, уменьшение поверхностного натяжения и т.д.” [1]. Среднее содержание фтора по Виноградову [2] в земной коре составляет 660 × 10–4 мас. %, при этом оно колеблется от 100 × 10–4 мас. % в ультраосновных породах до 1400 × 10-4 мас. % в агпаитовых нефелиновых сиенитах и может достигать 3.5% и более во фторапатитах. Основными минералами, содержащими фтор, которые встречаются в значительных количествах в горных породах, являются: флюорит CaF2, фторапатит Ca10(PO4)6F2 и криолит Na3AlF6.
Для определения фтора используют преимущественно потенциометрию и ионную хроматографию, но при этом требуется разложение пробы. Ведутся поиски методов, позволяющих определять содержание фтора непосредственно в исходном образце без трудоемкой операции пробоподготовки. Одним из них является метод рентгенофлуоресцентного анализа (РФА), который давно занял прочное место среди других физико-химических методов определения химического состава вещества. В большинстве геологических институтов применение РФА для определения содержания петрогенных элементов стало традиционным. Вместе с тем выпускаемые в настоящее время спектрометры, благодаря появлению кристалл-анализаторов, эффективно работающих в длинноволновой области спектра, позволяют определять и более легкие элементы, включая фтор. Однако практика использования РФА для определения фтора в горных породах, судя по литературным данным, не получила широкого распространения. Следует упомянуть некоторые работы, в которых при определении фтора использовали способ сплавления пробы с тетраборатом лития. В работе [3] определяли фтор в шлаках сталелитейной промышленности. Диссертация [4] посвящена определению петрогенных элементов и фтора в рудах. В работе [5] получена градуировочная зависимость для определения фтора с использованием таких горных пород, как гранит, цеолит, сиенит и пр.
В работах [6–9] авторы использовали для РФА прессованные таблетки из порошкового материала. Этот способ привлекает своей простотой и низкой стоимостью. Положительные результаты определения фтора, входящего в состав криолита в алюминиевой матрице, получены в работе Джехта [6]. Фтор в фонолитах с помощью РФА определяли Шефер и Медуна [7]. Образцы готовили в виде прессованных таблеток с добавления 20% воска. Иорданис и соавт. [8] сообщили об определении фтора в буром угле. Однако Газулла с соавт. [9] на основе РФА полевых шпатов, полевошпатоидов, колеманитов и улекситов сделали вывод, что более точные результаты определения бора и фтора в геологических образцах достигаются методами атомно-эмиссионной спектрометрии с индуктивно связанной плазмой или потенциометрии.
В наиболее подробном обзоре работ в рассматриваемой области [10] также отмечено, что РФА редко используется на практике для определения фтора в горных породах и почвах.
Сложившуюся ситуацию можно объяснить рядом факторов, влияющих на погрешность определения фтора: эффектами микроабсорбционной неоднородности, одним из которых является обволакивание; проблемами, связанными с измерением интенсивности FKα-линии; и матричными эффектами, в том числе особенностями возбуждения рентгеновской флуоресценции фтора.
Смагунова и соавт. отмечали [11], что при истирании пробы может происходить обволакивание зерен, содержащих атомы фтора, другими компонентами смеси, которые истираются быстрее зерен, содержащих фтор, и это приводит к снижению интенсивности флуоресцентного излучения фтора. Влияние этого эффекта можно снизить, отмывая пробу этанолом. В более ранней работе [12] на примере Zn, Pb, Mn установлено, что даже при “мокром” истирании порошка с добавлением этанола не удается полностью освободиться от агрегирования и обволакивания крупных частиц более мелкими.
Проблемы, связанные с измерением интенсивности флуоресцентного излучения фтора, подробно рассмотрены в работе Плесова [10]. Автор на примере многочисленных экспериментов с использованием различных фторсодержащих соединений показал, что интенсивность флуоресценции фтора в почвах, горных породах, минералах и синтетических химикатах может меняться при повторных измерениях. Это относится как к сплавленным образцам, так и к спрессованным таблеткам. Сплавленный апатит, спрессованный грунт и сплавленный флюорит демонстрируют отчетливое увеличение интенсивности фтора; прессованный апатит, K2TiF6, топаз и грейзен – умеренное увеличение; сплавленный топаз и прессованный флюорит – стабильную интенсивность флуоресценции; и, наконец, интенсивность FKα-линии прессованного криолита резко уменьшается. После анализа целого ряда факторов, которые могут вызывать подобные тренды, в том числе влияние загрязнений поверхности образца, уровня вакуума в спектральной камере, величины тока рентгеновской трубки и т.д., автор работы [10] пришел к заключению, что изменение интенсивности линии FKα связано с эффектом радиолиза. Сделан вывод о том, что этот эффект является решающим препятствием в рентгенофлуоресцентной спектрометрии фтора в горных породах и почвах при использовании как сплавленных, так и прессованных излучателей.
Влияние матричных эффектов на интенсивность рентгеновской флуоресценции фтора исследовано в работе [13]. Измерения проводили с использованием таблеток с добавлением воска в качестве связующего вещества. Установлено, что измеренные интенсивности линии FKα при сопоставимом содержании фтора в системах Na3AlF6–NaCl и K2TaF7–K2ZrF6–NaCl отличаются в пять раз. После попыток объяснить это различие авторы пришли к выводу, что чем больше “тяжелых” элементов содержится в пробе, тем труднее оценить степень влияния эффектов ослабления и усиления на интенсивность флуоресцентного излучения фтора.
Нами ранее [14] экспериментально изучены особенности возбуждения флуоресценции линии FKα в присутствии петрогенных элементов (Na, Mg, Al, Si, Ca, Ti, Mn, Fe). Использовали смеси NaF и CaF2 с оксидами петрогенных элементов в соотношении 1 : 1. Установлено, что наибольший вклад в интенсивность линии FKα за счет дополнительного возбуждения флуоресцентным излучением вносит натрий. Изучено суммарное влияние фото- и Оже-электронов перечисленных выше элементов на интенсивность линии FKα. Зависимость полученного в этом случае прироста интенсивности флуоресцентного излучения фтора в пересчете на один процент содержания соответствующего петрогенного элемента от его атомного номера показала, что максимум приходится на область Z ≈ 20–22. Так как в работе отсутствовали данные по P, S и К, нельзя исключить вероятности того, что реальный максимум может быть смещен в сторону более легких элементов. Следует отметить, что этот эффект оказывает более значительное влияние на интенсивность флуоресценции фтора по сравнению с эффектом ослабления.
Экспериментально также установлено [14], что интенсивность линий FKα в смесях NaF с оксидами петрогенных элементов примерно в 2–2.5 раза превосходит интенсивность линий FKα для смесей флюорита (CaF2) с этими же оксидами. В смесях NaF + CaCO3 и CaF2 + Na2CO3 выявлена та же закономерность, хотя элементный состав (мас. %) и коэффициенты ослабления линии FKα в этих двух случаях практически совпадают. Сделан вывод, что отличительной особенностью фтора является зависимость выхода флуоресценции от вида химического соединения, в состав которого входит этот элемент.
Авторы работ [5, 10, 13] экспериментально также установили, что предпочтительнее строить отдельные градуировки для каждого фторсодержащего минерала.
Очевидно, что фтор является весьма проблемным элементом в РФА, особенно при использовании прессованных таблеток. Однако преимущества этого метода анализа настолько привлекательны, что целесообразно продолжать поиск вариантов решения этой задачи. В любом случае новые и неожиданные проблемы, возникающие при определении легких элементов рентгенофлуресцентным методом, должны стимулировать как попытки их теоретических интерпретаций на основе дальнейшего развития теории взаимодействия рентгеновского излучения с веществом, так и поиск оригинальных экспериментальных подходов к их решению.
Цель данной работы заключалась в экспериментальной оценке влияния на интенсивность линии FKα при использовании прессованных таблеток эффекта обволакивания, погрешностей, связанных с измерением интенсивности флуоресцентного излучения фтора и пробоподготовки, а также матричного состава пробы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали рентгеновский спектрометр Axios Advanced (PANalytical B.V., Нидерланды), оснащенный рентгеновской трубкой с Rh-анодом и толщиной окна 75 мкм, системой регистрации по Соллеру, кристаллами-анализаторами LiF200, LiF220 PE002-С, Ge111-С, PX1, коллиматорами, пропорциональными газоразрядными детекторами в проточном и отпаянном вариантах и сцинтилляционным счетчиком. Условия измерения интенсивности линии FKα приведены ниже. Фон измеряли с длинноволновой стороны линии.
| Линия | Режим рентгеновской трубки | Кристалл | Коллиматор | Пик линии, град | Фон, град |
| FКα | 30 мА/100 кВ | PX1 | 550 мкм | 43.1050 | +1.9824 |
Применяли стандартные образцы (CО) горных пород с содержанием фтора от 0.03 до 3.5%, а также природный флюорит и химические реактивы NaF, CaF2, Na3AlF6. В качестве вмещающих матриц использовали базальт с содержанием фтора ниже предела обнаружения и химический реактив SiO2. Пробы для анализа готовили путем прессования таблеток как с полистиролом (0.3 г пробы и 0.06 г С8Н8), так и без добавления связующего вещества на подложке из борной кислоты. В последнем случае использовали 0.3 г материала пробы. Диаметр излучателя составлял 20 мм.
Распределение фтора по срезу таблетки получали с помощью сканирующего электронного микроскопа MIRA3 фирмы “TESCAN” (Чехия) с детектором EDS OXFORD instruments X-MAS-80.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Эффекты микроабсорбционной неоднородности. Для снижения влияния эффекта обволакивания на интенсивность линии фтора необходимо оценить и по возможности устранить причины, его вызывающие. Изучили влияние на интенсивность линии FKα связующего вещества, часто применяемого при прессовании излучателей для РФА. В качестве такого компонента мы использовали полистирол (С8Н8). Соотношение по массе пробы к полистиролу составляло 5 : 1. Использовали пробы с высоким содержанием фтора: NaF и CaF2. В табл. 1 приведены средние значения интенсивностей линии FKα, полученные на таблетках после “сухого” истирания пробы с полистиролом (Iсух) и промывания пробы после “сухого” истирания (Iпр). В последнем варианте в растертую пробу добавляли этанол, пробу перемешивали в течение 5 мин стеклянной палочкой без приложения усилий, высушивали, собирали со стенок ступки и прессовали в таблетку. Так как в первых двух образцах отсутствуют другие компоненты, кроме фторсодержащих соединений, можно утверждать, что эффект обволакивания приводит к потере как минимум 10% интенсивности и обусловлен именно полистиролом. Вероятнее всего, это относится и к другим связующим, в частности к воску, который также широко применяют при прессовании таблеток [7, 10]. Аналогичные измерения для пробы горной породы с небольшим содержанием фтора (<1%) показали, что после промывания интенсивность увеличивается на 16%. Хотя в данном случае обволакивание фторсодержащих частиц может вызывать не только полистирол, но и компоненты, входящие в состав самой пробы, можно утверждать, что влияние эффекта обволакивания, обусловленного связующим веществом, значительно.
Таблица 1.
Интенсивности (тыс. имп/c) линии FКα после “сухого” истирания с добавлением полистирола и промывания этанолом после “сухого” истирания
| Вещество | Iсух. | Iпр. | Iпр/Iсух |
|---|---|---|---|
| NaF | 4.09 ± 0.13 | 4.64 ± 0.12 | 1.13 |
| CaF2 | 1.35 ± 0.06 | 1.46 ± 0.05 | 1.08 |
| Горная порода | 0.028 ± 0.002 | 0.033 ± 0.002 | 1.16 |
Смагунова и соавт. [11] использовали двухслойные таблетки на подложке из борной кислоты без применения связующего материала. Нами рассмотрены четыре способа приготовления двухслойных таблеток. В первом способе использовали “сухое” истирание порошковых проб; во втором – промывание пробы этанолом после ее “сухого” истирания; в третьем способе использовали “мокрое” истирание, в четвертом – промывание пробы после “мокрого” истирания. Оценили вклад, обусловленный эффектом обволакивания, в каждом из способов. Результаты сопоставления интенсивности FKα-линии, полученные для двух проб горных пород с различным содержанием фтора, представлены в табл. 2. Измерение для каждой пробы проводили три раза и использовали средние значения. Из табл. 2 следует, что изменение значений интенсивности линии фтора составляет от 0 до 12% (в зависимости от выбранного способа подготовки). Наименьшее влияние эффекта обволакивания соответствует второму и четвертому способам, т.е. промыванию этанолом после “сухого” или “мокрого” истирания. Следует отметить, что и в этих случаях нет гарантии полного отсутствия эффекта обволакивания, но снижение его влияния является бесспорным.
Таблица 2.
Интенсивности (тыс. имп/c) линии FKα при различных способах пробоподготовки и их отношение к интенсивности линии при “сухом” истирании пробы
| Проба | “Сухое” истирание | Промывание после “сухого” истирания | “Мокрое” истирание | Промывание после “мокрого” истирания | ||||
|---|---|---|---|---|---|---|---|---|
| Iсух | Icух/Icух | Iсух пр | Iсух пр/Iсух | Iмокр | Iмокр/Icух | Iмокр пр | Iмокр пр/Iсух | |
| 1 | 0.035 ± 0.003 | 1.0 | 0.037 ± 0.003 | 1.06 | 0.035 ± 0.002 | 1.0 | 0.038 ± 0.003 | 1.08 |
| 2 | 0.057 ± 0.004 | 1.0 | 0.064 ± 0.003 | 1.12 | 0.046 ± 0.003 | 0.81 | 0.064 ± 0.004 | 1.12 |
Таким образом, в данной работе экспериментально подтверждено наличие эффекта обволакивания при РФА фтора, который может приводить к уменьшению значения интенсивности линии FKα на 10% и более. Вклад эффекта можно снизить путем исключения связующего вещества и применения промывания пробы этанолом после ее истирания. Однако излучатели с использованием связующего вещества – более прочные и долговечные по сравнению с двухслойными таблетками, приготовленными на борной кислоте. В связи с этим мы использовали связующее вещество, но с промыванием пробы этанолом после ее “сухого” истирания. В этом случае отмечена наибольшая стабильность значений интенсивности флуоресцентного излучения фтора.
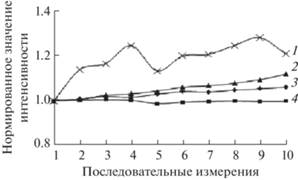
Погрешности измерения интенсивности и пробоподготовки. Для оценки влияния эффекта, связанного с изменением интенсивности линии FKα при многократном повторении измерений на прессованных таблетках, использовали схему экспериментов, предложенную в работе [10]. Выполнили по 10 измерений на прессованных таблетках из флюорита CaF2 (чистый природный с полистиролом), криолита Na3AlF6 (с полистиролом и без полистирола на борной кислоте) и СО апатитового концентрата (АК) (с полистиролом). Зависимости нормированных значений интенсивности FKα-линии (отношения к результату первого измерения) от порядкового номера измерения (измерения проведены в течение одного рабочего дня) приведены на рис. 1. Как и в работе [10], флюорит показывает стабильное значение интенсивности флуоресцентного излучения линии FKα; на апатите интенсивность увеличивается. На криолите интенсивность фтора не уменьшается, как в работе [10], а увеличивается со временем как на таблетках, спрессованных с полистиролом, так и без связующего вещества на борной кислоте.
Рис. 1.
Интенсивность линии FKα для десяти последовательных измерений апатита (1), криолита с полистиролом (2), криолита на борной кислоте (3) и флюорита (4).
Распределение фтора в поперечном разрезе таблетки после многократных экспозиций в рентгеновском спектрометре исследовали с помощью сканирующего электронного микроскопа. Использовали образец апатитового концентрата, содержащий 3.14% F, после увеличения интенсивности линии FKα на 20%. Съемку проводили в центре таблетки, на периферии и в средней точке. Все изображения оказались идентичными. Одно из них (в центре таблетки) приведено на рис. 2. Как видно, градиент распределения фтора от центра к поверхностям таблетки отсутствует, распределение фтора равномерно.
Рис. 2.
Изображение распределения фтора в поперечном сечении таблетки, полученное на сканирующем электронном микроскопе.

Проведенные исследования не позволяют сделать окончательный вывод о природе эффекта, связанного с изменением значений интенсивности при повторяющихся измерениях флуоресценции фтора. Однако его вклад в некоторых случаях составляет более 10%, что может приводить к систематическим погрешностям анализа. Использование первоначальных значений интенсивности, как показали наши эксперименты, позволяет добиться минимальной погрешности измерений. Кроме того, чтобы не подвергать пробу длительному облучению, сократили программу определения фтора (включены только “мешающие” петрогенные элементы), и ее продолжительность составила 6 мин.
В дальнейшем для оценки погрешности измерений использовали три первых значения измеренной интенсивности линии FKα (с учетом фона).
В общем случае случайная погрешность РФА определяется погрешностями пробоподготовки и измерения интенсивности на данном приборе при выбранных условиях анализа. Для расчета погрешностей, обусловленных действием этих факторов, и определения оптимального времени счета импульсов ни линии FKα нами выполнены измерения по схеме однофакторного дисперсионного анализа. Использовали прессованные таблетки с полистиролом (с промыванием). Так как в горных породах содержание фтора может меняться в довольно широких пределах, выбрали четыре диапазона, для каждого из которых подобрали соответствующий СО: 1) <0.05% (Му-3), 2) от 0.05 до 0.1% (ООКО-201), 3) от 0.1 до 1% (AP), 4) >1% (Mica-Mg). Из каждой пробы изготовили по три излучателя, и интенсивность линии FKα на каждом из них измеряли по три раза. Для проб первого и второго диапазонов измерения проводили при четырех временах экспозиции: 10, 20, 30 и 60 с, а для третьего и четвертого – при трех: 20, 30, 60 с. Выполнили расчеты для каждой экспозиции. Рассеяние значений интенсивности линии FKα вокруг ее среднего значения для каждой пробы определяет повторяемость (сходимость) измерений, связанную со статистической природой процесса и стабильностью работы аппаратуры, и характеризуется дисперсией $\left( {s_{{{\text{изм}}}}^{2}} \right)$ или коэффициентом вариации Vизм. Рассеяние средних значений интенсивностей, рассчитанных по трем измерениям каждой из трех параллельных проб, по отношению к их общему среднему определяет погрешность пробоподготовки и также характеризуется дисперсией $\left( {s_{{{\text{пп}}}}^{2}} \right)$ и коэффициентом вариации (Vпп) (табл. 3).
Таблица 3.
Результаты дисперсионного анализа
| Диапазон содержания фтора |
Время, с | $s_{{{\text{изм}}\,{\text{ср}}}}^{2}$ × 106 | Gmax | Vизм, % | s2пп × 107 | Vпп,% | V∑, % |
|---|---|---|---|---|---|---|---|
| <0.05% | 10 | 2.93 | 0.48 | 29.15 | 3.37 | 19.96 | 35.34 |
| 20 | 1.54 | 0.81 | 19.64 | 2.31 | 24.75 | 31.59 | |
| 30 | 1.07 | 0.49 | 17.98 | 0.225 | 20.81 | 27.52 | |
| 60 | 0.37 | 0.55 | 10.5 | 3.75 | 20.42 | 22.96 | |
| 0.05–0.1% | 10 | 0.846 | 0.65 | 10.86 | 1.41 | 14.34 | 17.99 |
| 20 | 0.435 | 0.58 | 7.5 | 1.69 | 12.43 | 14.52 | |
| 30 | 0.406 | 0.6 | 6.89 | 0.544 | 12.95 | 14.67 | |
| 60 | 0.398 | 0.57 | 6.0 | 0.609 | 12.63 | 13.68 | |
| 0.1–1% | 20 | 3.30 | 0.74 | 4.49 | 6.34 | 2.98 | 5.39 |
| 30 | 1.85 | 0.5 | 3.34 | 2.86 | 2.94 | 4.45 | |
| 60 | 0.537 | 0.66 | 1.81 | 7.03 | 2.96 | 3.48 | |
| >1% | 20 | 9.05 | 0.40 | 2.96 | 65.2 | 1.18 | 3.19 |
| 30 | 1.96 | 0.66 | 1.42 | 38.7 | 1.22 | 1.87 | |
| 60 | 1.51 | 0.66 | 1.29 | 61.7 | 1.25 | 1.79 |
Так как число степеней свободы, для которых рассчитывали дисперсии пробоподготовки для проб с различным содержанием фтора, при разных временах экспозиции одинаково (f = 2), то для оценки однородности этих дисперсий можно воспользоваться критерием Кохрена, который основан на законе распределения отношения максимальной выборочной дисперсии Gmax к сумме сравниваемых дисперсий:
Расчетное значение Gmax с учетом всех дисперсий выявило их однородность, так как Gmax = = 0.32 < Gmax (0.05, 2, 14) = 0.34. В этом случае для расчета коэффициента вариации, характеризующего погрешность пробоподготовки в каждом конкретном случае, можно использовать общее среднее значение дисперсий для данных измерений $s_{{{\text{ср}}}}^{2}$ = 1.563 × 10–6. Очевидно, что эта погрешность, выраженная в относительных единицах (коэффициент вариации), значительна для проб первого и второго диапазонов. Это объясняется низкой скоростью счета интенсивности при содержании фтора менее 0.1%, которая составляет единицы и десятки импульсов в секунду.
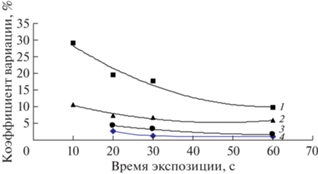
Однородность дисперсий, характеризующих погрешность измерения интенсивности, оценивали для всех СО в отдельности для каждой из экспозиций. По критерию Кохрена сравнивали дисперсии, получение для каждой из трех параллельных проб. Полученные расчетные значения Gmax представлены в табл. 4. Так как все Gmax < < Gmax (0.05, 2, 3) = 0.87, то сравниваемые дисперсии однородны, т.е. можно использовать среднее значение дисперсии для всех интервала содержания и всех экспозиций. В табл. 4 представлены средние значения дисперсий и коэффициентов вариации, характеризующие погрешность измерения интенсивности при различных временах измерений для всех СО. На рис. 3 показана зависимость коэффициента вариации (Vизм) от времени экспозиции для проб с различным содержанием фтора. Очевидно, что для проб с содержанием фтора более 0.1% можно ограничиться экспозицией 40 с, так как погрешность измерения интенсивности принимает постоянное значение. Для более низких содержаний: <0.1% и в особенности <0.05% время измерения должно составлять не менее 60 с. Эту экспозицию выбрали для программы определения фтора в образцах горных пород.
Таблица 4.
Случайные погрешности анализа и их суммы для фтора при экспозиции 60 с
| Содержание фтора, % |
СО | Vизм, % | Vпп, % | V∑, % | Vдоп, % |
|---|---|---|---|---|---|
| <0.05 | Mу-3 | 10.5 | 20.42 | 22.96 | 20 |
| 0.05–0.1 | ООКО-201 | 6.0 | 12.63 | 13.68 | 17 |
| 0.1–1.0 | AP-1 | 1.81 | 2.96 | 3.48 | 14–8 |
| >1.0 | MICA-MG | 1.29 | 1.25 | 1.79 | 8–6.5 |
Рис. 3.
Зависимость погрешности измерения интенсивности (Vизм, %) от времени экспозиции. 1 – Му-3, 2 – ООКО-201, 3 – АР, 4 – Mica-Mg.
Общая случайная погрешность анализа определяется как сумма дисперсий:
или в относительных единицах:В табл. 4 приведены значения суммарных и допустимых (Vдоп), регламентируемых для анализов третьей категории классификации [15], коэффициентов вариации. Видно, что при содержании фтора >0.1% случайные погрешности анализа, обусловленные пробоподготовкой и статистической природой интенсивности рентгеновского излучения, не превосходят допустимые. Следует еще раз заметить, что высокие погрешности измерений и пробоподготовки для низких содержаний фтора обусловлены низкими значениями скорости счета и связаны со статистическим характером излучения. Чтобы снизить эти погрешности, можно увеличить диаметр излучателя. Но это, в свою очередь, потребует более значительного количества материала как стандартных образцов, так и проб.
Погрешности анализа, связанные с градуировкой. Погрешность определения элементов рентгенофлуоресцентным методом, которая зависит от качества полученной градуировочной зависимости, определяется отклонением средних результатов анализа от их истинного значения. Для учета влияния матрицы можно использовать способ α-коррекции, алгоритм которого входит в пакет программ SUPER Q. Содержания компонентов рассчитывают по формулам:
где Ii испр = Ii(1 + $\sum {{{\alpha }_{{im}}}{{I}_{m}}} $); ci – содержание i-го элемента в пробе; Ii – измеренная интенсивность характеристического излучения i-го элемента (за вычетом фона); Im – измеренные интенсивности “мешающих” элементов, входящих в состав пробы; bi, di и αim – коэффициенты, определяемые с помощью стандартов. Программа SUPER Q позволяет также учитывать наложение линий.Интенсивность линии FKα существенно зависит от присутствующих в пробе петрогенных элементов и их содержания. Матричный эффект в этом случае, в первую очередь, связан с возбуждением линии фтора как флуоресцентным излучением этих элементов, так и их фото- и Оже-электронами. Этот эффект играет более значительную роль по сравнению с эффектом ослабления излучения. Влияние этих двух разнонаправленных факторов можно учесть с помощью уравнения α-коррекции. Способ же фундаментальных параметров не учитывает особенности возбуждения флуоресцентного излучения фтора и не может быть использован при его определении как в прессованных таблеток, так и в сплавленных образцах.
И, наконец, как отмечено выше, выход флуоресценции фтора зависит от вида соединения, в состав которого он входит, т.е. от фторсодержащего минерала в горной породе. И этот фактор невозможно учесть с помощью единого градуировочного уравнения. Тем не менее, используя имеющиеся СО с известным содержанием фтора, мы получили градуировочную зависимость, погрешность которой оценивается средним стандартным отклонением (s) 0.02%. Предел обнаружения составил 0.05%.
Надо признать, что при определении фтора в горных породах с применением полученной градуировки не может быть полной уверенности в достоверности полученных результатов. Использовали стандартные образцы различной природы, но, к сожалению, их минеральный состав, включая фторсодержащие минералы, как правило, неизвестен.
Выше отмечено, что многие авторы приходили к выводу о необходимости построения отдельных градуировок для каждого фторсодержащего минерала при использовании прессованных таблеток. Нами получены градуировки для основных фторсодержащих соединений: природного флюорита, стандартного образца апатита (АК) и синтетического криолита. В качестве матриц использовали базальт (с содержанием фтора ниже пределов обнаружения) и кремнезем. Содержание фтора в пробах менялось от 0.1 до 5%. С помощью уравнения α-коррекции для флюорита и апатита получили единое градуировочное уравнение для двух вмещающих матриц. Использовали поправки на Mg, Si, P, S и учли наложение линии FeLα. Погрешность градуировки составила 0.18%. Градуировка для криолита, входящего в состав также двух матриц, характеризуется значением s 0.11%. Использовали поправку на натрий и учли наложение линии FeLα. Полученные графики представлены на рис. 4.
Рис. 4.
Градуировочные зависимости для определения фтора, полученные на образцах криолита (1) и флюорита с апатитом (2).
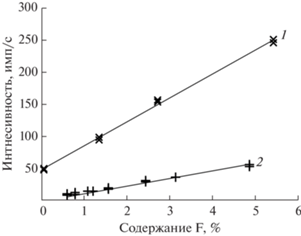
Получить общую градуировочную зависимость для всех трех соединений оказалось невозможным. Чтобы воспользоваться градуировками для этих основных фторсодержащих минералов, необходима информация о том, какой из них входит в состав пробы. Кроме того, помимо этих основных фторсодержащих минералов имеется достаточно большое количество других.
* * *
Таким образом, определение фтора в горных породах из прессованных таблеток с помощью РФА на данный момент является трудной задачей. Значительна погрешность, связанная с эффектом обволакивания. Наличие этого эффекта нельзя заранее предсказать и невозможно от него полностью освободиться, но можно снизить его влияние, промывая перед прессованием пробы в этаноле. Погрешности анализа, связанные с низким выходом флуоресценции в длинноволновой области спектра, а также влияние возможных трендов в изменении значений интенсивности при повторных измерениях линии FKα могут быть в какой-то степени минимизированы. Однако получение единого градуировочного уравнения для всех типов горных пород, в состав которых входят разные фторсодержащие минералы, невозможно. Это связано с необходимостью учета вида химического соединения, в состав которого входит фтор, так как этим определяется выход его флуоресценции. Этот эффект невозможно учесть, используя существующие способы расчета содержаний по данным РФА, в том числе эмпирическую α-коррекцию и способ фундаментальных параметров. Для каждого соединения фтора требуется построение отдельной градуировочной зависимости. Для этого необходимо располагать соответствующими наборами СО, а при определении фтора в пробе горной породы – информацией о ее минеральном составе, что не всегда представляется возможным. Кроме того, если в состав пробы входят различные фторсодержащие минералы, то задача определения фтора из прессованных таблеток становится еще более сложной.
Список литературы
Когарко Л.Н., Кригман Л.Д. Фтор в силикатных расплавах и магмах. М.: Наука, 1984. 125 с.
Виноградов А.П. Среднее содержание химических элементов в горных породах // Геохимия. 1962. № 7. С. 555.
Sato K., Tanaka I., Otssuki T. X-ray fluorescence analysis in the X-ray region of 0.4 to 7 nm: Application to determination of fluorine in slags // X-Ray Spectrom. 1979. V. 8. № 9. P. 68.
Симаков В.А. Рентгенофлуоресцентный аналих руд и продуктов их переработки при разведке месторождений. Дис. … докт. техн. наук. Москва, ВИМС, 2000. 252 с.
Gazulla F., Rodrigo M., Orduña M., Ventura M.J. Fluorine determination in glasses and glazes by WD-XRF // Eur. J. Glass Sci. Technol. A: Glass Technol. 2015. V. 56. № 3. P. 95.
Jecht U. Quantitative fluorine determination by X-ray fluorescence analysis // Chemiker-Zeitung. 1972. V. 96. № 2. P. 104.
Schäfer H.P., Meduna J.U. Routine determination of fluorine by XRF-analysis // Fresenius Z. Anal. Chem. 1987. V. 326. № 6. P. 558.
Iordanis A., Georgakopoulos A., Fillippidis A., Kassoli–Fournraki A. A correlation study of elements in lignite and fly ash generated in a power station // Int. J. Environ. Anal. Chem. 2001. V. 79. № 2. P. 133.
Gazulla M.F., Gomez M.P., Barba A., Orduña M. Chemical characterisation of geological raw materials used in traditional ceramic // Geostand. Geoanal. Res. 2004. V. 28. № 2. P. 203.
Pleßow A. X-ray-induced alteration of specimens as crucial obstacle in XRF spectrometry of fluorine in rocks and soils // X-ray Spertrom. 2013. V. 42. № 1. P. 19.
Смагунова A.Н., Широколобова И.С., Козлов В.А., Коржова Е.Н., Кривченко О.С., Шаулина Л.П., Финкельштейн А.Л. Разработка стандартного образца предприятия состава цементной сырьевой смеси // Стандартные образцы. 2014. № 3. С. 92.
Коржова Е.Н., Ставицкая М.В., Белозерова О.Ю. Хаптагаева Е.А., Смагунова А.Н. Изучение эффекта микроабсорбционной неоднородности при рентгенофлуоресцентном анализе ультрамелких частиц // Журн. аналит. химии. 2011. Т. 66. № 2. С. 175. (Korzhova E.N., Stavitskaya M.V., Belozerova O.Yu., Khaptagaeva E.A., Smagunova A.N. Effect of microabsorption heterogeneity in the X-ray fluorescence analysis of ultrafine particles // J. Anal. Chem. 2011. V. 66. № 2. P. 171.)
Boča M. Gurišová V., Šimko F. Some aspects of the wavelength dispersive X-ray determination of fluorine content in various matrices // J. Appl. Spectrosc. 2017. V. 84. №. 2. P. 324.
Кузьмина Т.Г., Тронева М.А., Ромашова Т.В. Роль петрогенных элементов в возбуждении рентгеновской флуоресценции фтора // Журн. аналит. химии. 2020. Т. 75. № 7. С. 636. (Kuz’mina T.G., Troneva M.A., Romashova T.V. role of petrogenic elements in the excitation of X-Ray fluorescence of fluorine // J. Anal. Chem. 2020. V. 75. № 7. P. 896.)
ОСТ 41-08-205-99. Стандарт отрасли. Методики количественного химического анализа. М.: ВИМС, 1999. 96 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии