

Журнал аналитической химии, 2022, T. 77, № 9, стр. 788-797
Спектрофотометрическое определение азорубина и понсо 4r в лекарственных препаратах с применением мицеллярной экстракции
Н. Б. Шестопалова a, *, Ю. А. Фомина a, Р. К. Чернова b
a Саратовский государственный медицинский университет
им. В.И. Разумовского Минздрава России, фармацевтический факультет
410012 Саратов,
ул. Большая Казачья, 112, Россия
b Саратовский государственный университет имени Н.Г. Чернышевского, Институт химии
410012 Саратов, ул. Астраханская, 183, Россия
* E-mail: shestopalovanb@yandex.ru
Поступила в редакцию 20.09.2021
После доработки 13.11.2021
Принята к публикации 15.11.2021
- EDN: MJDUCI
- DOI: 10.31857/S0044450222080138
Аннотация
Проведено сравнение возможностей экстракционных систем на основе полиоксиэтилированных алкилфенолов Тритона Х-100 и ОП-10 для извлечения синтетических пищевых красителей Азорубина и Понсо 4R при различных режимах фазового разделения: температуре помутнения и высаливании сульфатом натрия. Изучено влияние температуры, рН, концентрации ПАВ и электролита на физико-химические характеристики мицеллярной фазы и скорость установления фазового равновесия. Предложен простой и эффективный способ проведения экстракционной процедуры при “высаливании”. Определены оптимальные условия максимального извлечения красителей (R > 98%) в различных режимах: при температуре помутнения – Азорубина, при высаливании – Понсо 4R. Разработаны методики спектрофотометрического определения аналитов в сиропах и пастилках для рассасывания с предварительным мицеллярно-экстракционным концентрированием.
Современные лекарственные препараты (ЛП) представляют собой сложные терапевтические системы, содержащие наряду с действующими фармакологически активными веществами различные вспомогательные компоненты [1]. Синтетические пищевые красители (СПК) применяют для придания нужного цвета, обозначения терапевтической группы, маркировки дозы в сиропах, таблетках, капсулах, суспензиях [2]. Использование СПК в фармацевтической отрасли технологически и экономически оправдано их стабильностью при производстве и хранении, фото-, термо- и кислотоустойчивостью, низкой стоимостью [3].
Необходимость аналитического контроля содержания СПК в ЛП обусловлена высокими требованиями, предъявляемыми к безопасности не только действующих, но и вспомогательных веществ. Однако в Государственной Фармакопее РФ отсутствуют единые утвержденные методики определения СПК в лекарственных формах [4], несмотря на их возможное негативное влияние на здоровье человека [5–8], а содержание нормировано только в различной пищевой продукции [9]. Это становится особенно важным при регулярном приеме лекарств. На фармацевтическом рынке широко представлены препараты, в составе которых часто встречаются азокрасители Желтый “солнечный закат” FCF, Азорубин, Понсо 4R, Красный очаровательный АС [10], которые могут восстанавливаться до токсичных аминов [5].
Широко доступным и аппаратурно оснащенным методом, удовлетворяющим требованиям контроля качества фармацевтической продукции, является спектрофотометрия [11]. Однако применение этого метода для анализа углеводсодержащих ЛП (сиропы, таблетки и пастилки для рассасывания) затруднено оптической непрозрачностью растворов; требуется обязательная стадия предварительного отделения целевых аналитов от матрицы сложного состава. В связи с этим разработка простых и доступных подходов к эффективному извлечению СПК из ЛП, отвечающих современным требованиям экологической безопасности, экономической целесообразности и совместимых со спектрофотометрическим методом, является актуальной задачей.
Практический интерес представляют экстракционные системы на основе нетоксичных, малолетучих неионных поверхностно-активных веществ (нПАВ) как экологически безопасная альтернатива жидкостно−жидкостной экстракции органическими растворителями, соответствующая принципам “зеленой химии” [12]. В качестве экстрагентов в основном применяют полиоксиэтилированные алкилфенолы (Тритон Х-100, Тритон Х-114) и их промышленные аналоги (ОП-7, ОП-10), водные растворы которых образуют двухфазные системы при достижении определенной температуры (температуры помутнения, tпом) [13] или добавлении электролитов (“высаливание”) [12]. Выделяют два вида экстракции в зависимости от фактора, приводящего к разделению фаз в водных растворах указанных соединений. Если фазообразование происходит под действием температуры, то говорят об экстракции “в точке помутнения” (cloud point extraction, CPE) [14, 15], если только под действием неорганических солей – о водных двухфазных системах (aqueous biphasic systems, aqueous two-phase systems, ATPS) [16]. В последнем случае расслоение происходит при более низких температурах – 20–25°С.
Применению ПАВ-экстрагентов для извлечения СПК с последующим спектрофотометрическим определением посвящено ограниченное число работ, причем только с использованием технологии СРЕ [17–22]. Отсутствие систематических исследований не позволяет объективно оценить эффективность экстракционных систем в отношении этих аналитов, которая зависит не только от природы экстрагентов и извлекаемых соединений, но и от условий проведения экстракционной процедуры [15].
Цель настоящей работы состояла в сравнении эффективности экстракционных систем на основе Тритона Х-100 и ОП-10 для извлечения Азорубина и Понсо 4R по технологиям экстракции “в точке помутнения“ и "высаливания”; оптимизации проведения экстракционной процедуры при различных условиях фазового разделения; разработке простых и экономичных методик определения синтетических красителей в лекарственных препаратах экстракционно-спектрофотометрическим методом.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и аппаратура. Использовали Азорубин (Е122) (Sigma-Aldrich, США) и Понсо 4R (Е124) (Sigma-Aldrich, США); неионные ПАВ: Тритон Х-100 (Sigma-Aldrich, США) и ОП-10 (Реахим, Россия). Исходные растворы красителей (0.1 г/л) и нПАВ (20%) готовили растворением точных навесок в дистиллированной воде. Рабочие растворы СПК (1–10 мг/л) и нПАВ (1–10%) готовили разбавлением исходных растворов дистиллированной водой. В качестве высаливателя использовали безводный Na2SO4 (ч.д.а.), предварительно просушенный до постоянной массы, и его 1 М раствор, приготовленный растворением точной навески в дистиллированной воде. Необходимое значение рН создавали добавлением 0.1 М растворов НСl и NaOH.
Электронные спектры поглощения регистрировали на спектрофотометре SHIMADZU UV-1800 (Япония) с программным обеспечением UVProbe-2.31 в диапазоне 350–750 нм, l = 1 см. Кислотность среды контролировали рН-метром рХ-150 МП со стеклянным электродом и хлорсеребряным электродом сравнения. Взвешивание проводили на аналитических весах HR-200 (A&D, Япония, ±0.0001 г).
Хроматографическое определение проводили по методике [23] на высокоэффективном хроматографе SHIMADZU LC-20A (Япония) в обращенно-фазовом варианте с диодно-матричным детектором (UV SPD-20A). Для анализа применяли колонку Supelco LC 18 (150 × 4.66 мм) с размером частиц 5 мкм. В качестве подвижной фазы использовали смесь 0.05 М фосфатного буферного раствора (рН 4.5) с ацетонитрилом (80 : 20, по объему); скорость подачи элюента – 1 мл/мин; длина волны детектирования – 500 нм.
Методика эксперимента. Оптимальные параметры фазового расслоения определяли в двух режимах для систем состава нПАВ–Н2О и нПАВ–Н2О–Na2SO4, варьируя температуру, концентрации ПАВ и соли при постоянном объеме раствора – 10 мл. Межфазное распределение СПК контролировали спектрофотометрически. Эффективность экстракции оценивали по величине степени извлечения (R, %) [24], экстракционную процедуру – по объемам образующихся фаз, вязкости мицеллярной фазы и скорости ее формирования.
Режим 1, соответствующий технологии CPE: градуированные пробирки, содержащие необходимые компоненты (аликвоты 20%-ного раствора нПАВ, исходного раствора СПК, 1 М раствора Na2SO4 и дистиллированная вода до общего объема 10 мл) помещали в водяную баню и нагревали до необходимой температуры помутнения или расслоения, которую контролировали термометром. После охлаждения раствора фазы разделяли декантацией. На каждом этапе измеряли объемы образующихся фаз. Спектры поглощения мицеллярной фазы регистрировали после ее разбавления дистиллированной водой до 5 мл при нагревании до 40–50°С.
Режим 2, соответствующий технологии ATPS: в градуированные пробирки помещали необходимые компоненты: аликвоты исходных растворов нПАВ и красителя; навеску сухого безводного Na2SO4, необходимую для фазового разделения; дистиллированную воду до общего объема 10 мл. Полученную смесь тщательно перемешивали до полного растворения соли и выдерживали на водяной бане при 25°С 30 мин. По мере установления фазового равновесия фиксировали объемы образующихся фаз, которые в дальнейшем разделяли. Содержание красителей в мицеллярной фазе определяли после ее разбавления до 5 мл дистиллированной водой и измерения оптической плотности полученного раствора.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Основными этапами экстракционной процедуры с помощью нПАВ являются: растворение объекта анализа, содержащего аналит, в водном растворе нПАВ с концентрацией выше критической концентрации мицеллообразования (ККМ); инициирование фазового расслоения; отделение ПАВ-обогащенной фазы (мицеллярной фазы, МФ) с извлеченным аналитом от водной фазы (ВФ) с концентрацией нПАВ ниже ККМ; разбавление экстракта для последующего инструментального анализа [17–22]. Для аналитических целей на стадии фазового расслоения важное значение имеют скорость формирования фазы и ее объем, вязкость и компактность, а также ее оптическая прозрачность в УФ- и видимой областях спектра. Выбранные в качестве экстрагентов оксиэтилированные производные алкилфенолов, Тритон Х-100 и его промышленный аналог ОП-10 с общей формулой СnH2n+1С6Н4O(C2H4O)mH, имеют в структуре ароматический фрагмент, что приводит к поглощению в УФ-области, а при высокой концентрации ПАВ в МФ – увеличению оптической плотности ее водных растворов. Отсутствие поглощения выше 350 нм позволяет применять эти нПАВ для спектрофотометрического определения окрашенных соединений. Время установления фазового равновесия и физико-химические характеристики образующихся фаз зависят от факторов, приводящих к расслоению, поэтому оптимальные условия определяли для каждого режима; в качестве модельного соединения выбрали ОП-10.
Оптимизация параметров фазового расслоения при экстракции “в точке помутнения”. Индивидуальной характеристикой исследуемых нПАВ является температура помутнения (tпом) водных растворов (система нПАВ–Н2О), зависящая от строения углеводородного радикала и длины оксиэтиленовой цепи, концентрации ПАВ, присутствия различных добавок. Повышение температуры приводит к уменьшению прочности водородных связей между молекулами воды и оксиэтильными группами, дегидратации мицелл нПАВ и понижению их растворимости и, как следствие, к выделению в виде отдельной фазы. Однако достижение tпом на практике не приводит к расслоению системы и при охлаждении раствор снова становится гомогенным. Формирование компактной МФ, локализующейся в нижней части раствора, происходит только при нагревании до температуры выше tпом на 3–5°С (температура расслоения, tрас) [20]. Диапазоны температур, при которых наблюдается образование двухфазных систем исследуемых нПАВ (1–10% растворы), приведены в табл. 1. Растворы ОП-10 характеризуются более высокими tпом и tрас, чем Тритона Х-100, поскольку ОП-10 представляет смесь полимергомологов с различными примесями, образующимися в результате синтеза. Определение tпом водных растворов нескольких партий ОП-10, отличающихся датой изготовления и сроком хранения, показало, что варьирование основных физико-химических параметров фазового расслоения незначительно. Общими закономерностями для обоих нПАВ являются: уменьшение tрас, увеличение объема и скорости формирования МФ при увеличении концентрации нПАВ. Однако отделение МФ декантацией затруднительно вследствие ее невысокой вязкости.
Таблица 1.
Некоторые характеристики исследованных растворов неионных поверхностно-активных веществ
| нПАВ (содержание основного вещества) | n | m | tпом, °С | tрас, °С | ||
|---|---|---|---|---|---|---|
| водный раствор | при добавлении 0.2 М Na2SO4 | водный раствор | при добавлении 0.2 М Na2SO4 | |||
| Тритон Х-100 (98%) | 8–9 | 10 | 70–80 | 55–65 | 75–85 | 58–68 |
| ОП-10 (80%) | 8–10 | 10–12 | 80–90 | 65–75 | 85–95 | 69–79 |
Ранее установлено [21], что добавление в систему ОП-10–Н2О неорганических солей приводит к снижению tпом, причем наибольшее влияние оказывают соли щелочных металлов (фосфаты, карбонаты, сульфаты), особенно соли натрия. Изучение влияния Na2SO4 на параметры фазового расслоения 10%-ного раствора ОП-10 (система нПАВ–Н2О–Na2SO4) в диапазоне концентраций соли 0.1–0.5 М показало, что увеличение содержания электролита приводит не только к значительному уменьшению tпом (табл. 1), но и к образованию меньшей по объему и более вязкой МФ. Это объясняется дополнительной дегидратацией полиоксиэтиленовых цепей за счет гидратации ионов вводимых солей, т.е. “высаливающим” эффектом. Количество введенного в систему электролита влияет на локализацию МФ: при 0–0.2 М – в нижней части раствора, при 0.3–0.4 М – распределена во всем объеме, при 0.5 М – в верхней части раствора. Таким образом, в качестве оптимальной для дальнейших исследований выбрали концентрациию 0.2 М Na2SO4, при которой tпом и tрас 10%-ного раствора ОП-10 составляли соответственно (66 ± 1) и (69 ± 1)°С.
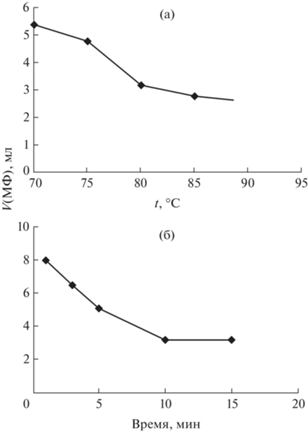
Установили, что температура влияет на скорость формирования и физико-химические характеристики МФ. Изучение влияния режима нагрева и охлаждения показало, что выдерживание в течение 5 мин водно-солевого раствора ОП-10 при температурах от 70 до 90°С приводит к уменьшению объема МФ (рис. 1а), а выдерживание при фиксированной температуре – установлению фазового равновесия в течение 10–15 мин (рис. 1б). При нагревании раствора до tрас со скоростью 1°С/мин с последующим быстрым охлаждением при 15°С формируется МФ объемом (3.2 ± 0.2) мл (из 10%-ного раствора ОП-10), а оптимальным является выдерживание растворов при 80°C в течение 10 мин также с последующим быстрым охлаждением, приводящее к образованию более вязкой, гелеобразной МФ объемом (2.8 ± 0.2) мл, не смешивающейся с ВФ в течение длительного времени и легко отделяющейся от нее декантацией. Однако высокая вязкость МФ не позволяет непосредственно измерять ее оптическую плотность, необходимы разбавление и гомогенизация при нагревании до 40–50°C.
Рис. 1.
Зависимость объема мицеллярной фазы в системе ОП-10–Н2О–Na2SO4 от температуры (а) и времени экспозиции (б) при 80°С. с(ОП-10) = 10%, с(Na2SO4) = 0.2 М, V0 = 10 мл.
Оптимизация параметров фазового расслоения при технологии ATPS. Единственным фактором, вызывающим расслоение при 20–25°С, является добавление сильного электролита – высаливателя. Ранее нами установлено [25], что добавление некоторых солей натрия вызывает расслоение водных растворов ОП-10 с концентрацией от 0.5 до 25% при 25°С; получен лиотропный ряд анионов-высаливателей: ${\text{PO}}_{4}^{{3 - }}$ > ${\text{HPO}}_{4}^{{2 - }}$ > ${\text{CO}}_{3}^{{2 - }}$ > ${\text{SO}}_{4}^{{2 - }}$ > > ${{{\text{H}}}_{{\text{2}}}}{\text{PO}}_{4}^{ - }$ > ${\text{HSO}}_{4}^{ - }$. Для выбранного высаливателя Na2SO4 расслоение 1%-ного раствора ОП-10 наблюдается при содержании соли в системе около 9%, расслоение 10%-ного раствора ОП-10 требует около 7%. Следует отметить, что при “высаливании” ПАВ-обогащенная фаза локализуется компактно в верхней части раствора; водная фаза, обогащенная солью, – в нижней части. Образующиеся МФ в режиме ATPS имеют несколько больший объем и меньшую вязкость по сравнению с режимом СРЕ, что можно объяснить наличием только “высаливания” в качестве фактора, дегидратирующего оксиэтиленовые цепи.
Помутнение раствора при соответствующих соотношениях нПАВ и соли (система нПАВ–Н2О–Na2SO4), установленное методом изотермического титрования, не приводит сразу к расслоению в системе, для некоторых концентраций его фиксировали только спустя сутки. Помутнение при высаливании также как и при СРЕ является обратимым процессом, понижение температуры приводит к гомогенизации системы. В связи с этим представляло интерес оценить влияние концентрации соли на фазовое расслоение. Исследование проводили для 10%-ного раствора ОП-10, помутнение которого наблюдается при добавлении 0.72 г Na2SO4. Установлено, что увеличение массы соли до 0.84 г приводит к уменьшению времени начала расслоения и более быстрому формированию МФ с меньшим объемом. Как видно из рис. 2, быстрее всего фазовое равновесие устанавливается при добавлении 0.80–0.84 г соли. При этом объем МФ остается практически неизменным спустя 5 мин после смешивания компонентов, а вязкость МФ позволяет разбавлять ее дистиллированной водой без дополнительного нагревания, что является оптимальным для этого режима. Увеличение массы добавляемой соли приводит к образованию мутной МФ.
Рис. 2.
Зависимость объема мицеллярной фазы от времени в системе ОП-10–Н2О–Na2SO4 при разных массах соли, г: (♦) – 0.72, (◻) – 0.74, (Δ) – 0.75, (■) – 0.84. t = 25°С; с(ОП-10) = 10%, V0 = 10 мл.

Основной трудностью при “высаливании” является разделение фаз, так как МФ с аналитом локализуется в верхней части раствора и отделение ее может приводить к смешиванию с ВФ. Для решения этой проблемы нами предложен способ проведения экстракционной процедуры “в шприцах” (“in-syringe”), схема которого представлена на рис. 3. В пробирку № 1 помещают исследуемый раствор, содержащий аналит, необходимое для расслоения количество соли, предварительно взвешенное с точностью ±0.0001 г и добавляют дистиллированной воды до общего объема 5 мл. Полученную смесь тщательно перемешивают до полного растворения соли и набирают в шприц объемом 10 мл. В пробирку № 2 помещают 5 мл раствора нПАВ необходимой концентрации и набирают в тот же шприц. Тщательно перемешивают содержимое шприца интенсивными движениями вверх–вниз в течение 1–2 мин до помутнения. Затем помещают шприц в водяную баню с контролируемой температурой 25°С и выдерживают необходимое время. После установления фазового равновесия верхний слой – ПАВ-обогащенная фаза с извлеченным аналитом, нижний слой – водная фаза. Разделение фаз проводят последовательно: сначала водную, а затем мицеллярную фазу удаляют из шприца в градуированные пробирки, измеряют их объемы. Для дальнейшего проведения инструментального анализа МФ разбавляют до необходимого объема подходящим растворителем.

Следует отметить, что такой способ проведения экстракции в режиме ATPS позволяет варьировать общий объем раствора, объемы солевого и ПАВ-раствора. Определяющим является соотношение ПАВ : соль, необходимое для образования двухфазной системы, которое должно быть предварительно установлено. Граница между фазами в прозрачных шприцах визуально хорошо фиксируется, при разделении фазы не перемешиваются. Дальнейшие исследования в режиме ATPS проводили предложенным способом.
Оценка эффективности экстракционных систем. Переход аналитов в МФ зависит от ряда факторов: природы аналита, рН среды, концентрации ПАВ, количества введенного в систему электролита. Азорубин и Понсо 4R относятся к сульфированным производным моноазокрасителей, в молекулах которых нафталиновые кольца связаны азогруппой, различаются только числом и положением сульфогрупп (схема 1 ).
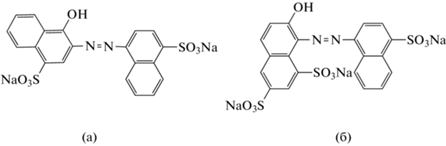
Схема 1 . Структурные формулы синтетических пищевых красителей Азорубина (а) и Понсо 4R (б).
Электронные спектры поглощения водных растворов (рис. 4а, 4б, спектр 1) в диапазоне 350−750 нм характеризуются наличием одной широкой полосы. Несмотря на одинаковый хромофорный фрагмент, форма полос и максимумы поглощения отличаются, что невозможно объяснить только числом сульфогрупп, которые отвечают в основном за растворимость соединений. Положение максимума поглощения Понсо 4R в более коротковолновой области (λmах = 507 нм), вероятно, связано с пространственным расположением азо- и одной из сульфогрупп, выведением фрагмента молекулы из плоскости сопряжения и затруднением π → π* перехода. В молекуле Азорубина нафталиновые ядра могут находится в одной плоскости с π-электронами азогруппы и образовывать единое электронное облако, что приводит к удлинению цепи сопряжения и сдвигу максимума поглощения в более длинноволновую область (λmах = 516 нм).
Рис 4.
Электронные спектры поглощения растворов Азорубина (а) и Понсо 4R (б) при различной кислотности среды и в присутствии неионного ПАВ: 1 – рН 6–8, 2 – рН 11–12, 3 – 10%-ный раствор ОП-10.
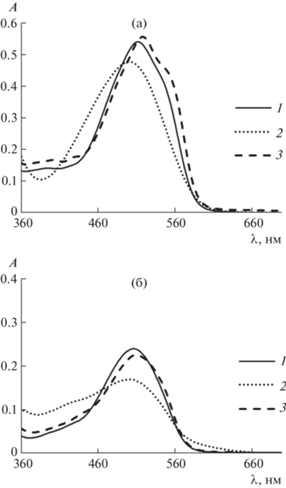
Изучение влияния кислотности среды на состояние исследуемых СПК в водном растворе показало, что при рН 1–10 они диссоциирует с образованием 2-зарядного (Е122) и 3-зарядного (Е124) анионов, форма электронных спектров поглощения остается практически неизменной. При рН > > 10 наблюдается гипсохромный сдвиг, изменение формы спектра и интенсивности поглощения, что связано с диссоциацией ОН-группы, приводящей к образованию 3-зарядного (Е122) и 4-зарядного (Е124) анионов (рис. 4а, 4б, спектр 2).
Изучение влияние мицелл ПАВ на состояние СПК в растворе (рН 6–8) показало небольшие батохромные сдвиги полос поглощения при сохранении формы спектров (рис. 4а, 4б, спектр 3). Изменение спектральных характеристик может свидетельствовать о взаимодействии функциональных групп красителей, сопряженных с их хромофорной системой (группа –ОН), с атомами кислорода полиоксиэтиленовых цепей нПАВ.
Оценку эффективности технологии экстракции “в точке помутнения” проводили в системах нПАВ–Н2О и нПАВ–Н2О–Na2SO4 для обоих экстрагентов при варьировании концентрации ПАВ, высаливателя и кислотности среды. Изучение влияния рН в диапазоне 1–13 показало, что добавление соответствующих количеств НСl, NaOH и красителей не изменяют параметры фазового расслоения. Системы состава нПАВ–Н2О в диапазоне рН 1–9 извлекают из водных растворов до 86% Е122 и 72% Е124 (табл. 2). При наличии в структуре одинаковых гидрофобных фрагментов такое различие в степенях извлечения может быть связано с разными зарядами анионов красителей: чем выше заряд, тем ниже степень извлечения. Это подтверждается уменьшением R до 79 и 65% соответственно для Е122 и Е124 при рН > 10, поскольку в этих условиях увеличивается заряд ионной формы каждого красителя.
Таблица 2.
Степени извлечения (%) Азорубина и Понсо 4R при различных режимах фазового расслоения (с(СПК) = 5 мг/л, с(ПАВ) = 10%, рН 6–8, n = 3, P = 0.95)
| Экстракционная система | Азорубин | Понсо 4R | ||
|---|---|---|---|---|
| СРЕ | ATPS | СРЕ | ATPS | |
| ОП-10–Н2О | 84 ± 2 | – | 70 ± 1 | – |
| Тритон Х-100–Н2О | 86 ± 1 | – | 72 ± 2 | – |
| ОП-10–Н2О–Na2SO4 | 96 ± 2 | 97 ± 1 | 83 ± 3 | 95 ± 1 |
| Тритон Х-100–Н2О–Na2SO4 | 97 ± 1 | 98 ± 1 | 85 ± 2 | 96 ± 2 |
Существенное влияние на эффективность извлечения оказывает концентрация высаливателя. Добавление Na2SO4 в концентрации 0.2 М увеличивает R для Е122 до 98%, а для Е124 – до 83%, что может быть связано с усилением гидрофобных взаимодействий ПАВ–СПК. Аналогичное влияние оказывает и температура, увеличение которой от 70 до 90°С приводит к более полному переходу СПК в МФ; при температуре 80°С и выше достигается максимально возможное значение R для каждого красителя.
Изучение влияния концентрации нПАВ в растворе (от 1 до 10%) показало, что в более концентрированных растворах R выше, т.е. большее количество мицелл способно солюбилизировать большее количество красителя (рис. 5). Соответственно, увеличение концентрации красителя приводит к уменьшению R, поэтому экстракцию целесообразнее проводить из разбавленных растворов более концентрированными растворами нПАВ. Следует отметить, что экстракционные возможности ОП-10 и Тритона Х-100 практически равнозначны.
Рис. 5.
Зависимость степени извлечения красителей Азорубина (1, 2) и Понсо 4R (3) от концентрации неионных ПАВ: 1 – Тритон Х-100; 2, 3 – ОП-10. с(СПК) = 5 мг/л, с(Na2SO4) = 0.2 М.

Оценку эффективности технологии высаливания проводили для наиболее гидрофильного Е124 при варьировании концентрации ОП-10, красителя и массы Na2SO4. Закономерности влияния этих факторов на R аналита аналогичны технологии СРЕ, наилучшими экстракционными характеристиками также обладали 10%-ные растворы нПАВ. Поскольку добавление электролита приводит к усилению гидрофобных взаимодействий ПАВ–СПК, увеличение массы высаливателя повышает концентрацию красителя в МФ и при добавлении 0.84 г соли R возрастает до 96% при концентрации красителя 5 мг/л. Из более разбавленных растворов (1 мг/л) Е124 извлекается практически полностью (R = 98–99%). В табл. 2 приведены степени извлечения Е122 и Е124 экстракционными системами в различных режимах фазового расслоения: для наиболее гидрофобного Азорубина практически полное однократное извлечение возможно при использовании как режима СРЕ, так и ATPS; для более гидрофильного Понсо 4R наилучшими экстракционными характеристиками обладает режим ATPS.
Экстракционно-фотометрическое определение красителей в лекарственных препаратах. При переходе в МФ форма электронных спектров поглощения СПК остается практически неизменной; это свойство использовали для их идентификации в анализируемых объектах спектрофотометрическим методом. С ростом концентрации СПК наблюдается увеличение оптической плотности раствора МФ. Зависимость между ее значениями в максимуме поглощения спектра каждого соединения и концентрациями красителей в растворе линейна, что использовали для количественного определения. Уравнения градуировочных зависимостей для режима ATPS, приведенные в табл. 3, показывают, что для целей экстракции возможности Тритона Х-100 и ОП-10 практически одинаковы. Можно отметить, что для Тритона Х-100 фазовое равновесие наступает быстрее, и образующиеся МФ несколько меньше по объему.
Таблица 3.
Уравнения градуировочных зависимостей оптической плотности раствора мицеллярной фазы (А) от концентрации (с, мг/л) красителей в водном растворе. (с(ПАВ) = 10%, m(Na2SO4) = 0.84 г, объем раствора – 10 мл, 25°С)
| Экстракционная система | СПК | λmax, нм | Градуировочная зависимость | R2 |
|---|---|---|---|---|
| ОП-10–Н2О–Na2SO4 | Е122 | 522 | А = 0.0675с + 0.0243 | 0.998 |
| ОП-10–Н2О–Na2SO4 | Е124 | 513 | А = 0.0573с + 0.0252 | 0.997 |
| Тритон Х-100–Н2О–Na2SO4 | Е122 | 522 | А = 0.0763с + 0.0261 | 0.998 |
| Тритон Х-100–Н2О–Na2SO4 | Е124 | 513 | А = 0.0691с + 0.0328 | 0.996 |
В качестве объектов анализа выбрали сиропы “Гексо Бронхо” (Франция) и “Либексин Муко” (Германия), растительные пастилки от кашля “Доктор МОМ” с малиновым вкусом (Индия). Выбор способа проведения экстракционной процедуры обусловлен не только природой аналита (для более гидрофильных соединений предпочтителен режим ATPS), но и матрицей анализируемого объекта (состав может влиять на локализацию МФ. Оценили возможности применения способа СРЕ и ATPS. В режиме СРЕ установлена инверсия фаз. В связи с этим для анализа выбрали режим ATPS, в качестве экстрагента – Тритон Х-100.
Определение Понсо 4R в сиропах. Аликвоту сиропа (1–3 мл) помещали в пробирку и добавляли дистиллированной воды до общего объема 5 мл. К полученному раствору добавляли 0.84 г (точная навеска) безводного Na2SO4, смесь тщательно перемешивали до полного растворения соли. Полученный раствор набирали в шприц объемом 10 мл. В этот же шприц набирали 5 мл 20%-ного раствора Тритона Х-100. Смесь в шприце тщательно перемешивали до помутнения (1–2 мин) и выдерживали на водяной бане течение 30 мин при 25°С. Водную фазу отделяли, а мицеллярную фазу помещали в мерную колбу емк. 5 мл, добавляли дистиллированную воду до метки, перемешивали и измеряли оптическую плотность полученного раствора. Краситель идентифицировали сравнением спектров поглощения растворов мицеллярной фазы после экстракции и рабочего раствора красителя Понсо 4R. Количественное определение проводили по градуировочной зависимости, аналитическая длина волны – 513 нм.
Результаты определения Понсо 4R в сиропах от кашля и данные проверки правильности [23] представлены в табл. 4. Полученные данные подтвердили пригодность режима ATPS для отделения красителя от матрицы сиропов, вспомогательные компоненты не оказывали мешающего действия. Сопоставление результатов двух методов по F- и t-критериям подтвердило отсутствие систематической погрешности.
Таблица 4.
Результаты (мг/л) определения Понсо 4 R в лекарственных сиропах методами мицеллярно-экстракционно-фотометрическим и ВЭЖХ (n = 3, P = 0.95, tтабл = 2.78, Fтабл = 19.2)
| Анализируемый сироп | Мицеллярно- экстракционно- фотометрический метод |
ВЭЖХ | tэксп | Fэксп | ||
|---|---|---|---|---|---|---|
| Хср ± ΔХ, мг/л | sr | Хср ± ΔХ, мг/л | sr | |||
| “Гексо Бронхо” | 39.8 ± 6.2 | 0.04 | 39.5 ± 2.0 | 0.01 | 0.35 | 3.2 |
| “Либексин Муко” | 38.6 ± 7.5 | 0.05 | 40.2 ± 2.9 | 0.02 | 1.49 | 2.6 |
Определение Азорубина в пастилках для рассасывания. Одну пастилку (m ≈ 2.5 г) растворяли при перемешивании в 25 мл дистиллированной воды. Полученный раствор анализировали аналогично описанной экстракционной процедуре для сиропов. Краситель идентифицировали сравнением спектров поглощения растворов мицеллярной фазы и рабочего раствора красителя Азорубина. Для количественного определения использовали градуировочную зависимость, аналитическая длина волны – 522 нм.
Правильность определения проверяли методом введено–найдено, результаты представлены в табл. 5. Статистическая обработка результатов определения показала возможность определения Азорубина в пастилках для рассасывания с погрешностью, не превышающей 5%.
* * *
Таким образом, установлено, что на эффективность извлечения синтетических красителей Азорубина и Понсо 4R из водных растворов существенно влияют режим фазового разделения и концентрация высаливателя. Наилучшими экстракционными характеристиками обладают системы состава нПАВ–Н2О–Na2SO4 при использовании технологии “высаливания”. Оптимизированы условия проведения экстракции по технологии СРЕ путем добавления электролита, снижающего температуру помутнения; выдерживания растворов при фиксированной температуре, на 10°C превышающей температуру расслоения; охлаждения при 15°С. Для экстракционной процедуры по технологии ATPS определено количество высаливателя, обеспечивающее быстрое установление фазового равновесия при 25°С. Для этого режима предложен способ экстракции “в шприцах”, позволяющий просто и эффективно проводить как саму процедуру фазового расслоения в небольших объемах, так и разделение фаз. Разработаны экспрессные методики, позволяющие применять экологически безопасные и доступные неионные ПАВ Тритон Х-100 и ОП-10 для экстракционно-спектрофотометрического определения Азорубина и Понсо 4R в лекарственных препаратах.
Список литературы
Меньшутина Н.В., Мишина Ю.В., Алвес С.В. Инновационные технологии и оборудование фармацевтического производства. Т. 1. М: БИНОМ, 2016. 328 с.
Чуешов В.И., Гладух Е.В., Сайко И.В., Ляпунова О.А., Сичкарь А.А., Крутских Т.В., Рубан Е.А., Черняев С.В. Технология лекарств промышленного производства. Винница: Нова Книга, 2014. 696 с.
Сарафанова Л.А. Пищевые добавки: Энциклопедия. Спб: ГИОРД, 2003. 688 с.
Государственная фармакопея Российской Федерации. В 4-х тт. XIV изд. http://www.femb.ru/femb/pharmacopea.php (дата обращения 17.09.2021).
Смирнов Е.В. Пищевые красители. Справочник. СПб: Профессия, 2009. 352 с.
Песня Д.С., Романовский А.В., Прохорова И.М. Исследование токсического и генотоксического эффектов синтетических пищевых красителей Allium test // Ярославский педагогический вестник. 2012. Т. 3. № 3. С. 86.
Титова Н.Д. Пищевые добавки как алиментарные аллергены // Иммунопатология, аллергология, инфектология. 2008. № 2. С. 41.
Рудометова Н.В. безопасность пищевых продуктов: контроль применения синтетических красителей // Пищевая промышленность. 2010. № 12. С. 64.
Гигиенические требования по применению пищевых добавок. Санитарно-эпидемиологические правила и нормативы. СанПиН 2.3.2.1293-03. М.: Минздрав России, 2003.
Государственный реестр лекарственных средств: https://grls.rosminzdrav.ru/grls.aspx (дата обращения 17.09.2021).
Руководство по инструментальным методам исследования при разработке и экспертизе качества лекарственных препаратов / Под ред. Быковского С.Н., Василенко И.А., Харченко М.И., Белова А.Б., Шохина И.Е., Дориной Е.А. М.: Перо, 2014. 656 с.
Петров Б.И., Леснов А.Е. Современное состояние экстракционного метода: Учебное пособие. СПб: Лань, 2018. 356 с.
Кущевская Н.Ф., Дорощук В.А., Гобачевский А.Н. Мицеллярно-экстракционное концентрирование микрокомпонентов фазами неионных ПАВ при температуре помутнения // Химия и технология воды. 2008. Т. 30. № 5. С. 551.
Yazdi A.S. Surfactant-based extraction methods // Trends Anal. Chem. 2011. V. 30. № 6. P. 918.
Paleologos E.K., Giokas D.L., Karayannis M.I. Micelle-mediated separation and cloud-point extraction // Trends Anal. Chem. 2005. V. 24. № 5. P. 426.
Hatti-Kaul R. Aqueous two-phase systems // Mol. Biotechnol. 2001. V. 19. P. 269.
Pourreza N., Rastegarzadeh S., Larki A. Determination of Allura red in food samples after cloud point extraction using mixed micelles // Food Chem. 2011. V. 126. P. 1465.
Pourreza N., Ghomi M. Simultaneous cloud point extraction and spectrophotometric determination of carmoisine and brilliant blue FCF in food samples // Talanta. 2011. V. 84. P. 240.
El-Shahawi M.S., Hamza A., Al-Sibaai A.A., Bashammakh A.S., Al-Saidi H.M. A new method for analysis of sunset yellow in food samples based on cloud point extraction prior to spectrophotometric determination // J. Ind. Eng. Chem. 2013. V. 19. № 2. P. 529.
Чернова Р.К., Шестопалова Н.Б., Козлова Л.М. Фазовое разделение в системе (ОП-10)–Н2О и “cloud point” экстракция некоторых красителей // Изв. Саратовского ун-та. Сер. Химия. Биология. Экология. 2012. Т. 12. Вып. 3. С. 32.
Чернова Р.К., Шестопалова Н.Б., Козлова Л.М. Некоторые аспекты влияния электролитов на фазовое разделение и “cloud point” экстракцию азорубина в системе (ОП-10)–Н2О // Изв. Саратовского ун-та. Сер. Химия. Биология. Экология. 2012. Т. 12. Вып. 4. С. 11.
Güray T., Menevşe B., Yavuz A.A. Determination of optimization parameters based on the Box-Behnken design for cloud point extraction of quinoline yellow using Brij 58 and application of this method to real samples // Spectrochim. Acta A: Mol. Biomol. Spectrosc. 2020. V. 243. Article 118800.
Руководство Р.4.1.1672-03 по методам контроля качества и безопасности биологически активных добавок к пище. М.: Федеральный центр госсанэпиднадзора Минздрава России, 2004. 240 с.
Основы аналитической химии. Т. 1 / Под ред. Золотова Ю.А. М.: Высшая школа, 1996. 384 с.
Шестопалова Н.Б., Чернова Р.К. Влияние солей натрия на фазовое разделение в системе (ОП-10)–Н2О // Вестн. Тамбовского гос. техн. ун-та. 2014. Т. 20. № 2. С. 322.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии