

Журнал аналитической химии, 2023, T. 78, № 1, стр. 80-89
Определение глифосата, его метаболита и глюфосината в продукции растительного происхождения методом хромато-масс-спектрометрии
А. В. Сорокин *
Всероссийский государственный центр качества и стандартизации лекарственных средств
для животных и кормов (ВГНКИ)
123022 Москва, Звенигородское ш., 5, Россия
* E-mail: alex_sorokin@list.ru
Поступила в редакцию 25.02.2022
После доработки 19.04.2022
Принята к публикации 27.04.2022
- EDN: KLABFO
- DOI: 10.31857/S0044450222120167
Аннотация
Разработана селективная методика хромато-масс-спектрометрического определения глифосата, аминометилфосфоновой кислоты и глюфосината в сырье растительного происхождения. Предел количественного определения глифосата составляет 0.1 мг/кг, глюфосината и аминометилфосфоновой кислоты 0.4 мг/кг. Методика основана на извлечении определяемых соединений из образца деионизованной водой, подкисленной соляной кислотой, твердофазной очистке с последующей дериватизацией 9-флуоренилметоксикарбонил хлоридом и дополнительной очистке дериватов на сорбенте со слабыми катионообменными свойствами. Оптимизированы процедуры экстракции и очистки экстрактов. Изучено влияние компонентов матрицы на хроматографическое разделение и детектирование глифосата на хромато-масс-спектрометрах различных типов. Валидация разработанной методики, показала, что относительная расширенная неопределенность лежит в диапазоне от 15 до 25%.
Глифосат (ГФ) и глюфосинат (ГЛ) являются неселективными гербицидами, применяемыми для борьбы с сорной растительностью при выращивании сельскохозяйственных культур. Данные гербициды способны накапливаться в тканях растений и загрязнять пищевую цепочку, особенно через генетически модифицированные культуры [1, 2]. В 2017 г. Международное Агентство по изучению рака (IARC) отнесло ГФ к группе веществ “потенциально канцерогенных для человека” (группа 2А). Глифосат запрещен к применению в ряде стран из-за его устойчивости в почве и донных отложениях [3]. Сообщалось [4], что препарат Раундап, содержащий ГФ в качестве активного компонента, может вызывать проблемы во время беременности, что подтверждалось обработкой им линии клеток плаценты человека. Цитотоксический эффект, который в перспективе может привести к раку, наблюдался при обработке клеточной линии буккального эпителия [5]. Препараты на основе ГФ вызывают хронические эффекты: гепаторенальные, тератогенные, туморогенные, а также нарушение эндокринной функции [6]. В тканях растений ГФ метаболизируется до аминометилфосфоновой кислоты (АМФК) (схема 1 ) [3].
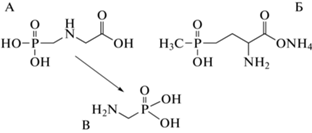
Схема 1 . Структурные формулы глифосата (А), глюфосината (Б) и аминометилфосфоновой кислоты (В).
Для контроля безопасности сырья растительного происхождения в Российской Федерации ТР ТС 015/2011 устанавливает максимально допустимые уровни (МДУ) содержания ГФ и ГЛ. МДУ ГФ: 0.3 мг/кг в подсолнечнике и кукурузе; 3.0 мг/кг в зерне хлебных злаков; 0.15 мг/кг в рисе и сое. МДУ ГЛ: 0.4 мг/кг в подсолнечнике, гречихе, просе, рапсе, зерне хлебных злаков и бобовых. Наличие установленных норм и возможная опасность от присутствия ГФ, ГЛ и АМФК в сырье растительного происхождения предопределяют необходимость создания селективной методики для контроля их остаточного содержания. Определение данных соединений возможно методами иммуноферментного анализа (ИФА), высокоэффективной жидкостной хроматографии с УФ-детектированием (ВЭЖХ-УФ), газовой хроматографии (ГХ) с масс-спектрометрическим детектированием (ГХ-МС) и ВЭЖХ с тандемным масс-спектрометрическим детектированием (ВЭЖХ-МС/МС). ИФА-методики характеризуются низким пределом обнаружения (ПО) ГФ (0.05–0.12 мкг/л) [7–9], ориентированы на анализ природных, поверхностных, питьевых вод и не позволяют проводить совместное определение ГФ, АМФК и ГЛ. Тем не менее существует способ определения ГФ тест-полосками (Glyphosate Strip Kit, Eurofins Abraxis) в экстрактах кукурузы и сои с ПО 0.04 мг/кг, а так же ИФА-набор (Glyphosate ELISA AOAC Test Kit, Eurofins Abraxis) для определения ГФ в растительном сырье с обязательным подтверждением результатов более селективным методом. В целом, применение ИФА для анализа сырья растительного происхождения затруднительно, так как присутствие микроэлементов, липидов и сахаров может приводить к ложноположительным результатам анализа [10–12].
Применение метода ВЭЖХ-УФ подразумевает обязательную дериватизацию молекул ГФ, АМФК и ГЛ, так как в них отсутствуют хромофорные группы, и они плохо удерживаются на большинстве хроматографических колонок с обращенной фазой. Достигнутые ПО ГФ таким методом могут составлять 0.01–0.3 мг/кг [13–17], а АМФК – 0.05 мг/кг [18]. Описано совместное определение ГФ, АМФК и ГЛ в природных водах [19]. Однако из-за дериватизации первичных аминов, содержащихся в экстрактах совместно с определяемыми соединениями, специфичность метода снижается, а интерпретация результатов анализа усложнена. Определение ГФ и АМФК методами ГХ и ГХ-МС также предполагает проведение дериватизации. Заявляемый предел количественного определения (ПКО) ГФ и АМФК методом ГХ-МС в образцах растительного сырья может достигать 0.05 мг/кг при работе в режиме мониторинга выделенных ионов (SIM) [20]. Описан способ ГХ-определения ГФ на уровне 0.01 мг/кг в образцах почвы с азотно-фосфорным детектором [21], а также в биологических жидкостях человека [22]. Следует отметить, что ГФ и ГЛ относятся к группе В3b [23], поэтому для их количественного определения и подтверждения результатов целесообразно применять селективный метод, обеспечивающий не менее трех точек идентификации [24], например ВЭЖХ-МС/МС.
Некоторые ВЭЖХ-МС/МС-методики позволяют определять ГФ и АМФК с ПКО 0.3–0.4 мг/кг, что существенно выше установленного в РФ МДУ. При этом определяемые соединения не дериватизируют, а хроматографическое разделение осуществляют на колонках типа Zorbax Eclipse RDB C8 [25] и Sielc Obelisc N [26]. В первом случае время выхода определяемых соединений составляет менее минуты при применении карбоната аммония в подвижных фазах. Во втором случае возможна нестабильность в работе хроматографической колонки, кроме того, еe отличают высокая стоимость и недолговечность. Описан способ определения ГФ на уровне от 0.02 мг/кг при использовании колонки Click TE–Cys с модифицированной цистеином цвиттер-ионной гидрофильной фазой (HILIC) [27]. Такой способ включает длительную стадию пробоподготовки, а хроматографическая колонка имеет высокую стоимость и требует контроля стабильности работы. Возможность определения АМФК и ГЛ авторами не изучена. Описан способ [28] совместного определения ГФ, АМФК и ГЛ в растительном сырье на уровне 0.1 мг/кг. Несмотря на достаточно низкий ПКО, сообщается о значительном подавлении сигнала АМФК компонентами матрицы. Градуировочную зависимость строили на очищенных экстрактах, полученных в ходе пробоподготовки, а не в начале процедуры, не смотря на ее простоту. В РФ разработана методика [29 ] определения ГФ и АМФК в растительном сырье с заявленным ПКО 0.025 мг/кг. Извлечение ГФ и АМФК из образца осуществляют метанолом, несмотря на низкую растворимость в нем данных соединений (растворимость ГФ в воде при 20°С 10–12 г/л, в метаноле <10 мг/л). Экстракт очищают вымораживанием, а хроматографическое разделение проводят на ионообменной хроматографической колонке (DIONEX IonPac AS-11-HC) с обязательной регенерацией для сохранения свойств и ресурса. Градуировочную зависимость по данной методике получают, анализируя серию чистых растворов стандартов, что может существенно искажать результаты анализа. Не предусмотрена коррекция потерь определяемых соединений в ходе пробоподготовки за счет применения их изотопно-меченного аналога. Следует отметить, что ГФ обладает способностью связываться с активными центрами стекла, что может негативно сказываться на результатах анализа при использовании несиланизированных расходных материалов [30].
Цель данной работы – разработка селективной методики совместного определения ГФ, АМФК и ГЛ методом ВЭЖХ-МС/МС в сырье растительного происхождения, лишенной перечисленных выше недостатков, с применением дериватизации определяемых соединений, последующей очистки дериватов методом твердофазной экстракции (ТФЭ) и колонок с обращенной фазой на стадии хроматографического разделения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы, материалы, оборудование. Применяли метанол 99% (CAS 67-56-1), ацетонитрил 99% (CAS 75-05-8), муравьиную кислоту 99% (CAS 64-18-6), ацетон 99% (CAS 67-64-1), ацетат аммония 99% (CAS 631-61-8), 9-флуоренилметоксикарбонил хлорид 97% (ФМОК) (CAS 28920-43-6), глифосат 95% (CAS 1071-83-6), глюфосинат аммония 95% (CAS 77182-82-2), АМФК 95% (CAS 1066-51-9), Глифосат-2-13C,15N 95% (ГФ-Ist) (CAS 285978-24-7), изопропанол 99.5% (CAS 67-63-0) (Merck, Германия); соляную кислоту 37% (CAS 7647-01-0), натрия тетраборат гексагидрат 99.5% (CAS 1303-96-4), натрия гидроксид 97% (CAS 1310-73-2), эфир диэтиловый 99% (CAS 60-29-7), раствор аммиака 25%-ный (CAS 1336-21-6), кислоту уксусную 99% (CAS 64-19-7) (ТД “Химмед”, Россия). Использовали картриджи для ТФЭ марок Oasis HLB, WCX и MCX с 60 мг сорбента объемом 3 мл (Waters, США). Для получения деионизованной воды использовали систему очистки воды Millipore (Merck, Германия); для экстракции и перемешивания – шейкеры Reax 2 и Reax Control (Heidolph, Германия); для концентрирования экстрактов и дериватизации – модуль Pierce Reacti-Therm III (Thermo, США). Для количественного определения использовали квадрупольно-времяпролетный масс-спектрометр Maxis (Bruker, Германия) с хроматографом ACQUITY (Waters, США), а также масс-спектрометр QTRAP 6500 (Sciex, США) с хроматографом 1290 Infinity II LC (Agilent, США).
Экстракция. Исходные растворы и смеси рабочих растворов вносимых стандартов готовили в деионизованной воде. Концентрация исходных растворов составляла 0.5 мг/мл, рабочих рас-творов – 0.1 и 0.01 мг/мл, рабочего раствора ГФ-Ist – 0.2 мг/мл. Перед экстракцией проводили гомогенизацию объектов исследования и отбирали по 4 г гомогенатов в полипропиленовые пробирки емк. 50 мл. В образцы вносили аликвоты рабочих растворов определяемых соединений (для построения градуировочной зависимости) и 50 мкл рабочего раствора ГФ-Ist. Экстракцию проводили 25 мл деионизованной воды в течение 30 мин на переворачивающемся встряхивателе. После этого к содержимому пробирки приливали 0.16 мл 12 М HCl и продолжали экстракцию еще в течение 30 мин. Пробирку с содержимым центрифугировали при 4750 об/мин и 20°С в течение 30 мин.
Очистка экстрактов и дериватизация. Сорбент картриджа для ТФЭ Oasis HLB активировали и уравновешивали последовательным пропусканием 2 мл метанола и деионизованной воды. На сорбент наносили 0.8 мл полученного экстракта и позволяли стечь в слив. Помещали под картридж приемную пробирку емк. 15 мл и вносили на слой сорбента еще 1 мл экстракта. К 1 мл очищенного экстракта добавляли 1 мл боратного буферного раствора (рН 10.5–11), перемешивали, вносили 1 мл раствора ФМОК с концентрацией 3 мг/мл в ацетоне. Пробирку закрывали крышкой, содержимое перемешивали и выдерживали при 40–50°С в течение 30 мин. После дериватизации к охлажденному до комнатной температуры раствору приливали 2 мл диэтилового эфира, встряхивали, и отбрасывали верхний органический слой после разделения фаз центрифугированием.
Концентрирование и финальная очистка методом твердофазной экстракции. Экстракт концентрировали до 1–1.2 мл при 45–50°С в токе воздуха. Остаток разбавляли в три раза деионизованной водой и подкисляли 10 мкл 12 М HCl. Полученный раствор перемешивали и центрифугировали при 4750 об/мин и 10°С в течение 15 мин. Сорбент картриджа для ТФЭ Oasis WCX активировали и уравновешивали последовательным пропусканием 2 мл метанола и 2 мл раствора 5%-ной муравьиной кислоты в деионизованной воде. На сорбент наносили полученный экстракт и позволяли стечь в слив. После этого сорбент последовательно промывали 2 мл 5%-ной муравьиной кислоты в деионизованной воде, 1.5 мл 30%-ного метанола c 5%-ной муравьиной кислотой. Под картридж помещали приемную пробирку емк. 15 мл и вносили на слой сорбента 3 мл 9%-ной деионизованной воды c 1%-ным раствором аммиака в метаноле. Полученный элюат концентрировали при 45–50°С до 0.3 мл, разбавляли до 1 мл раствором, содержащим 20% метанола и 1% уксусной кислоты в деионизованной воде, центрифугировали при 4750 об/мин и 5–10°С в течение 10–15 мин и использовали для анализа.
Условия хроматографического разделения и детектирования. Использовали режим отрицательной ионизации. Подвижными фазами являлись: 20 мМ раствор ацетата аммония в деионизованной воде (А) и 20 мМ раствор ацетата аммония в метаноле (Б).
При работе на хромато-масс-спектрометре Maxis/ACQUITY хроматографическое разделение осуществляли на колонке BEH C18 (100 мм × × 1.0 мм, размер зерна сорбента 1.7 мкм) (Waters, США) в режиме градиентного элюирования: 0 мин – 10% Б, 0 – 1 мин до 20% Б, 1 – 6 мин до 95% Б, 6 – 8 мин 95% Б, в 8.1 мин переключение на 10% Б, уравновешивание до 14 мин. Скорость потока элюента 0.1 мл/мин, температура 30°С. Общие параметры масс-спектрометра Maxis: температура испарителя источника 350°С, напряжение на капилляре 1.0 кВ, напряжение в источнике 400 В, напряжение заряда 300 В, давление газа-распыления 400 кПа, расход газа осушения 8 л/мин, температура газа осушения 200°С, напряжение в отсеке торможения ионов 40 В, время транспортировки ионов 30 мкс, время накопления ионов 10 мкс, скорость сканирования 2 Гц. Измерения проводили в режиме тандемной масс-спектрометрии высокого разрешения (HRMS/MS).
При работе на хромато-масс-спектрометре QTRAP 6500/1290 Infinity хроматографическое разделение осуществляли на колонке Eclipse Plus C18 RRHD (50 × 2.1 мм, размер зерна сорбента 1.8 мкм) (Agilent, США). Условия градиентного элюирования: 0–3 мин 30% Б, к 8.5 мин до 5% А, 8.5–9.5 мин 5% А, к 10 мин градиент к 30% Б, до 14 мин уравновешивание колонки. Скорость потока элюента 0.3 мл/мин, температура 30°С. Общие параметры масс-спектрометра QTRAP 6500: скорость сканирования 80 мкс, температура источника 500°С, напряжение в источнике 4500 В, входной потенциал 10 В. Режим сканирования – мониторинг заданных реакций (MRM). Остальные параметры работы масс-спектрометров указаны в табл. 1.
Таблица 1.
Параметры работы масс-спектрометров и время выхода определяемых соединений
| Регистрируемое соединение | Исходный ион, m/z |
Ионы-продукты/фрагменты, m/z | Время выхода, мин | Энергия соударений, эВ |
|---|---|---|---|---|
| Maxis/ACQUITY (BEH C18) | ||||
| ГФ–ФМОК | 390.08 | 168.009/149.997/124.02 | 4.8 | 22 |
| АМФК–ФМОК | 332.08 | 110.001/135.981 | 5.6 | |
| ГЛ–ФМОК | 402.12 | 180.044/206.024 | 5.2 | |
| ГФ-ISt–ФМОК | 392.08 | 170.0104 | 4.8 | |
| QTRAP 6500/1290 Infinity (Eclipse Plus C18 RRHD) | ||||
| ГФ–ФМОК | 390.1 | 150.2/124.2 | 4.7 | –30/–32 |
| АМФК–ФМОК | 332.1 | 136.2/110.2 | 6.3 | –17/–12 |
| ГЛ–ФМОК | 402.1 | 180.2/206.2 | 5.5 | –15/–4 |
| ГФ-ISt–ФМОК | 392.1 | 152.2/126.2 | 4.7 | –10/–12 |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Детектирование и хроматографическое разделение. Одно из преимуществ гибридного анализатора перед системой тройного квадруполя – возможность работы в режиме высокого разрешения, позволяющая корректно интерпретировать полученный результат. Недостаток таких масс-спектрометров – сравнительно низкая чувствительность. При определении ГФ с помощью хромато-масс-спектрометра Maxis/ACQUITY за основной ион-фрагмент принимали депротонированную молекулу ГФ (С3H7NO5P с m/z 168.009 ± 0.005), которая образовывалась при разрушении деривата ГФ−ФМОК (С18H18NO7P – 391.082 г/моль). Ионы-фрагменты с m/z 149.99 и 124.02, интенсивность которых была значительно ниже, использовали для подтверждения (рис. 1).

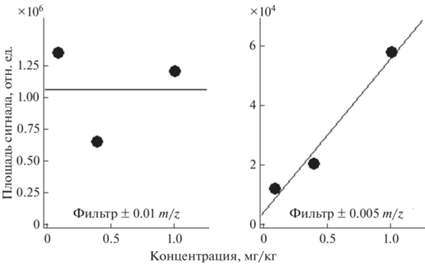
При определении ГФ наблюдался матричный эффект в виде иона, мешавшего количественному определению. Данный эффект устраняли с помощью применения фильтров для обработки масс-хроматограмм: не грубее ± 0.005 m/z. Для поддержания точности определения значений m/z проводили периодическую калибровку масс-спектрометра смесью гидроксида натрия с муравьиной кислотой в изопропаноле. Пример влияния матрицы на сигнал ГФ показан на рис. 2, а результат применения фильтра на рис. 3. Подобный матричный эффект отсутствовал для ионов-фрагментов с m/z 149.99, 124.02 и при определении АМФК c ГЛ.
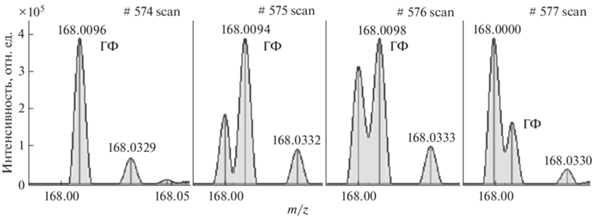
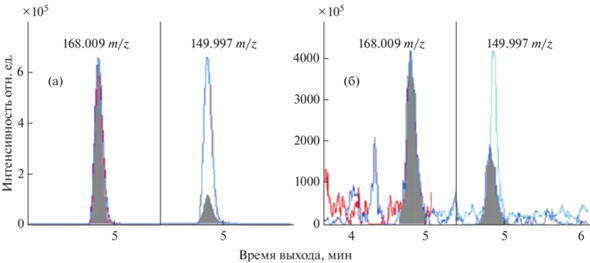
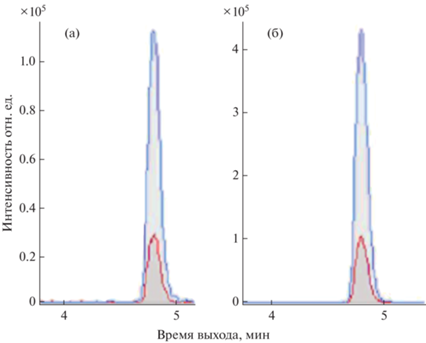
Чувствительность масс-спектрометра QTRAP 6500 позволяет использовать в качестве основного иона-продукта для ГФ ион с m/z 150.2. Такой подход позволяет преодолеть описанную выше проблему, неразрешимую на данном типе масс-спектрометра. При этом подтверждающим ионом-продуктом является ион с m/z 124.02. Несмотря на это при работе на QTRAP 6500 также наблюдали значительное влияние компонентов матрицы на сигнал ГФ, однако его удалось устранить с помощью хроматографической колонки Eclipse Plus C18 RRHD, откорректировав метод разделения введением изократического участка (30% подвижной фазы Б) в течение 3 мин с момента ввода образца в инжектор. Пример масс-хроматограмм, полученных при начальном способе разделения и после его изменения, приведен на рис. 4. Дальнейшее увеличение времени изократического участка программы разделения приводило к уширению хроматографических пиков АМФК и ГЛ с потерей их высоты.
Рис. 4.
Масс-хроматограммы разделения глифосата и компонента матрицы, при первоначальном способе хроматографирования (а), после его оптимизации (б).

Рутинное определение ГФ, ГЛ и АМФК целесообразно осуществлять на масс-спектрометрах с тройным квадруполем благодаря их распространенности, доступности, широкому линейному динамическому диапазону и достаточной чувствительности.
Экстракция. В качестве экстрагентов образцов сырья растительного происхождения опробовали деионизованную воду, подкисленную соляной кислотой (А) и смесь метанол−вода (1 : 1), подкисленную муравьиной кислотой до содержания 1% (Б). К навескам гомогенатов с внесенными в них аликвотами рабочих растворов и раствора ГФ-Ist добавляли по 25 мл растворов А и Б и проводили экстракцию как указано выше. После процедуры экстракции образцы очищали на картриджах Oasis HLB, дериватизировали, концентрировали и очищали на картриджах Oasis WCX в соответствии с описанной процедурой. По результатам данного эксперимента в качестве экстрагента выбрали деионизованную воду, подкисленную в процессе экстракции соляной кислотой, так как разница в относительной интенсивности сигналов составляла более 150 раз. Масс-хроматограммы ГФ, извлеченного деионизованной водой с соляной кислотой и подкисленной смесью метанола с водой, представлены на рис. 5. Объем 12 М HCl для подкисления экстракта подбирали экспериментально. Рассматривали следующие объемы (в скобках приведена конечная концентрация соляной кислоты в экстракте): 0.1 мл (0.05 М), 0.13 мл (0.06 М), 0.16 мл (0.076 М), 0.18 мл (0.086 М), 0.2 мл (0.096 М) и 0.3 мл (0.14 М). Наибольшая полнота извлечения достигалась при добавлении 0.13–0.18 мл 12 М HCl с максимумом при 0.16 мл. Увеличение объема соляной кислоты приводилo к образованию мелкодисперсной фракции образца, мешающей дальнейшему анализу и неотделяемой центрифугированием или фильтрацией в имеющихся условиях.
Рис. 5.
Масс-хроматограммы глифосата, извлеченного деионизованной водой с соляной кислотой (а) и смесью метанола, воды и муравьиной кислоты (б).
Очистка экстрактов. При выборе способа очистки экстракта перед дериватизацией сравнивали осаждение компонентов матрицы ацетонитрилом и пропускание экстракта через картридж Oasis HLB. Для этого в подкисленные экстракты вводили аликвоты растворов стандартов и ГФ-Ist и перемешивали. В первом случае к 1 мл экстракта приливали 1 мл ацетонитрила и перемешивали, центрифугировали при 4750 об/мин и 10°С в течение 15 мин; 1 мл очищенного экстракта использовали для дериватизации. Во втором случае активировали и уравновешивали сорбент картриджа, наносили 1 мл экстракта и позволяли ему протечь в приемную пробирку для дериватизации. Дериватизацию проводили по описанной выше схеме. Применение ацетонитрила положительно сказалось на чистоте образцов, однако относительная интенсивность сигнала была в четыре раза ниже, чем при использовании Oasis HLB (рис. 6). Дериватизация определяемых соединений без очистки приводила к снижению относительной интенсивности сигнала в среднем на 40−50%.
Рис. 6.
Масс-хроматограммы глифосата, прошедшего очистку ацетонитрилом (а); с помощью картриджа Oasis HLB (б).
После стадии дериватизации, путем жидкость-жидкостной экстракции диэтиловым эфиром и последующего концентрирования, удается удалить из раствора дериватов остатки ацетона. За счет этого определяемые соединения лучше удерживаются на сорбенте на второй стадии ТФЭ, кроме того, повышается относительная интенсивность сигнала в среднем на 5–10%. В случае отказа от этапа концентрирования, после жидкость-жидкостной экстракции следует доводить экстракт до 4 мл деионизованной водой, подкислять 10 мкл раствора 12 М HCl, перемешивать и центрифугировать при 4750 об/мин и 5–10°С в течение 15–20 мин. После этого полученный раствор можно наносить на активированный и уравновешенный сорбент картриджа ТФЭ для продолжения процедуры пробоподготовки.
При выборе способа очистки экстракта после дериватизации сравнивали сорбенты картриджей Oasis WCX, Oasis MCX и Oasis HLB (вторичное применение). Протокол очистки состоял из активации сорбента метанолом и уравновешивания водой с 5% муравьиной кислоты, нанесения экстракта, промывки 5%-ной муравьиной кислотой, элюирования смесью 9%-ной деионизованной воды с 1%-ным аммиаком в метаноле. Относительная интенсивность сигналов, полученных при очистке на Oasis WCX, выше, чем на MCX в 2.1 раза для АМФК, в 1.25 раза для ГЛ и в 2.5 раза для ГФ. Вторичное применение Oasis HLB позволяет количественно определять ГФ, АМФК и ГЛ, однако значение коэффициента вариации, рассчитанное для площади пика внутреннего стандарта серии образцов, достигает 30–40%.
Изучaли возможность дополнительной промывки картриджей с нанесенными на сорбент определяемыми соединениями раствором метанола в деионизованной воде с 5% муравьиной кислоты перед стадией элюирования. Для этого на сорбент картриджей после прохождения экстракта и 2 мл раствора 5%-ной муравьиной кислоты в деионизованной воде наносили по 1.5 мл 10, 20, 30, 40 и 50%-ных растворов метанола в деионизованной воде с 5% муравьиной кислоты. После этого определяемые соединения элюировали и завершали пробоподготовку, как указано выше. Установлено, что определяемые соединения надежно удерживаются на сорбентах вплоть до применения 30–40%-ного метанола в деионизованной воде с 5% муравьиной кислоты на стадии промывки.
Валидацию разработанной методики выполняли на хромато-масс-спектрометре QTRAP 6500/1290 Infinity. Валидируемая схема пробоподготовки соответствовала описанной выше и включала в себя этап очистки экстрактов на Oasis HLB перед дериватизаций, устранение остатка ацетона концентрированием, а также очистку дериватов на Oasis WCX. В качестве матрицы для проведения валидации использовали смесь молотой сои, шрота и пшеничной муки 1 : 1 : 1, не содержащих определяемых соединений; 4 г на каждый градуировочный уровень. Всего провели четыре эксперимента с изменяющимися факторами 1) время, 2) оператор. Аналитическая серия каждого эксперимента состояла из чистого образца (холостой опыт), шести уровней градуировки (0.1/0.4/1.0/2.0/5.0/10.0 мг/кг), двух образцов контроля качества (QC) с добавкой, эквивалентной третьему градуировочному уровню (G3), и шести образцов с добавками, соответствующими градуировочным уровням, выполненных в параллели. Критерии, подтверждающие пригодность разработанной методики для решения поставленной задачи: достигнутый коэффициент корреляции R ≥ 0.99; значения правильности для каждого градуировочного уровня не более ±15%; разброс результатов анализа образцов QC не более 15% по отношению к G3. Предел обнаружения ГФ по данной методике составил 0.01 мг/кг, ГЛ и АМФК – 0.04 мг/кг. Предел количественного определения: ГФ – 0.1 мг/кг, ГЛ и АМФК – 0.4 мг/кг. Специфичность методики проверяли в ходе валидации путем анализа 20 образцов соевого шрота и пшеничной муки. Мешающих влияний компонентов матрицы на сигнал не обнаружили. Значения относительного среднеквадратического отклонения повторяемости для ГФ, АМФК и ГЛ не превышали 10%. Показатель воспроизводимости находился в диапазоне от 8 до 15%; предел повторяемости от 11 до 22%. Значения относительной расширенной неопределенности U при Р = 0.95 достигали 25% на первых уровнях добавок с последующим уменьшением до 15–17% по мере увеличения определяемой концентрации соединений. Градуировочные зависимости описываются линейными уравнениями: для ГФ y = 0.496x + 0.0269 (R = 0.9998); для АМФК y = 4.77x + 0.0124 (R = 0.9967); для ГЛ y = = 29.6x + 1.58 (R = 0.998).
* * *
Разработана селективная методика определения ГФ, АМФК и ГЛ, основанная на применении экстрагента, учитывающего свойства аналитов, дериватизации и применении двухэтапной ТФЭ. Методика воспроизводима в большинстве лабораторий, оснащенных хромато-масс-спектрометром с хроматографической колонкой на основе обращенно-фазового сорбента (C18). Достигнутые ПКО ГФ, АМФК и ГЛ в сырье растительного происхождения ниже МДУ, установленных ТР ТС 015/2011. Описанная методология позволяет количественно определять ГФ, АМФК и ГЛ и с более низкими ПКО, вплоть до 0.02–0.05 мг/кг. Однако при этом необходимо применять функцию “взвешивание” при построении градуировочных зависимостей с использованием соответствующего программного обеспечения и смещать содержания аналитов в градуировочных растворах в область более низких концентраций, концентрировать образец перед анализом до меньшего объема. Разработанную методику применили для контроля импортируемого сырья из стран Латинской Америки. Установили, что среднее содержание ГФ в генномодифицированной сое превышает установленный МДУ в 20 и более раз. В целом содержание ГФ и АМФК в таких образцах может достигать 3.7 и 2.8 мг/кг соответственно [31]. Разработанную методику применили для анализа пищевых продуктов, реализуемых на территории РФ, и выявили проблемы, связанные с содержанием ГФ в таких объектах, как гречка (до 2.78 мг/кг), крупы бобовых (до 4 мг/кг) и сложносоставные товары (до 0.54 мг/кг).
Список литературы
Arregui M.C., Lenardón A., Sanchez D., Maitre M.I., Scotta R., Enrique S. Monitoring glyphosate residues in transgenic glyphosate-resistant soybean // Pest. Manag. Sci. 2004. V. 60. P. 163.
Bai S.H., Ogbourne S.M. Glyphosate: environmental contamination, toxicity and potential risks to human health via food contamination // Environ. Sci. Pollut. Res. 2016. V. 23. № 19. P. 18988.
Singh S., Kumar V., Datta S., Wani A.B., Dhanjal D.S., Romero R., Singh J. Glyphosate uptake, translocation, resistance emergence in crops, analytical monitoring, toxicity and degradation: A review // Environ. Chem. Lett. 2020. V. 18. P. 663.
Richard S., Moslemi S., Sipahutar H., Sipahutar H., Benachour N., Seralini G. Differential effects of glyphosate and Roundup on human placental cells and aromatase // Environ. Health Perspect. 2005. V. 113. № 6. P. 716.
Koller V.J., Fürhacker M., Nersesyan A., Mišík M., Eisenbauer M., Knasmueller S. Cytotoxic and DNA-damaging properties of glyphosate and Roundup in human-derived buccal epithelial cells // Arch. Toxicol. 2012. V. 86. № 5. P. 805.
International Agency for Research on Cancer. Some Organophosphate Insecticides and Herbicides. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Lyon. France: International Agency for Research on Cancer, 2017. V. 112.
Mörtl M., Németh G., Juracsek J., Darvas B., Kamp L., Rubio F., Székács A. Determination of glyphosate residues in Hungarian water samples by immunoassay // Microchem. J. 2013. V. 107. P. 143.
Byer J.D., Struger J., Klawunn P., Todd A., Sverko E. Low cost monitoring of glyphosate in surface waters using the ELISA method: An evaluation // Environ. Sci. Technol. 2008. V. 42. № 16. P. 6052
Rubio F., Veldhuis L.J., Clegg B.S., Fleeker J.R., Hall C.J. Comparison of a direct ELISA and an HPLC method for glyphosate determinations in water // J. Agric. Food Chem. 2003. V. 51. № 3. P. 691.
Shan G. Immunoassays in Agricultural Biotechnology. John Wiley & Sons, 2011. 350 p.
Singh G., Velasquez L., Brady B., Koerner T., Huet A.C., Delahaut P. Development of an indirect competitive ELISA for analysis of alternariol in bread and bran samples // Food Anal. Methods. 2018. V. 11. № 5. P. 1444.
Vicini J.L., Jensen P.K., Young B.M., Swarthout J.T. Residues of glyphosate in food and dietary exposure // Compr. Rev. Food. Sci. Food. Saf. 2021. V. 20. № 5. P. 5526.
Nedelkoska. T.V., Low G.K.C. High-performance liquid chromatographic determination of glyphosate in water and plant material after pre-column derivatisation with 9-fluorenylmethyl chloroformate // Anal. Chim. Acta. 2004. V. 511. P. 145.
Fitri F., Muhamad H., Omar D., Asib N. A rapid liquid chromatography method for determination of gylphosate in crude palm oil with fluorescence detection // J. Chromatogr. Sep. Tech. 2017. V. 8. № 1. P. 346.
Sun L., Kong D., Gu W., Guo X., Tao W., Shan Z., Wang Y., Wang N. Determination of glyphosate in soil/sludge by high performance liquid chromatography // J. Chromatogr. A. 2017. V. 1502. P. 8.
Sharma1 P.O., Pholphana N., Rangkadilok1 N. Development of simple and sensitive HPLC method for determination of glyphosate residues in soybean // Nep. J. Environ. Sci. 2015. V. 3. P. 21.
Hernandez F., Hidalgo C., Sancho J.V. Determination of glyphosate residues in plants by precolumn derivatization and coupled-column liquid chromatography with fluorescence detection // J. AOAC Int. 2000. V. 83. № 3. P. 728.
Kaczyński P., Lozowicka B. Liquid chromatographic determination of glyphosate and aminomethylphosphonic acid residues in rapeseed with MS/MS detection or derivatization/fluorescence detection // J. Open Chem. 2015. V. 13. № 1. P. 1011.
Pires N., Passos C., Morgado M., Mello D., Infante C., Caldas E. Determination of glyphosate, AMPA and glufosinate by high performance liquid chromatography with fluorescence detection in waters of the Santarém Plateau, Brazilian Amazon // J. Environ. Sci. Health B. 2020. V. 55. № 9. P. 794.
Alferness P.L., Iwata Y. Determination of glyphosate and (aminomethy1)phosphonic acid in soil, plant and animal matrices, and water by capillary gas chromatography with mass-selective detection // J. Agric. Food Chem. 1994. V. 42. № 12. P. 2751.
Hu J.Y., Chen C.L., Li J.Z. A simple method for the determination of glyphosate residues in soil by capillary gas chromatography with nitrogen phosphorus // J. Anal. Chem. 2008. V. 63. № 4. P. 371.
Hori Y., Fujisawa M., Shimada K., Hirose Y. Determination of the herbicide glyphosate and its metabolite in biological specimens by gas chromatography-mass spectrometry. A case of poisoning by Roundup herbicide // J. Anal. Toxicol. V. 27. № 3. P. 162.
2002/657/EC: Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results (Text with EEA relevance) (notified under document number C(2002) 3044) // Official J. 2002. V. 221. P. 36.
Council Directive 96/23/EC of 29 April 1996 on measures to monitor certain substances and residues thereof in live animals and animal products and repealing Directives 85/358/EEC and 86/469/EEC and Decisions 89/187/EEC and 91/664/EEC // Official J. 1996. V. 125. P. 10.
Helio A.M., Lebre D.T., Wang A.Y., Pires M.A.F., Bustillos O.V. An alternative and fast method for determination of glyphosate and aminomethylphosphonic acid (AMPA) residues in soybean using liquid chromatography coupled with tandem mass spectrometry // Rapid Commun. Mass Spectrom. 2009. V. 23. № 7. P. 1029.
Botero-Coy A.M., Ibáñez M., Sancho J.V., Hernández, F. Direct liquid chromatography–tandem mass spectrometry determination of underivatized glyphosate in rice, maize and soybean // J. Chromatogr. A. 2013. V. 1313. P. 157.
Junjie D., Gehui J., Gaowa J., Aijin S., Zhimou G., Bing Y., Yang J., Jingyu Y., Xinmiao L. Determination of underivatized glyphosate residues in plant-derived food with low matrix effect by solid phase extraction-liquid chromatography-tandem mass spectrometry // Food Anal. Methods. 2016. V. 9. № 10. P. 2856.
Chamkasem N., Harmon T. Direct determination of glyphosate, glufosinate, and AMPA in soybean and corn by liquid chromatography/tandem mass spectrometry // Anal. Bioanal. Chem. 2016. V. 408. № 18. P. 4995.
МИ-ВЛ-1-03-2018. Методика измерений массовой доли глифосата, его метаболита аминометилфосфоновой кислоты и малеинового гидразида в продукции растительного происхождения методом высокоэффективной жидкостной хроматографии с тандемным масс-спектрометрическим детектированием. М.: Федеральный центр оценки безопасности и качества зерна и продуктов его переработки, 2019. 19 с.
Goscinny S., Unterluggauer H., Aldrian J., Vincent H., Masselter S. Determination of glyphosate and its metabolite AMPA (Aminomethylphosphonic Acid) in cereals after derivatization by isotope dilution and UPLC-MS/MS // Food Anal. Methods. 2012. V. 5. № 5. P. 1177.
Сорокин А., Некрасов Д., Батов И., Петров А., Киш Л. Глифосат в сырье растительного происхождения и кормах // Комбикорма. 2022. № 3. С. 58.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии