

Журнал аналитической химии, 2023, T. 78, № 11, стр. 1019-1031
Планарная хроматография – метод аналитического контроля композитных фармацевтических препаратов на основе N-виниламидов
В. Д. Красиков a, *, И. И. Малахова a, Ю. Г. Сантурян a, Е. Ф. Панарин a
a Институт высокомолекулярных соединений Российской академии наук
В.О.
199004 Санкт-Петербург, Большой просп., 31, Россия
* E-mail: lenchrom@hq.macro.ru
Поступила в редакцию 22.11.2022
После доработки 07.04.2023
Принята к публикации 07.04.2023
- EDN: MTOQRZ
- DOI: 10.31857/S0044450223110099
Аннотация
Методом планарной хроматографии разработаны способы аналитического контроля процессов получения биомедицинских полимеров и их комплексов с биологически активными веществами, позволивших создать ряд пролонгированных противовирусных, противоопухолевых, антимикробных и антисептических препаратов.
Планарная (тонкослойная) хроматография (ТСХ) является разновидностью жидкостной хроматографии (ЖХ), поэтому основные механизмы ЖХ реализуются и в ТСХ – адсорбционный, распределительный (нормально- и обращенно-фазовый), ионообменный, ион-парный обращенно-фазовый, эксклюзионный (гель-проникающий, ГПХ), хиральный [1, 2].
На практике разделение в ЖХ веществ обычно протекает по смешанным механизмам. Например, адсорбционная хроматография сопровождается распределительной на слабоактивных сорбентах, содержащих воду; на разделение по адсорбционному механизму может оказывать влияние ГПХ-механизм; на эксклюзионную влияют адсорбционные взаимодействия. В ТСХ, в отличие от ЖХ, все процессы осложняются влиянием паровой фазы. Это может приводить к возникновению тонких взаимодействий между веществом (сорбатом), элюентом и сорбентом, что позволяет разделить близкие по структуре вещества и сложные многокомпонентные смеси, которые при реализации чистых механизмов ЖХ разделить невозможно.
Тонкослойная хроматография позволяет быстро и эффективно анализировать сложные по составу смеси веществ в процессах производства и аналитического контроля качества фармацевтических препаратов [3–5]. В конце 20 века доля фармацевтических, медицинских и аналитических исследований с использованием ТСХ составляла более 50%, однако в настоящее время она существенно снизилась [5, 6]. При этом метод ТСХ имеет ряд особенностей, которые важны для анализа лекарственных средств [7, 8]. В аппаратурном отношении ТСХ гораздо проще колоночной ЖХ, поэтому этот вид анализа является самым простым в исполнении и доступным [9, 10]. В то же время, несмотря на кажущую простоту, возможен одновременный анализ более 100 образцов в варианте инструментальной высокоэффективной планарной n-мерной хроматографии [11, 12]; легко осуществить двумерное разделение [13, 14]; а для обнаружения отдельных пятен можно использовать специфические чувствительные цветные реакции [15, 16]. Хроматографическое разделение и детектирование в ТСХ, как правило, представляют собой процессы, разделенные во времени, что дает возможность детектировать пятна при необходимой длине волны, несмотря на то, что элюент также может сильно поглощать свет в этом диапазоне УФ-спектра [17, 18]. Пластины для ТСХ одноразовые, следовательно, не требуют регенерации [10, 19]. Кроме того, в анализе все шире используют высокоэффективную ТСХ в сочетании с другими методами, такими как УФ- и ИК-фурье спектроскопия, хромато-масс-спектрометрия и др. [20, 21].
Широкое использование ТСХ обязано появлению в семидесятых годах прошлого века убедительной теории хроматографических процессов в жидкостной хроматографии [22–24], позволившей создать вариант современной высокоэффективной инструментальной количественной ТСХ [25].
Теория адсорбционной ТСХ [26] создана в результате синтеза трех теоретических концепций: теории Киселева [27], теории Болджера–Майклса [28] и корреляционной теории Снайдера [22, 25]. Эти исследования позволили сформулировать основные критерии проведения хроматографического эксперимента с учетом оптимизации по скорости ($u$), чувствительности (δ = cm/q где q – количество вносимого вещества, cm – средняя его концентрация по толщине слоя адсорбента) и разрешению хроматографических зон (${{R}_{s}}$).
Так, ТСХ, являясь двумерным хроматографическим процессом [29], может быть описана дифференциальным уравнением:
(1)
$\begin{gathered} \frac{{\partial {{c}_{m}}}}{{\partial t}} = \left[ {u + (1--{{R}_{f}}){{\tau }}\frac{{\partial u}}{{\partial t}}} \right]{{R}_{f}}\frac{{\partial {{c}_{m}}}}{{\partial x}} = \\ = {{R}_{f}}{{D}_{{l,х}}}\frac{{{{\partial }^{2}}{{c}_{m}}}}{{\partial {{x}^{2}}}} + {{R}_{f}}{{D}_{{l,y}}}\frac{{{{\partial }^{2}}{{c}_{m}}}}{{\partial {{y}^{2}}}} + \left( {1--{{R}_{f}}} \right){{R}_{f}}{{u}^{2}}{{\tau }}\frac{{{{\partial }^{2}}{{c}_{m}}}}{{\partial {{x}^{2}}}}, \\ \end{gathered} $Для эффективного хроматографического разделения необходимо создание таких условий, при которых размывание (σ) компонентов смеси существенно слабее эффекта их разделения. В зависимости от условий хроматографирования вклад в размывание зоны различных факторов меняется. При малых размерах частиц сорбента (dp – диаметр частиц) главную роль играет размывание, связанное с профилем скорости (если ∆dp/dp велико) и молекулярной диффузией, при больших dp – массоперенос.
Размывание при скорости анализа ($u$) существенно зависит от значений dp, длины хроматографической системы (l) и давления в ней ($\Delta p$). Указанные параметры связаны между собой соотношением Дорси–Карменуа: для ТСХ $\Delta p = 0$, тогда $u = \xi \frac{{{{d}_{p}}}}{l}$, ($\xi $ – структурно гидродинамический коэффициент). Скорость элюции (u) определяется капиллярными силами и зависит от пробега элюента ${{R}_{l}}~$ по хроматографической пластинке.
(2)
$u = \frac{{{{V}_{m}}^{3}}}{{15{{{(1--{{V}_{m}})}}^{2}}}}\frac{{{{d}_{p}}}}{{\eta {{R}_{s}}}}\left( {{{\lambda cos}}\theta \pm \frac{{{{d}_{p}}~{{\rho }}g~{{R}_{l}}}}{{12}}} \right),$(3)
$с\left( {x,y,t} \right) = {{с}_{m}}{\text{exp}}\left\{ {--\frac{1}{2}\left[ {\frac{{{{{\left( {x--u{{R}_{f}}t} \right)}}^{2}}}}{{{{\sigma }}_{x}^{2}}} + \frac{{\left( {{{y}^{2}}} \right)}}{{{{\sigma }}_{y}^{2}}}} \right]} \right\},$(4)
${{\sigma }}_{x}^{2} = 2{{R}_{f}}\left[ {{{D}_{l}},x\left( {t--{{t}_{0}}} \right) + \left( {1--{{R}_{f}}} \right){{\tau }}~{}_{{{{t}_{o}}}}^{{{{\wr }^{t}}}}{{u}^{2}}\left( {t{\kern 1pt} '} \right)dt{\kern 1pt} '} \right]~,$(6)
${{с}_{m}} = \frac{q}{{4{{\pi }}{{{\left\{ {{{R}_{f}}{{D}_{l}},y\left( {t--{{t}_{0}}} \right)\left[ {{{R}_{f}}{{D}_{l}},x\left( {t--{{t}_{0}}} \right) + \left( {1--{{R}_{f}}} \right){{R}_{f}}{{\tau }}\smallint _{{{{t}_{0}}}}^{t}{{u}^{2}}\left( {t{\kern 1pt} '} \right)dt{\kern 1pt} } \right]} \right\}}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}.$Используя уравнения (3)–(6), можно найти дисперсию размывания хроматографических зон ${{\sigma }}_{x}^{2}$, ${{\sigma }}_{y}^{2}$ и ${{с}_{m}}$ для горизонтальной, восходящей и нисходящей ТСХ. Численное сравнение этих величин показывает, что они примерно равны. Поэтому, чувствительность ТСХ (δ), как следует из (6), можно существенно повысить при ${{R}_{f}} \to 0$.
Анализируя соотношения для дисперсий хроматографического размывания ${{\sigma }}_{x}^{2}$, ${{\sigma }}_{y}^{2}$, ${{\sigma }}_{ \bot }^{2}$ (совокупность ${{\sigma }}_{x}^{2}$ и ${{\sigma }}_{ \bot }^{2}$ определяет размывание вдоль оси х – уравнения (4), (5)), можно использовать следующие приемы по выбору оптимальных операционных условий ТСХ: увеличение скорости ($u$), чувствительности (δ) анализа и разрешения (${{R}_{s}}$) хроматографических зон осуществляется за счет уменьшения хроматографического размывания и регулирования продольной и горизонтальной диффузии (δy и δx); уменьшение неравномерности структуры адсорбционного слоя приводит к уменьшению профиля скорости ($\Delta u \to 0$) по толщине слоя и уменьшает дополнительное размывание хроматографической зоны по оси х; повышение чувствительности детектирования хроматографического пятна реализуется уменьшением размера стартового пятна, дисперсия которого ${{{{\sigma }}}^{2}}$ аддитивно складывается с дисперсией размывания при нанесении образца и его движении по пластинке.
С момента возникновения ТСХ и до настоящего времени вопросы выбора разделительных систем и взаимодействия вещества с адсорбентом и растворителем рассматривались в двух аспектах: с позиции так называемых элюотропных рядов (ε°) Снайдера, которые позволяют распределить растворители по величине Rf определенного вещества, и оценки вклада отдельных функциональных групп вещества в величину Rf для определения системы растворителей [30]. Основой для рассмотрения этих взаимодействий может служить объединённая теория Киселева, Снайдера и Болджера–Майклса [30–32]. Эти теории исходят из того, что в зависимости от природы взаимодействующих молекул и функциональных групп сорбента между ними действуют разные силы: неспецифические (главным образом Ван-дер-Ваальсовы, дисперсионные) и специфические (диполь-дипольные взаимодействия, водородные связи).
Элюирующая способность растворителя определяется суммой всех сил его взаимодействия с хроматографируемым веществом и характеризуется селективностью и элюирующей силой (I). Вследствие того, что эти взаимодействия могут протекать в противоположных направлениях, учет относительной полярности и элюирующей силы элюента может иметь лишь предварительный информативный характер. Тем не менее эти принципы положены в основу ряда подходов к целевому выбору растворителей и созданию подходящего сложного элюента [10, 31, 32].
Используя данные Снайдера [30, 32], наиболее общий подход к подбору элюентов в ТСХ предложил Найреди с сотр. [33] на основе модели “Призмы” [1, 33, 34] (трехмерная модель Снайдера, где элюирующая сила растворителя коррелирует с его селективностью) [35]. Согласно этому подходу, опирающемуся на приведенные выше теории, подбор растворителей проводят в два этапа. На первом этапе после тестирования растворимости и поведения исследуемой смеси в отдельных разных группах растворителей подбирают условия для получения Rf в пределах 0.2–0.8. При этом для понижения Rf мало- и среднеполярных веществ к избранному растворителю добавляют неполярный гексан, а для повышения – предпочтительно воду. На втором этапе оптимизируют селективность определенной комбинацией из выбранных растворителей. Далее к выбранному оптимальному по селективности и элюирующей силе элюенту можно добавлять небольшие количества модификатора (аммиак, уксусная кислота, вода) для поджатия “хвостов” сильно вытянутых хроматографических зон.
Для определения микропримеси с максимальной чувствительностью (Rf → 0) применяют хроматографический процесс, который обеспечивает нахождение микрокомпонентов вблизи стартового пятна, в то время как макрокомпонент уходит вперед и его хроматографический “хвост” не мешает определению микрокомпонентов. Для этой цели лучше всего подходит процесс элютивно-вытеснительной хроматографии. Многокомпонентный элюент при движении по пластинке с активным сорбентом разделяется на зоны по законам фронтальной полизональной хроматографии. Для разделения веществ, сильно различающихся по адсорбционным характеристикам, используют сильные градиенты в полизональной ТСХ, а близкие по свойствам вещества разделяют в условиях вытеснительной (фронтальной) хроматографии и слабого градиента. Таким образом, варьируя состав элюента и концентрацию второго (третьего) компонента элюента (вытеснителя), получают системы, позволяющие определять микрокомпонент в зоне позади фронта вытеснителя.
Используя теоретические закономерности тонкослойной хроматографии и эмпирические приемы подбора элюентов Снайдера и Найреди, можно достаточно успешно применять ТСХ при разработке методов аналитического контроля качества сложных препаратов в фармацевтических производствах.
Общие критерии оценки лекарств, так называемый фармацевтический контроль качества подлинности (прежде всего, состав, однородность, идентичность, чистота, химическая и биологическая доступность), включают качественные и количественные оценки (терапевтическую ценность) активных ингредиентов фармацевтического продукта, наличие примесей и консервантов в продаваемых препаратах.
При выборе объектов исследования учитывали, что медицинские полимеры-носители биологически активных веществ (БАВ) должны обладать уникальным комплексом биологических свойств, прежде всего низкой токсичностью [36], биологической совместимостью [37] и высокой комплексообразующей активностью [38]. Они могут быть использованы в качестве носителей обширного класса веществ, в том числе таргентных препаратов для визуализации и лечения новообразований [39]. Принципиальная возможность создания препаратов пролонгированного действия основывается на модификации целевых лекарственных веществ анионного типа поликатионами [40]. В качестве поликатионов часто используют водорастворимые карбоцепные сополимеры на основе водорастворимых поли-N-виниламидов, которые с 40-х годов прошлого века нашли широкое применение в различных областях фармацевтики и медицины [41].
Определение комплексов поли-N-виниламидов и его сополимеров с БАВ с помощью традиционной высокоэффективной жидкостной хроматографии (ВЭЖХ), высокоэффективной газовой и жидкостной хроматографии с масс-спектрометрическим и другими способами детектирования достаточно сложно и не всегда дает исчерпывающие результаты, поскольку некоторые компоненты смесей могут необратимо сорбироваться на колонках ВЭЖХ [42]. Исходя из этого неудивительно, что, несмотря на наличие мощных современных физико-химических методов анализа [43–45], метод тонкослойной хроматографии рекомендован к обязательному использованию американской и европейской фармакопеями. В этой связи использование современных методов планарной хроматографии для массового анализа полимерных БАВ представляется наиболее многообещающим.
Цель настоящей работы – оценка возможностей ТСХ для аналитического контроля ключевых стадий получения ряда противовирусных, бактериальных, антисептических, онкологических и фармацевтических препаратов, созданных на основе комплексов синтетических водорастворимых медицинских полимеров (N-виниламидов) с БАВ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали реактивы х. ч. или ос. ч. производства Sigma Aldrich (США); Merck (Германия); Вектон, Нева-Реактив и Криохром (Россия). Для приготовления водных растворов использовали дистиллированную воду. Мономеры N-винилпирролидон и N-винилформамид (Fluka, Германия) очищали вакуумной перегонкой (Ткип 334 К/339.9 Па, $n_{D}^{{20}}$ 1.4920; Ткип 342 К/399.9 Па, $n_{D}^{{20}}$ 1.5117 соответственно).
В качестве адсорбента в ТСХ-анализах использовали силикагель (SiO2) – полярный гидрофильный сорбент. Известно, что в водных средах молекулы воды легко связываются с поверхностью SiO2 с образованием многослойного гидратного слоя (“шубы”), который удаляется до нижнего адсорбционного слоя активированием – нагреванием до 120°С [46]. Так как адсорбционная активность SiO2 (активность по Брокману) зависит от содержания в нем воды и количества свободных силанольных групп, ее меняли прогреванием пластинок при 90–100°С с доведением активности по Брокману до II (количества адсорбционной воды не более 10%) и предельным гидроксилированием поверхности SiO2. Последнее достигали обработкой кремнезема 2 M HCl для получения максимального количества силанольных групп за счет раскрытия силоксановых связей SiO2. И наоборот, при необходимости гидроксильные группы деактивировали добавлением незначительного количества (~2 об.%) диэтиламина.
Для ТСХ-анализа использовали аналитические или высокоэффективные пластины “Sorbfil” ПТСХ-П-А, ПТСХ-П-В-УФ 10 × 15 см (Россия) с кремнеземным сорбентом КСК с объeмом и размером пор Vп = 0.9–1.1 см3/г, dп = 9.0–11.2 нм, удельной поверхностью Sуд = 350–450 м2/г и размером частиц dp = 5–17 мкм и dp = 8–12 мкм для аналитических и высокоэффективных пластинок соответственно, с силикагелевым связующим и полимерной подложкой. Предварительно слой силикагеля на пластине очищали в ацетоне, пропуская растворитель в хроматографической камере до верхнего края пластины. После этого пластину высушивали в сушильном шкафу.
Растворы образцов наносили при помощи микрошприца на специальном термостолике при 50–70°С (НЦ “Ленхром”, Россия). Фронт элюента пропускали на расстояние около 8 см от нижнего края пластины, далее пластину извлекали и высушивали от растворителя в сушильном шкафу при 90–100°С.
Аналит идентифицировали спектрофотометрически при помощи УФ-кабинета (НЦ “Ленхром”, Россия) при длинах волн λn 254 и 365 нм и колориметрически (методом проявления в парах йода в течении 1 ч). Проявленную хроматограмму извлекали из камеры, накрывали кварцевым стеклом (предотвращая обесцвечивание) и выполняли расчеты на приборе для количественной ТСХ – денситометре-флюориметре ДенСкан (НЦ “Ленхром”, Россия), используя программу DENS (Ленхром, Россия) [47].
Сополимеры-носители лекарственных веществ, полученные свободнорадикальной сополимеризацией исходных мономеров N-винилпирролидона (N-ВП) и N-винилформамида (N-ВФА) (превращение I) с последующим снятием формильной защиты (в присутствии ортофосфорной кислоты) с образованием сополимера N-винилпирролидона с N-виниламином (N-ВА) [N-ВП]n–[N-ВА]m (превращение II), представлены на схеме 1 [48].
Содержание остаточных мономеров в процессе сополимеризации определяли методом ТСХ. Снятие формильной защиты в сополимерах [N-ВП]n-[N-ВФА]m проводили в присутствии ортофосфорной кислоты [49]. Полноту снятия формильной защиты определяли ТСХ-методом по количеству образовавшейся в результате реакции муравьиной кислоты (схема 1 , превращение II).
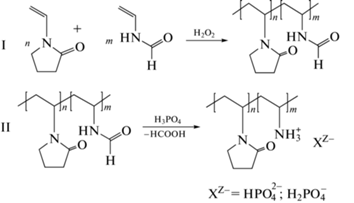
Схема 1 . Синтез сополимера [N-ВП]n-[N-ВА]m – радикальная сополимеризация N-ВП с N-ВФА (I); снятие формильной защиты (II).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Критерии оптимизации хроматографического процесса. При разработке методов разделения промежуточных продуктов и конечных лекарственных препаратов использовали следующий набор технологий ТСХ:
– элюирование проводили методом восходящей хроматографии с использованием восходящего режима ТСХ на “сухом” и “мокром” слоях сорбента с целью изменения адсорбционной активности силикагеля за счет корреляции количества геминальных парных силанольных групп; содержание свободных силанольных гидроксильных групп увеличивали путем протонирования поверхности кремнезема вплоть до достижения их максимального количества 8–9 мкмол/м2, т.е. величина силанольного числа (константа Киселева–Журавлева) ${{ \propto }_{{{\text{OH}}}}}\,\,\sim 4.9~~$ OH-групп/нм2;
– применяли элютивный (для микрокомпонента) и фронтальной (для макрокомпонента) с многокомпонентным растворителем постоянного состава варианты хроматографии для достижения максимальной чувствительности анализа (${{R}_{f}} \to 0$);
– с целью уменьшения стартового размывания пятен вещества наносили на пластинку при градуировке по чувствительности ТСХ-анализа в количестве не более 0.5, 1.0 и 1.5 мкл в точки, располагающиеся одна относительно другой на расстоянии 8 мм;
– с целью уменьшения размывания за счет краевых эффектов хроматографии и нивелирования эффекта изменения скорости элюции на начальных стадиях образцы наносили на расстоянии 1 см от краев пластинки;
– удаляли излишки растворителя путем прогревания пластинок при нанесении аналита, что позволяет уменьшить площадь пятна за счет размывания (σx, σx) наносимой пробы;
– выбирали растворители, в которых определяемые примеси в аналите хроматографически малоподвижны (Rf → 0);
– стартовые точки веществ располагали на минимальном расстоянии от линии погружения пластинки в растворитель, где скорость растворителя достаточно мала и, следовательно, вклад второго члена уравнения (4) в величину размывания $\left( {{{\sigma }}_{x}^{2}} \right)$ снижается.
Критерии выбора элюентов для ТСХ-анализа фармпрепаратов на основе водорастворимых полимеров-носителей биологически активных веществ. Подбор и оптимизацию элюентов [50–52] для исследования хроматографического поведения сополимеров проводили с учетом силы растворителей и их элюотропных рядов, которые можно классифицировать на основе возрастающей силы элюирования (ε°), связанной с поверхностным натяжением (γ) и вязкостью растворителей (ƞ) [33, 53, 54]. Характеристики адсорбционной активности растворителей, использованных в работе, для силикагеля (сорбент II типа по Киселеву) [55] приведены в табл. 1.
Таблица 1.
Характеристика растворителей для тонкослойной хроматографии на кремнеземных сорбентах
| Подвижная фаза | Коэффициент скорости K* | Диэлектрическая константа ε (20°С) | Элюирующая сила ε°** | Индекс полярности P ' ** |
|---|---|---|---|---|
| Кислоты (уксусная, муравьиная кислота) | – | Очень большая | 6.0 | |
| Вода | 76.45 | 80.3 | Большая | 10.2 |
| (Метанол), этанол | (53.6) | (32.6) | 0.68 | 4.3, (5.1) |
| Ацетон | 12.7 | 20.7 | 0.47 | 5.1 |
| Этилацетат | 9.2 | 6.0 | 0.38 | 4.4 |
| Хлороформ | 90 | 4.81 | 0.26 | 4.1 |
| н-Бутанол | 8.9 | 17.8 | 0.34 | 3.9 |
| н-Гексан | 9.2 | 1.9 | 0.01 | 0 |
* K – скорость распространения пятна растворителя на 1 мм2 площади поверхности сорбента; ** ε°, P' – безразмерные эмпирические параметры растворителя [35].
Перечисленные в табл. 1 растворители использовали в виде бинарных или тройных смесей для создания подвижной фазы (элюента) при разделении приведенных ниже смесей фармацевтических веществ и полупродуктов их синтеза. В табл. 2 приведена сила (St) элюента, рассчитанная из сил индивидуальных компонентов (Si) и объeмных долей этих компонентов (φi) в смеси: St = ΣSiφi [1].
Таблица 2.
Характеристики элюентов для тонкослойной хроматографии фармацевтических препаратов
| Объект анализа | Растворитель | Сила растворителя Si** |
Объемная доля компонента φi, об. % | Сила элюента St** |
|---|---|---|---|---|
| Контроль определения остаточных мономеров при сополимеризации N-ВП и N-ВФА | Хлороформ | 4.1 | 60 | 4.97 |
| Этилацетат | 4.4 | 27 | ||
| 25%-ный водн. р-р NH3 | 10.2 | 13 | ||
| Контроль выделения муравьиной кислоты в процессе кислотного гидролиза сополимера [N-ВП]9 и [N-ВФА]1 | Этанол | 4.3 | 80 | 5.48 |
| 25%-ный водн. р-р NH3 | 10.2 | 4 | ||
| Вода | 10.2 | 16 | ||
| Контроль остаточных мономеров при получении противотуберкулезных препаратов на основе сополимера ВП-6-КАКК | Этанол | 4.3 | 78 | 5.59 |
| 25%-ный водн. р-р NH3 | 10.2 | 10 | ||
| Вода | 10.2 | 12 | ||
| Экспресс-определение содержания лактонов D(+)-глюкаровой кислоты в процессе получения противоопухолевого препарата “Поглюкар” | Этанол | 4.3 | 80 | 5.48 |
| 25%-ный водн. р-р NH3 | 10.2 | 4 | ||
| Вода | 10.2 | 16 | ||
| н-Бутанол | 3.9 | 50 | 6.0 | |
| Ледяная уксусная к-та | 6.0 | 25 | ||
| Вода | 10.2 | 25 | ||
| Анализ противовирусных препаратов Полирем (Ремантадин), а также Метилурацил на основе антибактериальных полимерных соединений | Этилацетат | 4.4 | 68 | 6.16 |
| Муравьиная к-та | 9.6 | 16 | ||
| Вода | 10.2 | 16 | ||
| Анализ содержащего наносеробро полимера-антисептика Повиаргол | Ацетон | 5.1 | 96 | 5.31 |
| 3 М HCl | 10.2 | 4 |
** Si, St – безразмерные эмпирические параметры элюента [1].
Незначительное различие в значениях силы элюента, которые лежат в интервале 5.5 ± 0.5, указывает на то, что в этих системах сорбент–элюент–элюат преобладают взаимодействия между определяемым веществом и определенным типом сорбента. Можно полагать, что использование других типов кремнеземов, не говоря уже о бумаге или целлюлозе, приведет к значительному изменению силы элюента.
Контроль примесей мономеров, гомополимеров, катализаторов в продуктах полимеризации биомедицинских сополимеров. При использовании изложенных выше приемов проведения планарной хроматографии удалось осуществить постадийный контроль продуктов синтеза ряда лекарственных препаратов. Характерным примером могут служить методы ТСХ-контроля процессов получения пролонгированных биомедицинских лекарственных средств на основе водорастворимых поли-N-виниламидов, важным представителем которых является поли-N-винилпирролидон и его сополимеры.
Для биомедицинских полимеров важно не только молекулярно-массовое распределение, полидисперстность, композиционная однородность, но и отсутствие примесей мономеров, катализаторов и других низкомолекулярных веществ. С целью выявления низкомолекулярных соединений и гомополимеров в продуктах сополимеризации разработан ряд планарных экспрессных методов, например контроль остаточных мономеров при получении сополимера [N-ВП]n−[N-ВФА]m (схема 1 ).
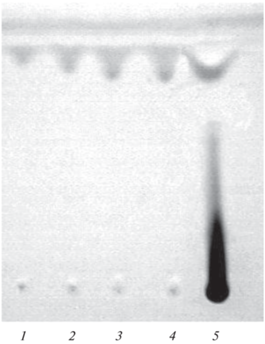
Хроматографическую ТСХ-систему – трехкомпонентный элюент (хлороформ (Si = 4.1)–этилацетат (Si = 4.4)–водный раствор аммиака (Si = 10.2)) выбирали таким образом, чтобы подвижность сополимера [N-ВП]n−[N-ВФА]m в этих условиях была равна нулю (Rf = 0). Это позволяет вести контроль остаточных количеств мономеров N-ВП (Rf = 0.84) и N-ВФА (Rf = 0.58) в реакционной среде в зависимости от продолжительности полимеризации (рис. 1).
Рис. 1.
ТСХ реакционной смеси при сополимеризации N-винилпирролидона и N-винилформамида (мольное соотношение мономеров 9 : 1): 1 – исходная смесь; 2 – 0.5 ч от начала процесса; 3 – 1.0 ч; 7 – 1.5 ч; 8 – 2 ч; 9 – 3 ч; 4–6 – градуировочные смеси N-винилпирролидона и N-винилформамида. Элюент: хлороформ–этилацетат–25%-ный водный раствор аммиака (9 : 4 : 2). Детектирование: пары йода.

Количественные расчеты по хроматограммам проводили на видеоденситометре-флюориметре “ДенСкан” (НТЦ “Ленхром”, Россия). Динамика изменения концентрации мономеров в реакционной среде при мольном соотношении мономеров в сополимере 1 : 1 показана в табл. 3.
Таблица 3.
Динамика изменения концентрации мономеров в реакционной среде (n = 5, P = 0.95)
| Параметр измерения | № опыта | ε, % | Коэффициент корреляции r | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||
| Время от начала синтеза, ч | 0 | 0.5 | 1 | 1.5 | 2.0 | 3.0 | ||
| Содержание ВП, мас.% | 18.0 | 12.8 | 7.3 | 3.38 | 0.27 | 0.27 | 3.6 | 0.9825 |
| Содержание ВФА, мас.% | 11.5 | 1.62 | 0.99 | 0.126 | 0.008 | 0.008 | 1.80 | 0.9946 |
Процесс кислотного гидролиза и получения сополимера [N-ВП]n−[N-ВА]m (рис. 2) контролировали методом ТСХ по выделению муравьиной кислоты (схема 1 , превращение II). Предложенная методика позволила контролировать хроматографическую зону муравьиной кислоты (фактор удерживания Rf = 0.9) с учетом того, что основное количество сополимера [N-ВП]n−[N-ВА]m сорбируется на старте (Rf = 0).
Рис. 2.
Хроматограмма реакционной смеси при гидролизе сополимера [N-винилпирролидон]9–[N-винилформамид]1: 1, 2, 3, 4 – муравьиная к-та 0.5, 1.0, 1.5 и 2.0 мкл соответственно; 5 – реакционная смесь по окончании гидролиза. Элюент: этанол–25%-ный раствор аммиака–вода (80 : 4 : 16). Детектирование: пары йода.
Задача определения примесей катализаторов и не вступивших в реакцию мономеров возникла при получении новой генерации полимерных противотуберкулезных препаратов с использованием в качестве полимерных носителей водорастворимых карбоксилсодержащих сополимеров на основе N-виниламидов (N-ВП) с 6-кротоноиламинокапроновой кислотой (6-КАКК) (схема 2 ).
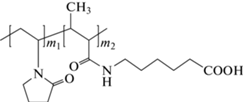
Схема 2 . Структурная формула сополимера N-ВП–6-КАКК.
В случае синтеза таких сополимеров с варьируемым гидрофильно-гидрофобным балансом возникает проблема очистки их от гидрофобного мономера (6-КАКК), который трудно удалить методами диализа или переосаждения.
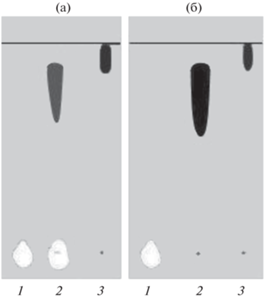
Данные ТСХ показывают, что сополимер ВП–6-КАКК после очистки с помощью препаративной хроматографии на сорбенте Sephadex G20 не содержит примеси 6-КАКК (рис. 3). В этом случае в качестве элюента использовали смесь этанол–вода–25%-ный раствор аммиака (10 : 1.2 : 1.6, по объему). Детектирующим специфическим реактивом служил раствор бромкрезолового зеленого в этиловом спирте с рН 8–9 (создавали добавлением раствора NaOH). В этих условиях зона 6-КАКК движется с Rf = 0.1–0.2, сополимер – с Rf = 0.7, а ПВП с ММ = 40 000 – с Rf = 0.9.
Рис. 3.
Хроматограммы сополимера винилпирролидон (ВП)–6-кротоноиламинокапроновая кислота (6-КАКК) до (а) и после очистки (б). 1 – 6-КАКК, 2 – сополимер ВП–6-КАКК, 3 – поливинилпирролидон 40 000 Да.
Тонкослойная хроматография лекарственных противоопухолевых препаратов мочевого пузыря. С помощью ТСХ удалось существенно упростить аналитический контроль при производстве ряда противоопухолевых препаратов. Известно, что одной из причин возникновения опухолей мочевого пузыря является накопление канцерогенных метаболитов в его слизистой. Канцерогенные вещества выводятся из организма с мочой в виде водорастворимых коньюгатов с D-глюкуроновой кислотой. Однако на путях их экскреции из мочевого пузыря присутствует фермент β-глюкуронидаза (β-ГА), который расщепляет коньюгаты, высвобождая D-глюкуроновую кислоту и свободный липидорастворимый канцерогенный метаболит, накапливающийся в слизистой оболочке, вызывая развитие злокачественных опухолей [56].
Известны селективные ингибиторы β-глюкуронидазы. В ряде работ показано, что специфическими конкурентными ингибиторами активности β-ГА являются производные D(+)-глюкаровой (сахарной) кислоты, в частности 1,4- и 6,3-глюкаролактоны (1,4-ГЛ и 6,3-ГЛ), однако они быстро выводятся из организма [57]. Полимерные комплексы с лекарственными препаратами в большинстве случаев позволяют снизить их токсичность и увеличить продолжительность действия. В качестве полимеров-носителей лактонов D(+)-сахарной кислоты выбраны сополимеры N-ВП с N-ВА. Разработанные хроматографические методы экспресс-определения содержания лактонов D(+)-глюкаровой кислоты в процессе получения полимерных комплексов позволили установить, что в слабощелочном элюенте (рН 7.8–7.9) комплекс с глюкаролактонами достаточно устойчив (рис. 4).
Рис. 4.
Хроматограмма полимерного комплекса [N‑винилпирролидон]n−[N-виниламин]m с 1.4- и 6.3-глюкаролактонами: 1 – сополимер [N-винилпирролидон]n−[N-виниламин]m; 2 – смесь 1.4- и 6.3-глюкаролактонов; 3 – полимерный комплекс [N-винилпирролидон]n−[N-виниламин]m с 1,4- и 6,3-глюкаролактонами. Элюент: этанол–25%-ный водный р-р аммиака–вода (8.0 : 0.4 : 1.6). Детектирование: УФ 365 нм.

Показано что при длительном хранении в составе полимерного комплекса появляются два новых компонента с Rf = 0.6 и Rf = 0.44, предположительно соответствующие 1,4- и 6,3-дилактону и D-глюкаровой кислоте. Подобранная хроматографическая система н-бутанол–уксусная кислота–вода (2 : 1 : 1) позволяет разделить, идентифицировать и определить компонентный состав реакционной смеси, полученной в результате синтеза. Из данных ТСХ видно, что образующиеся полимерные комплексы с 1,4- и 6,3-лактонами в этих условиях разделяются на ряд компонентов. Зона с ${{R}_{f}} = 0.54$ относится к 6,3-лактону; зона с ${{R}_{f}} = 0.78$ – к 1,4-лактону, что доказывается нанесением на ТСХ-пластину индивидуальных 1,4- и 6,3-лактонов в качестве реперных образцов. Полимерный комплекс в данных хроматографических условиях выделяет свободные ГЛ, при этом сополимер-носитель сорбируется на старте с Rf = 0 (рис. 5). Распад комплекса при рН 5.5 с выделением свободных ГЛ крайне важен для ингибирования β-ГА in vivo, активность которой особенно высока в слабокислой среде.
Рис. 5.
ТСХ-хроматограмма полимерного комплекса [N-винилпирролидон]n−[N-виниламин]m с 1,4- и 6,3-глюкаролактонами: 1 – 6,3-лактон; 2 – образец диализованного полимерного комплекса; 3 – образец 2 после длительного хранения при 23°С; 4 – 1,4-лактон; 5 – полимер-носитель [N-винилпирролидон]n−[N-виниламин]m; 6 – D-глюкаровая кислота. Элюент: н‑бутанол–уксусная кислота–вода (2 : 1 : 1), рН 5.5. Детектирование: пары йода. Чувствительность метода – 0.25 мкг.

Планарная хроматография противовирусных и антимикробных препаратов. Другим примером применения метода ТСХ могут служить исследования новых противовирусных препаратов широкого спектра действия с сополимерным носителем, способных защищать не только от гриппа, но и от других ОРВИ, а также от микроорганизмов, вызывающих постгриппозные осложнения. Такую задачу целесообразно решать путем синтеза антибактериальных полимерных соединений с иммобилизацией (прикреплением) к их молекулам известных противогриппозных препаратов. Типичным представителем подобных препаратов является “Катапол” (синонимы: “Пиреноксин”, “Pirenoxine”) – полимерный комплекс диметилбензилалкиламмония хлорида (соль четвертичного аммониевого основания) с сополимером N-винилпирролидона с кротоновой кислотой (схема 3 ).
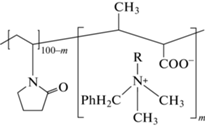
Схема 3 . Структурная формула “Катапола” (сополимер N-винилпирролидона с кротоновой ки лотой + диметилбензилалкиламмония хлорид).
Связывание катионных ПАВ с ионогенными сополимерами N-винилпирролидона с образованием нанодисперсных систем мицеллярного типа с размером частиц от 10–150 нм и более приводит к существенным изменениям их свойств. Так, токсичность снижается в 1.5–2 раза, а кожно-раздражающее действие – в 10 раз. Путем оптимизации химического строения полимера-носителя, его молекулярной массы, надмолекулярной структуры комплекса на основе сополимера N-винилпирролидона с кротоновой кислотой и диметилбензилалкиламмония хлоридом (катамин АВ) создан полимерный антисептик “Катапол”, разрешенный для медицинского применения (per. № 91/146/7). Наночастицы “Катапола” активно взаимодействуют с цитоплазматической мембраной бактерий, разрушают безоболочные и оболочные вирусы: коронавирусы, ротавирусы, аденовирусы, вирус гриппа AH1N1, проявляя высокое бактерицидное и вирулицидное действие при концентрациях 0.005–0.5% [58].
На основе “Катапола” разработан новый препарат “Полирем” – полифункциональное полимерное производное “Ремантадина”, обладающий разными видами биологической активности, полученный путем присоединения к полимерной матрице (полимерная соль N-винилпирролидона и кротоновой кислоты) методом ионного связывания двух биологически активных низкомолекулярных веществ – противовирусного препарата “Ремантадина” (α-метил-1-адамантилметиламина гидрохлорида) и антисептика “Катамина АБ” (диметилбензилалкиламмония хлорида) (схема 4 ).
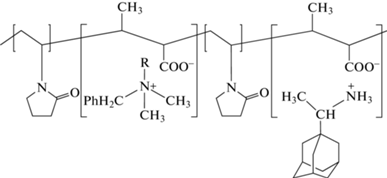
Схема 4 . Структурная формула комплексного препарата “Полирем Ремантадин” (“Катапол” с “Катамином АВ”).
Исследования показали, что наличие терминальных заряженных групп в макромолекуле приводит к полной адсорбции аналита на кремнеземной ТСХ-пластине в водной среде. Увеличения хроматографической подвижности можно достичь лишь путем деактивации гидроксильных групп силикагеля и увеличения доли гидрофобных взаимодействий элюента и аналита. Первое достигается за счет образования водородных связей при взаимодействии силанольных групп сорбата с кротоновой кислотой. Второе условие выполняется при добавлении этилацетата в состав смешанного элюента. Именно такие компоненты смешанного элюента выбирали с учетом элюотропных рядов (ε°) и результатов расчета силы смешанного растворителя (St) на основании индивидуальных компонентов (Si), объемных долей этих растворителей, их протонодонорных и протоноакцепторных свойств согласно Шталю. Как видно из рис. 6, такой подход позволяет осуществлять разделение полимерных солей “Ремантадина” и “Катапола” с высокими разрешающей способностью и селективностью (Rs = 1.25, St = 1.12). Условия проведения анализа и детектирования позволяют не только разделить все компоненты синтезируемых образцов, но и определить их с достаточной степенью точности.
Рис. 6.
Хроматограмма разделения полимерных солей “Ремантадина” с “Катаполом”. 1–3 – градуировочные смеси “Катапола” и “Ремантадина”; 4, 5 – образцы 37, 20 и 40 мкг соответственно; 6, 7 – образцы 38, 20 и 40 мкг соответственно. Элюент: этилацетат–муравьиная кислота–вода (6 : 1.5 : 1.5). Детектирование: пары йода (чувствительность по “Ремантадину” – 2 мкг, по “Катаполу” – 1 мкг). Rf “Ремантадина” – 0.83, Rf “Катамина” – 0.93.
“Катапол” позволил получить целую гамму препаратов с антимикробной и противовирусной активностью для ветеринарии. Например, препарат “Мекапол”, являющийся фармацевтитеской субстанцией с комбинированной антимикробной и репаративной активностью представляет собой коньюгат “Катапола” с “Метилурацилом” (C5H6N2O2) (схема 5 ).
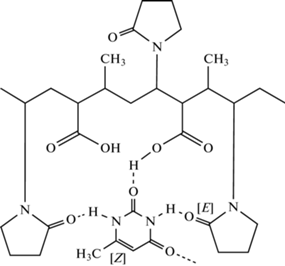
Схема 5. Структурная формула коньюгата и полимера-носителя с “Метилурацилом”.
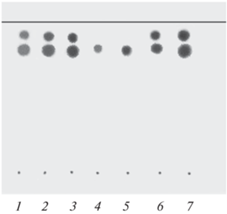
Методом ТСХ удалось установить присутствие в препарате исходных компонентов “Катамина” (КТМ) и “Метилурацила”.
Разработаны методики ТСХ-определения в “Мекаполе” исходного “Катамина” и “Метилурацила” (МУ). Для анализа использовали описанный выше трехкомпонентный элюент этилацетат–муравьиная кислота–вода (6 : 1.5 : 1.5). В качестве проявляющего агента применяли йод или лампу с длинной волны УФ-365 нм для проявления метилурацила (рис. 7).

Количество “Катамина” и “Метилурацила” в составе “Мекапола” (МКП) определяли на денситометре по калибровкам по индивидуальным соединениям. Количество полимера-носителя “Совикана”, оставшегося на “старте”, рассчитывали по следующей формуле: РСВК = РМКП – (РМУ + РКТМ), где РМКП – количество препарата “Мекапол” в 1.0 мкл раствора, нанесенного на пластину, мкг; РМУ – количество “Метилурацила” в “Мекаполе”, полученное в результате денситометрии, мкг; РКТМ – количество “Катамина” в “Мекаполе”, полученное в результате денситометрии, мкг; РСВК – рассчитанное количество полимера-носителя, входящего в состав “Мекапола”, мкг. Таким образом, ТСХ позволяет контролировать состав компонентов в медицинском препарате “Мекапол”.
В случае лекарственного препарата “Повиаргол” на основе поливинилпирролидона (ПП) – антисептика широкого спектра действия на основе Ag0 – планарная хроматография позволяет оценивать остаточное содержание наноразмерного металлического серебра (Ag), стабилизированного медицинским ПВП (ММ 12 000 ± 3000 Да), – (C6H9NO)n·Ag. Прекурсором при получении “Повиаргола” является нитрат серебра, а ПВП выполняет роль полимерного стабилизатора. Вследствие резкого снижения потенциала ионизации серебра у нанокластеров с размерами 1–2 нм (на 1.5 эВ по сравнению с массивным серебром) с развитой поверхности частиц в металлополимерном кластере легче генерируются ионы серебра, обеспечивающие антимикробный эффект, существенно превышающий обычные антимикробные свойства изделий из массивного серебра. В то же время пролонгированное действие “Повиаргола” в организме обусловлено тем, что нанокластеры серебра являются своеобразной депонированной формой ионного серебра, которое постоянно генерируется по мере его связывания с биологическими субстратами. ТСХ позволяет оценить кинетику процесса восстановления ионов серебра (рис. 8), при этом определяли остаточное содержание катиона Ag+ в реакционной смеси. Чувствительность методики при проявлении 0.1 М водном раствором сульфида натрия составила 1–2 мкг.
Рис. 8.
Хроматограмма определения примеси ионов серебра в 10%-ном водном растворе “Повиаргола”. 1, 2, 4, 5 – 0.25, 0.5, 0.75 и 1.0 мкл р-ра “Повиаргола” соответственно; 3 – 0.6 мкг сAgNO3. Элюент: ацетон–3 М НСl (9.3: 0.4). Детектирование: 0.1 М водный раствор Na2S. Пластины: ПТСХ-П-А, обработанные 6 М водным раствором HCl.

* * *
Разработаны элюирующие системы, условия проведения анализа и детектирования, позволяющие не только разделить все компоненты сложных композиций на кремнеземных тонкослойных пластинах, но и выполнить их определение с достаточной степенью точности. Показано, что, несмотря на многообразие специфических и неспецифических взаимодействий, возникающих в хроматографических системах сорбент–сорбат–элюент, элюирующая сила элюента, достаточная для разделения многокомпонентных смесей на кремнеземном сорбенте, лежит в интервале I ≈ 5–6. Приведены примеры оптимизации условий хроматографических разделений для ряда фармацевтических препаратов, полученных на основе водорастворимых полимерных N-виниламидов, таких как противоопухолевый препарат “Поглюкар” – комплекс сополимера N-винилпирролидона с N-виниламином и 1,4-/6,3-лактонами D(+)-сахарной кислоты; антимикробный и антибактериальный (противовирусный) препарат “Катапол” – сополимер N-ВП с кротоновой кислотой; “Мекапол” – компонентный препарат на основе “Катапола” с комбинированной антимикробной и репаративной активностью; “Ремантадин” – комплексный препарат на основе “Катапола” и антисептика “Катамина АБ” (диметилбензилалкиламмоний хлорид); “Повиаргол” – металлополимерная композиция антисептика на основе N-ВП и Ag0.
Список литературы
Йорк Г. Руководство по современной тонкослойной хроматографии. Пер. с англ. / Под ред. Ларионова О.Г. М., 1994. 311 с.
Гейсс Ф. Основы тонкослойной хроматографии. В 2-х тт. Пер. с англ. М., 1988. 407 с.
Ravi Sankar P., Hema Sri P., Eswarudu M.M., Srinivasa Babu P., Maha Lakshmi V., Lakshmi Tirupati M. High-performance thin layer chromatography in the pharma industry // High Technol. Lett. 2021. V. 27. № 73. P. 63.
Sethi P.D. Sethi’s HPTLC, High Performance Thin Layer Chromatography. Quantitative Analysis of Pharmaceutical Formulations. New Delhi, India: CBS Publishers & Distributors, 2013. V. 1–3.
Attimarad Mahesh, Mueen Ahmed K.K., Aldhubaib Bandar E., Harsha Sree. High-performance thin layer chromatography: A powerful analytical technique in pharmaceutical drug discovery // Pharm. Methods. 2011. V. 2. № 2. P. 71. https://doi.org/10.4103/2229-4708.84436
Weins C., Hauck H.E. Advances and developments in thin layer chromatography // LC-GC Int. 1996. V. 9. P. 710.
Шаршунова М., Шварц В., Михалец Ч. Тонкослойная хроматография в фармации и клинической биохимии. В 2-х частях. М.: Мир, 1980. 607 с.
Wagner H., Bladt. S. Plant Drug Analysis. A Thin Layer Chromatography Atlas. 2nd Ed. Berlin: Springer-Verlag, 1996. 384 p.
CAMAG. Instrumental thin-layer chromatography. Switzerland: Camag, 2019. 48 p. https://www.camag.com/sites/default/files/Virtual%20Analytical%20Summit%202022/CAMAG_TLC%202019_web.pdf (22.11.2022).
Кирхнер Ю. Тонкослойная хроматография. В 2-х тт. Пер. с англ. М.: Мир, 1981. 616 с.
Ravisankar P., Lokapavani Ch., Devadasu, Deval Rao G. HPTLC: A versatile method for rapid analysis of pharmaceutical formulations and comparison with other chromatographic techniques and its applications // Indian J. Res. Pharm. Biotechnol. 2014. V. 2. № 3. P. 1209.
Spangenberg B., Poole C.F., Weins Ch. Quantitative Thin-Layer Chromatography. A practical survey. Heidelberg: Springer, 2011. 388 p. https://doi.org/10.1007/978-3-642-10729-0
Jupille T., Perry J. Programmed multiple development in thin-layer chromatography // Science. 1976. V. 194. № 4262. P. 288. https://doi.org/10.1126/science.968483
Berezkin V.G., Kulakova N.Y., Khrebtova S.S. Three-dimensional thin-layer chromatography on plates with open and closed adsorption layers // J. Planar Chromatogr. Mod. TLC. 2009. V. 22. № 5. P. 313. https://doi.org/10.1556/JPC.22.2009.5.1
Герасимов А.В., Малахова И.И., Красиков В.Д. Идентификация окрашенных веществ в тонкослойной хроматографии с применением компьютерной обработки // Журн. прикл. химии. 2000. Т. 73. № 10. С. 1640.
Малахова И.И., Беляев А.Н., Красиков В.Д. Тонкослойная хроматография ряда переходных металлов // Сорбционные и хроматографические процессы. 2015. Т. 15. № 6. С. 802.
Kalasz H., Bathori M. Pharmaceutical applications of TLC // LC GC Europe. 2001. V. 14. № 5. P. 311.
Ferenczi-Fodor K., Lauko A., Wiskidenszky A., Vegh Z., Jszaszy K. Chromatographic and spectroscopic investigation of irreversible adsorption in conventional TLC and HPTLC and in OPLC // J. Planar Chromatogr. 1999. V. 12. P. 30.
Reich E., Schibli A. Stationary phases for planar separations – Plates for modern TLC // LC GC. 2015. V. 23. P. 58.
Ludanyi K., Gomory A., Klebovich I., Monostory K., Vereczkey L., Ujszaszy K., Vekey K. Application of TLC-FAB mass spectrometry in metabolism research // J. Planar. Chromatogr. Mod. TLC. 1997. V. 10. № 2. P. 90.
Brzezinka H., Dallakian P., Budzikiewicz H. Thin-layer chromatography and mass spectrometry for screening of biological samples for drugs and metabolites // J. Planar Chromatogr. 1999. V. 12. № 2. P. 96.
Snyder L.R. Principles of Adsorption Chromatography. N.Y.: M. Dekker, 1968. 413 p.
Беленький Б.Г., Виленчик Л.З. Хроматография полимеров. М.: Химия, 1978. 344 с.
Belenky B.G., Gank1na E.S., Tennikov M.B., Vilenchik L.Z. Fundamental aspects of adsorption chromatography of polymers and their experimental verification by thin-layer chromatography // J. Chromatogr. 1978. № 147. P. 99. https://doi.org/10.1016/S0021-9673(00)85121-3
Снайдер Ллойд Р., Киркленд Джозеф Дж., Долан Джон У. Введение в современную жидкостную хроматографию. М.: Техносфера, 2020. 960 с.
Беленький В.Г., Нестеров В.В., Ганкина Э.С. Теория тонкослойной хроматографии. II. Cравнение теории с экспериментальными результатами // Журн. физ. химии 1968. Т. 42. № 11. С. 2876.
Киселев A.B., Яшин Я.И. Газо-адсорбционная хроматография. М.: Наука, 1967. 256 с.
Balger J.C., Michaels A.S. Molecular structure and electrostatic interaction at polymer-solid interface / Interface Conversion for Polymer Coatings / Eds. Weiss P., Dale Cheever G., American Elsevier Publishing Co., 1968. P. 3.
Belenky B.G., Nesterov V.V. Gankina E.S. Smirnov M.M. A dynamic theory of thin layer chromatography // J. Chromatogr. 1967. V. 31. P. 360. https://doi.org/10.1016/s0021-9673(01)86084-2
Snyder L.R. Classification of the solvent properties // J. Chromatogr. Sci. 1978. V. 16. P. 223.
Киселев А.В., Лыгин В.И. Энергетические и спектральные проявления специфической молекулярной адсорбции / Основные проблемы теории физической адсорбции. М.: Наука, 1970. С. 132.
Pfaab G. The mobile phase in thin-layer chromatography / Modern Thin-layer Chromatography / Eds. Larionov O., Kolomiets L. Russian Academy of Science, Moscow, 1994, p. 85 (translated into Russian).
Nyiredy Sz., Evdelmeier C.A., Meier B. TLC mobile phase optimization procedure using the “PRIZMA” model // Planta Med. 1985. P. 241.
Красиков В.Д. Современная планарная хроматография // Журн. аналит. химии. 2003. Т. 58. № 8. С. 792.
Snyder L.R. Classification of the solvent properties // J. Chromatogr. Sci. 1978. V. 16. P. 223.
Prabha S., Dorothy R., Jancirani S., Rajendran S., Singh G., Senthil K.S. Recent advances in the study of toxicity of polymer-based nanomaterials // Nanotoxicity. 2020. P. 143. https://doi.org/10.1016/b978-0-12-819943-5.00007-5
Duncan R. Polymer conjugates as anticancer nanomedicines // Nat. Rev. Cancer. 2006. V. 6. P. 688. https://doi.org/10.1038/nrc1958
Li I., Alsehli M., Scotti L., Tullius Scotti M., Tsai S.T., Yu R.S., Chen J.C. Progress in polymeric nano-medicines for theranostic cancer treatment // Polymers. 2020. V. 12. № 3. P. 598. https://doi.org/10.3390/polym12030598
Gorshkov N.I., Murko A.Y., Zolotova Y.I., Nazarova O.V., Krasikov V.D., Panarin E.F., Shatik S.V. Introduction of ${{{\text{Re}}\left( {{\text{CO}}} \right)_{{\text{3}}}^{{\text{ + }}}} \mathord{\left/ {\vphantom {{{\text{Re}}\left( {{\text{CO}}} \right)_{{\text{3}}}^{{\text{ + }}}} {^{{{\text{99}}}}{\kern 1pt} {\text{mTc}}\left( {{\text{CO}}} \right)_{{\text{3}}}^{{\text{ + }}}}}} \right. \kern-0em} {^{{{\text{99}}}}{\kern 1pt} {\text{mTc}}\left( {{\text{CO}}} \right)_{{\text{3}}}^{{\text{ + }}}}}$ organometallic species into vinylpyrrolidone-allyliminodiacetate copolymers // Polymers. 2021. T. 13. № 11. P. 1823 https://doi.org/10.3390/polym1311183210.3390/polym13111832
Panarin E.F., Solovskij M.V., Zaikina N.V., Afinogenov G.E. Biological activity of cationic polyelectrolytes // Macromol. Chem. Suppl. 1985. V. 9. P. 25. https://doi.org/10.1002/MACP.1985.020091985104
Панарин Е.Ф., Лавров Н.Л., Соловский М.В., Шальнова Л.И. Полимеры – носители биологически активных веществ. СПб: ЦОП “Профессия”, 2014. 304 с.
Красиков В.Д. Основы планарной хроматографии. СПб.: Химиздат, 2005. 232 с.
Janvi S., Rajendra K. A Critical review on analytical methods for recently approved FDC drugs: Pregabalin and Etoricoxib // Crit. Rev. Anal. Chem. 2020. P. 1. https://doi.org/10.1080/10408347.2020.1855411
Отто М. Современные методы аналитической химии. М.: Техносфера, 2021. 656 с.
Майер В.Р. Практическая высокоэффективная жидкостная хроматография. М.: Техносфера, 2017. 408 с.
Unger K.K. Porous Silica, Its Properties and Use as Support in Column Liquid Chromatography (Journal of Chromatography Library. V. 16). Amsterdam: Elsevier: 1979. 863 p.
Абилев С.К., Вайсбург А.Ф., Дегтярь В.Г., Ежова И.Е., Лукина Г.П., Тарасов А.П., Тяглов Б.В., Дегтерев Е.В., Крылов В.И., Новицкий А.П., Полубенцева М.И., Малахова И.И., Красиков В.Д. Видеоденситометр “Денсискан” для количественной тонкослойной хроматографии // Биотехнология. 1993. № 5. С. 44.
Кирш Ю.Э., Семина Н.В., Калниньш К.К., Шаталов Г.В. Радикальная сополимеризация N-винилпирролидона и N-винилформамида // Высокомолекулярные соединения. Б. 1996. Т. 38. № 11. С. 1905.
Gu L., Zhu S., Hrymak A.N. Acidic and basic hydrolysis of poly(N-vinylformamide) // J. Appl. Polym. Sci. 2002. V. 86. P. 3412. https://doi.org/10.1002/app.11364
Nurok D. Strategies for optimizing the mobile phase in planar chromatography // Chem. Rev. 1989. V. 89. № 2. P. 363. https://doi.org/10.1021/cr00092a007
Kirkland J.J., Glajch J.L. Optimization of mobile phase for multisolvent gradient elution liquid chromatography // J. Chromatogr. 1983. V. 255 P. 27. https://doi.org/10.1016/S0021-9673%2801%2988271-6
Schoenmaker P.J. Optimization of chromatographic selectivity – A guide to method development. Ch. 5 / Optimization Procedures (Journal of Chromatography Library. V. 35). Amsterdam, Oxford, New York, Tokyo: Elsevier, 1986. P. 170. https://doi.org/10.1016/S0301-4770(08)60419-4
Nyiredy Sz., Evdelmeier C.A., Sticher O. “PRIZMA” Optimierung der mobilen Phase in der praparativen CLC, GIT Supplement. 1987. № 4. P. 208
Снайдер Л.Р., Киркленд Д.Д., Доланд Д.У. Введение в современную жидкостную хроматографию. М.: Техносфера, 2020. 960 с.
Лисичкин Г.В. Модифицированные кремнеземы в сорбции, катализе и хроматографии. М.: Химия, 1985. 246 с.
Vlasov N.N., Pliss G.B. The role of beta-glucuronidase in the tumor process and cancer prevention // Probl. Oncol. 1990. V. 36. № 9. P. 1037.
Redinbo M.R., Mani S., Williams A., Scott J., Yeh L.A., Wallace B.D., Lane K.T. Selective beta-glucuronidase inhibitors as a treatment for side effects of camptothecin antineoplastic agents. U.S. Patent US8557808B2, 15 October 2013.
Панарин Е.Ф. Биологически активные полимерные наносистемы // Изв. АН. Сер. хим. 2017. № 10. С. 1812.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии