

Журнал аналитической химии, 2023, T. 78, № 6, стр. 538-545
Экспрессное газохроматографическое определение фенола и крезолов в воде методом экстракционного вымораживания
В. Н. Бехтерев *
Сочинский государственный университет, инженерно-экологический факультет
354000 Сочи, ул. Пластунская, 94, Россия
* E-mail: vic-bekhterev@yandex.ru
Поступила в редакцию 05.10.2022
После доработки 12.11.2022
Принята к публикации 12.11.2022
- EDN: KZDTHY
- DOI: 10.31857/S0044450223040059
Аннотация
Разработана экспрессная методика определения фенола и изомеров крезола в питьевой, морской и загрязненной бытовой сточной воде, основанная на одностадийном извлечении аналитов с помощью экстракционного вымораживания в условиях центрифугирования пробы. В комбинации с газовой хроматографией методика обеспечивает минимальные пределы обнаружения каждого из фенолов в 10 мл воды на уровне 0.0005 мкг/мл. Продолжительность пробоподготовки не превышает 30 мин, расход экстрагента этоксиэтана 1.4 мл. В сравнении с традиционно применяемыми на этапе предварительной подготовки пробы жидкостной и твердофазной экстракцией, предложенная методика проще и более экологична. Исключены этапы фильтрования проб и обезвоживания экстрактов, минимизировано количество химической посуды.
В последнее время в мире обостряется ситуация с антропогенной загрязненностью водной среды. Для оперативного выявления и локализации источников загрязнений требуется проведение большого количества исследований. В этой связи важен поиск экспрессных, надежных, недорогих способов определения примесей в воде.
Способы определения в питьевой воде низкомолекулярных фенолов должны обладать низкими пределами обнаружения в связи с их высокой токсичностью и, как следствие, низкими предельно допустимыми концентрациями (ПДК). Так, ПДК фенола в воде хозяйственно-бытового назначения в Российской Федерации составляет 0.001 мг/л [1]. Несмотря на значительный прогресс в развитии инструментальных методов анализа, при таких нормативах содержания фенолов в воде процедура анализа должна включать этап предварительного концентрирования аналитов.
Миниатюризация жидкостной экстракции начала развиваться в конце ХХ в. с целью оптимизации пробоподготовки в химическом анализе, уменьшения количества используемых токсичных растворителей [2]. Однако на пути развития этого направления возникли трудности, связанные, прежде всего, с обеспечением удовлетворительной воспроизводимости результатов, упрощением процедуры и снижением стоимости анализа.
Цель настоящего исследования – разработка экспрессной методики определения фенола и изомеров крезола в воде, включая питьевую пресную, морскую, а также довольно сложный объект химического анализа – сточную бытовую канализационную воду на примере локальной очистной системы. В качестве основного метода использовали широко представленную в лабораториях газовую хроматографию (ГХ), которая является высокочувствительным, надежным, селективным аналитическим инструментом [3]. На этапе пробоподготовки применяли способ экстракционного вымораживания в режиме центрифугирования (ЭВЦ) образца, характеризующийся экспрессностью, простотой выполнения и высокой эффективностью извлечения целевых органических веществ, в том числе гидрофильных, из водных сред [4–6].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты. Использовали ГСО 7270-96 “СО состава фенола в этаноле”, СОП 030603 ER-SPH 6 “о-крезол”, СОП 0307-03 ER-SPH 7 “п-крезол”, СОП 0308-03 ER-SPH 8 “м-крезол”; ацетонитрил Сорт 0 (ООО “НПК Криохром”, Россия) и этоксиэтан х.ч., ТУ 2600-001-43852015-10 (ООО “Кузбассоргхим”, Россия). Все реактивы были аналитической чистоты.
Аппаратура. Для приготовления модельных и стандартных растворов органических веществ, определения массы получаемого экстракта использовали аналитические весы Mettler Toledo XS 205DU. Исследовали получаемые экстракты и стандартные смеси на газовом хроматографе Кристаллюкс-4000М (Россия) с пламенно-ионизационным детектором (ПИД) и капиллярной колонкой длиной 60 м, внутренним диаметром 0.53 мм, стационарная фаза ZB-WAX, толщина пленки 1.00 мкм (Phenomenex, США).
Методика эксперимента. Процедура проведения экстракционного вымораживания в условиях одновременного центрифугирования описана в работе [6]. Исследуемые образцы модельных водных растворов аналитов готовили добавлением к воде в стеклянной виале емк. 12 мл необходимого количества стандартного раствора аналита в ацетонитриле или этоксиэтане, используемых в качестве экстрагентов. Регулировали рН добавлением водного раствора серной кислоты (1 : 1). Флакон герметично закрывали завинчивающейся пробкой. Интенсивно перемешивали в течение 1 мин и помещали в ротор криоэкстрактора ЭВЦ-2 (Россия) для кристаллизации водной части образца и получения экстракта в виде незамерзающей части образца. Условия проведения процедуры ЭВЦ: температура –(30 ± 2)°С, скорость вращения ротора 2000–4000 об./мин (центробежное ускорение 400–1650 g), продолжительность процедуры 25 мин. После этапа ЭВЦ верхний, жидкий органический слой (экстракт) отделяли декантацией, взвешивали и подвергали ГХ-анализу. При необходимости концентрировали упариванием на воздухе при комнатной температуре, контролируя степень концентрирования взвешиванием экстракта на аналитических весах.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Анализу имеющегося арсенала методов экстракции и пробоподготовки в доступной научной литературе посвящено достаточное количество обзоров [2, 7–11]. Используемые в настоящее время в Российской Федерации официальные методики газохроматографического определения фенолов в водах базируются на их предварительном концентрировании жидкостной экстракцией этилацетатом в нативном виде с промежуточной процедурой реэкстракции [12] или после дериватизации путем бромирования [13]. При этом для загрязненных сточных вод этапу экстракции предшествует стадия перегонки с паром. Вместе с тем нельзя не согласиться с авторами работы [14], утверждающими, что многостадийность пробоподготовки существенным образом отражается на точности определения, особенно при низких содержаниях аналитов. Следует добавить, что бромирование фенолов [13] или переведение их в ацетильные производные [14] также повышает риск роста погрешности результатов анализа, поскольку не исключено образование побочных продуктов реакции и изомеров. На направление и глубину протекания дериватизации влияет множество факторов, в том числе температура, качество реактивов, матрица пробы и прочее. Кроме того, поскольку при этом применяется большое количество лабораторной посуды, трудоемкость данного этапа дополнительно связана и с необходимостью поддержания ее чистоты. Указанные недостатки снижают производительность лаборатории.
Официальный метод [15], использующий твердофазную экстракцию (ТФЭ), еще более трудоемок. Для проведения одного анализа, включая пробоподготовку и получасовое хроматографирование (ВЭЖХ), требуется 15 ч. Необходима предварительная мембранная фильтрация пробы, оптимизация работы ТФЭ-картриджей, так как их свойства зависят от партии поставки и от ионного фона матрицы пробы, присутствия соэкстрактивных веществ [16]. Существенным недостатком данного способа является и тот факт, что применяемые ТФЭ-картриджи, мембранные фильтры являются одноразовым расходным материалом. Утилизация подобных отходов после использования строго регламентирована, что в совокупности с их ценой отражается на стоимости исследований. Кроме того, это не соответствует принципам “зеленой химии”.
Принимая во внимание перечисленные выше недостатки официальных методов, а также опыт использования ЭВЦ в качестве этапа пробоподготовки при определении органических кислот [17] и оснований [6], мы предложили новый способ определения фенолов в воде. На этапе изолирования аналитов из водной матрицы применяли ЭВЦ. К настоящему времени способ ЭВЦ реализован в серийно выпускаемом криоэкстракторе ЭВЦ-2 (ООО НПФ “Метахром”/ООО “Химбиомед”, Россия).
При оптимизации процедуры пробоподготовки в качестве сосудов, в которых выполняли ЭВЦ, использовали стеклянные многоразовые виалы емк. 12 мл (диаметр 18.5 мм, высота 66 мм) типа LLG-Screw Neck Vials for Storage Purposes/ND15 (National Scientific, кат. № 7.616 655) с плоским дном. В настоящем исследовании применяли ротор с посадочными отверстиями именно под указанные виалы. Однако криоэкстрактор комплектуется также ротором, позволяющим использовать пенициллиновые флаконы. Установлено, что смена ротора и переход от использования виал к применению пенициллиновых флаконов не отражаются на эффективности экстракции изучаемых аналитов в диапазоне скоростей вращения ротора от 2000 до 4000 об./мин и температуре экстракции 30 ± 2°С.
Ранее полученные результаты извлечения органических кислот и фенолов из воды с помощью экстракционного вымораживания (ЭВ) без режима центрифугирования показали, что наиболее эффективными экстрагентами для таких гидрофильных аналитов являются полярные растворители, например ацетонитрил, диэтиловый эфир, спирты [18, 19]. Однако одним из важных требований к экстрагентам в случае применения газовой хроматографии на заключительном этапе анализа является отсутствие воды в анализируемых экстрактах, негативно влияющей на работоспособность капиллярной колонки.
Теоретические основы метода ЭВ базируются на большом массиве экспериментальных данных. Предложен критерий оценки эффективности экстракции целевых компонентов из воды данным методом [6, 17–19] – коэффициент распределения аналита *Keq между кристаллической поверхностью образующегося льда и добавленным незамерзающим растворителем (жидкая фаза). В модели ЭВ это угловой коэффициент линейной зависимости концентрации аналита сорг в получаемом экстракте от величины Mo/Vэкстр, где Mo это масса аналита в исходной пробе, а Vэкстр – объем экстрагента [6, 17–19]. Показано, что эта величина является индивидуальным параметром аналита, зависящим от его физико-химических свойств, а также природы экстрагента. Чем больше *Keq, тем выше эффективность извлечения аналита из воды. Кроме того, при использовании ацетонитрила *Keq практически равен степени извлечения аналита из воды, поскольку ее растворимость в данном экстрагенте при –30°С не превышает 4% [20].
Установили, что при экстракционном вымораживании изучаемых аналитов в ацетонитрил в условиях центрифугирования пробы коэффициент *Keq составил для фенола 0.95 ± 0.09, о-крезола 1.0 ± 0.1, м-крезола 0.88 ± 0.09 и п-крезола 1.0 ± ± 0.1. Полученные параметры в пределах погрешности совпадают с ранее установленными в условиях отсутствия воздействия поля центробежных сил [19]. Таким образом, центрифугирование образца не влияет на эффективность экстракционного вымораживания фенолов в ацетонитрил. Подобное поведение аналитов выявлено для наиболее гидрофильных членов гомологического ряда одноосновных карбоновых кислот: коэффициент *Keq возрастал с переходом в режим центрифугирования во время экстракционного вымораживания, лишь начиная с масляной кислоты и далее [17]. Кроме того, коэффициент *Keq практически не изменялся для уксусной и пропионовой кислот [17]. Вместе с тем режим ЭВЦ дает возможность снизить объемную долю экстрагента в исходной смеси в случае ацетонитрила до 6%, что позволяет достичь 16-кратного концентрирования фенолов в экстракте в результате одноразовой процедуры экстракции.
Однако, как показал эксперимент, при такой степени концентрирования с учетом применения ГХ-ПИД предел определения указанных фенолов в воде составляет 0.006 мг/л, что превышает их ПДК [1]. Предел определения не удалось снизить и путем объединения параллельно получаемых ацетонитрильных экстрактов нескольких проб с последующим упариванием (в криоэкстракторе ЭВЦ-2 восемь посадочных мест).
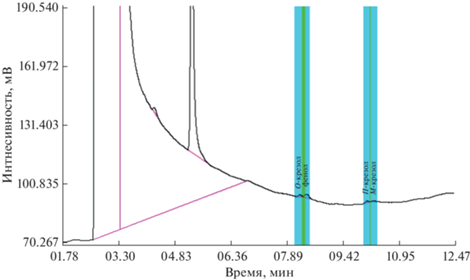
Поскольку в экстракте после процедуры ЭВЦ содержится до 4% воды, удаление ацетонитрила ведет к увеличению ее доли в экстракте. Это, в свою очередь, сопровождается существенным уширением фронта растворителя на хроматограмме и ростом фонового сигнала детектора (рис. 1), что в итоге снижает чувствительность детектирования аналитов.
Рис. 1.
Хроматограмма объединенного ацетонитрильного экстракта четырех проб водопроводной воды с добавкой фенолов (содержание каждого 0.01 мкг/мл), полученного способом однократного ЭВЦ и дополнительно упаренного в 4.5 раза. Условия пробоподготовки: объем пробы 11 мл, объем экстрагента 0.7 мл, центрифугирование в течение 25 мин при 4000 об/мин и –(30 ± 2)°С.
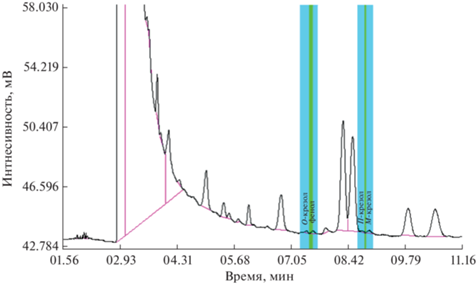
В качестве другого экстрагента опробовали этоксиэтан. При –30°С после ЭВЦ в нем содержится значительно меньше воды, поскольку даже при комнатной температуре растворимость воды в эфире не превышает 2.7% [21]. Кроме того, время удерживания эфира (~2.85 мин) в хроматографической колонке меньше, чем ацетонитрила (~2.93 мин), что заметно улучшает условия детектирования фенолов. Как видно из рис. 2, к моменту их появления на хроматограмме фронт растворителя (эфира) фактически снижается до фонового уровня. Кроме того, угловой коэффициент линейной зависимости концентрации аналита в экстракте (сорг) от величины Mo/Vэкстр при ЭВЦ, характеризующий эффективность экстракции, при замене ацетонитрила на эфир существенно возрастает. Для фенола коэффициент *Keq достиг значения 2.89, а в случае изомеров крезола для о-крезола – 2.82, м-крезола – 2.68, п-крезола – 2.61. Полученный результат, по-видимому, обусловлен способностью диэтилового эфира, который, как известно, обладает заметными свойствами основания Льюиса, образовывать с фенолами как с кислотами оксониевые соли за счет межмолекулярной водородной связи. Свидетельство этому – батохромный сдвиг полосы поглощения при 205 нм в УФ-спектре фенола в растворе этоксиэтана [22]. Благодаря этому повышается эффективность экстракции: коэффициент распределения аналита *Keq между получаемым жидким экстрактом и поверхностью образующегося льда при переходе от ацетонитрила к эфиру возрастает практически в три раза.
Рис. 2.
Хроматограмма эфирного экстракта, полученного однократной процедурой ЭВЦ из морской воды с добавкой фенолов (содержание каждого 0.001 мкг/мл), дополнительно упаренного в 3.3 раза. Условия пробоподготовки: объем пробы 10 мл, объем экстрагента 1.4 мл, центрифугирование в течение 25 мин при 2000 об./мин и –30 ± 2°С.
Следует отметить, что именно значительное повышение эффективности ЭВЦ при замене ацетонитрила этоксиэтаном дало возможность достичь предела определения фенолов в воде на уровне 0.0025 мг/л в результате однократной процедуры экстракции из объема пробы 10 мл в 1.4 мл эфира (табл. 1). Кроме того, за счет одностадийности и отсутствия каких-либо дополнительных операций во время процедуры пробоподготовки (фильтрования, осушки экстрактов и пр.) обеспечивается хорошая повторяемость результатов анализа. Относительное стандартное отклонение не превышает 20%. Получаемый объем эфирного экстракта составляет более 0.25 мл (масса 0.194 ± ± 0.008 г), что позволяет дополнительно его сконцентрировать путем упаривания, контролируя кратность взвешиванием на аналитических весах. В итоге установили, что разработанный способ после упаривания экстракта обеспечивает предел определения фенола и изомеров крезола в воде 0.0005 мг/л (для каждого аналита в отдельности). Достигнутый результат открывает перспективы применения разработанного метода в целях оперативного санитарно-гигиенического контроля качества воды.
Таблица 1.
Предел определения фенола в воде в результате однократного экстракционного вымораживания в условиях центрифугирования образца (ЭВЦ) в комбинации с газовой хроматографией
| Вода | Соотношение вода : этоксиэтан (мл) | Масса экстракта, г | Предел определения в воде, cнвод, мг/л | Стандартное отклонение, s(x) (n = 8) |
|---|---|---|---|---|
| Водопроводная (минерализация 0.1 г/л) |
10 : 1.4 | 0.194 ± 0.009 | 0.0025 | ±0.0003 |
| 10 : 1.0 | 0.08 ± 0.061 | 0.002 | ±0.0016 | |
| 10 : 0.7 | 0 | – | – | |
| Морская (минерализация 16.1 г/л) | 10 : 1.4 | 0.34 ± 0.042 | 0.003 | ±0.0005 |
| 10 : 1.0 | 0.092 ± 0.075 | 0.002 | ±0.0017 | |
| 10 : 0.7 | 0.012 ± 0.009 | 0.001 | ±0.0008 | |
| Сточная, локальной очистной системы |
10 : 1.4 | 0.31 ± 0.041 | 0.005 | ±0.0007 |
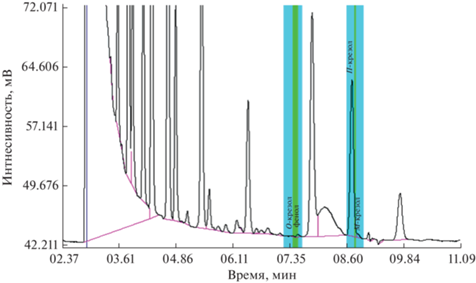
Особенно это актуально при мониторинге бытовых сточных вод. Помимо огромной массы растворенных антропогенных органических и неорганических веществ, они содержат коллоидные частицы и биологические объекты (бактерии, грибки и пр.). Все это усложняет применение жидкостной и твердофазной экстракции для предварительной подготовки проб к анализу инструментальными методами. Метод ЭВЦ обеспечивает возможность напрямую, без каких-либо предварительных манипуляций анализировать такого рода пробы (см. фотоизображение исходной пробы и получаемого экстракта на рис. 3). Исследование образцов сточных вод методом добавок показало, что достигнутый предел определения фенолов составляет 0.005 мг/л (табл. 1). Его можно снизить путем контролируемого взвешиванием концентрирования экстракта, объем которого составляет более 0.4 мл (масса более 0.3 г.). Как видно из хроматограммы ЭВЦ-экстракта реальной пробы сточной воды локальной очистной системы одной из минигостиниц г. Сочи, в ней присутствуют изучаемые фенолы в заметных количествах (рис. 4). Наличие множества других хроматографических пиков на рис. 4 свидетельствует о значительной загрязненности пробы другими веществами, в том числе органическими. Вместе с тем из рис. 4 следует, что выбранные условия хроматографирования обеспечивают возможность надежного детектирования данных аналитов.
Рис. 3.
Фотоизображение образца пробы бытовых сточных вод локальной очистной системы в ходе этапа пробоподготовки с помощью ЭВЦ перед ГХ-анализом. (а) – исходные пробы бытовых сточных вод локальной очистной системы гостиницы; (б) – проба после этапа ЭВЦ; (в) – отделенный эфирный экстракт (слева) и кристаллическая часть пробы (справа) после ЭВЦ.

Рис. 4.
Хроматограмма эфирного экстракта пробы бытовых стоков локальной очистной системы одной из мини-гостиниц г. Сочи, полученного методом однократного ЭВЦ. Условия пробоподготовки: объем пробы 10 мл, объем экстрагента 1.4 мл, центрифугирование в течение 25 мин при 2000 об/мин и –(30 ± 2)°С.
Установлено, что минерализация при переходе от водопроводной пресной воды (до 1 г/мл) к морской (14–16 г/л) существенно не влияет на предел определения фенолов (табл. 1). Обнаружен даже некоторый высаливающий эффект, поскольку при прочих равных условиях (объем пробы 10 мл, объем этоксиэтана 1.4 мл, скорость вращения ротора 2000 об/мин) масса получаемого экстракта составила 0.341 ± 0.042 г. Из табл. 1 следует также, что предлагаемый способ определения изучаемых фенолов в питьевой, морской воде из Черного моря (бассейн дельфинария) с уменьшением доли экстрагента в смеси дает возможность их детектирования на уровне 0.001 мг/л, т.е. на уровне ПДК. Однако за счет роста вариабельности массы (объема) получаемого экстракта снижается воспроизводимость результатов анализа, увеличивается стандартное отклонение.
Таким образом, предложена новая экспрессная методика определения фенола и изомеров крезола в воде, в том числе питьевой водопроводной и морской, а также в бытовых сточных водах, основанная на однократном извлечении аналитов с помощью ЭВЦ в диэтиловый эфир. Схема анализа представлена на рис. 5. Процедура исследования методом добавок включает следующие этапы:
Рис. 5.
Схема газохроматографического определения фенолов в воде методом экстракционного вымораживания.
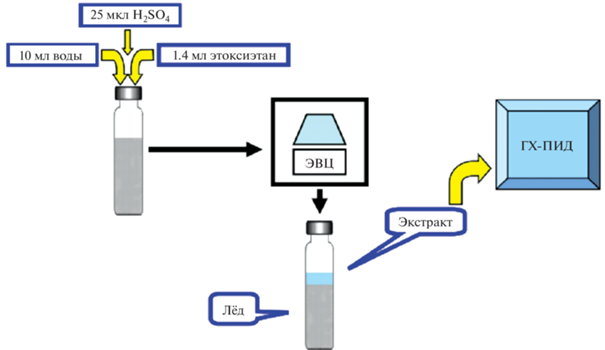
1) в первую виалу (проба) помещают 10 мл пробы воды, добавляют 1.4 мл этоксиэтана и 0.025 мл 50%-ной серной кислоты в воде;
2) во вторую виалу (проба с добавкой) помещают 10 мл пробы воды, добавляют 1.4 мл стандартного раствора фенолов в этоксиэтане и 0.025 мл 50%-ной серной кислоты в воде;
3) плотно закрыв виалы завинчивающимися пробками, после одноминутного интенсивного встряхивания их помещают диаметрально в ротор криоэкстрактора;
4) проводят экстракционное вымораживание в условиях центрифугирования при скорости вращения ротора 2000 об./мин и –30°С в течение 25 мин;
5) по окончании ЭВЦ образовавшиеся жидкие экстракты отделяют путем декантации в пенициллиновые флаконы, оценивая взвешиванием их количества;
6) проводят ГХ-определение фенолов в полученных экстрактах, при необходимости концентрируют упариванием, контролируя массу экстрактов взвешиванием. Содержание фенолов в пробе спр (мкг/мл) рассчитывают в соответствии с известным алгоритмом метода добавок:
(1)
${{c}_{{{\text{пр}}}}} = {{{{S}_{{{\text{пр}}}}}{{m}_{{{\text{доб}}}}}} \mathord{\left/ {\vphantom {{{{S}_{{{\text{пр}}}}}{{m}_{{{\text{доб}}}}}} {[({{S}_{{{\text{доб}}}}}--{{S}_{{{\text{пр}}}}}){{V}_{{{\text{пр}}}}}]}}} \right. \kern-0em} {[({{S}_{{{\text{доб}}}}}--{{S}_{{{\text{пр}}}}}){{V}_{{{\text{пр}}}}}]}},$Общая продолжительность анализа, включая этап хроматографирования экстрактов (ГХ-ДИП), составляет менее 1 ч, в том числе продолжительность пробоподготовки не превышает 30 мин. Метод обеспечивает возможность определения указанных фенолов на уровне ПДК в питьевой воде.
Преимущества предложенной методики:
1) пробоподготовка осуществляется в одну стадию без каких-либо дополнительных процедур (фильтрование проб, осушка экстрактов и прочее);
2) процедура проста, отсутствуют особые требования к квалификации специалиста;
3) методика универсальна, так как последовательность операций одинакова для питьевой, морской воды, бытовой сточной воды;
4) низкая стоимость в отношении расходных материалов, химической посуды и реактивов (необходимый объем экстрагента диэтилового эфира 1.4 мл на пробу, не используются дополнительные материалы, в том числе осушители, фильтры и прочее);
5) методика отвечает требованиям “зеленой химии” ввиду того, что не используются полимерные и иные специально утилизируемые материалы, а также минимизирован объем экстрагента;
6) подготовка проб к ГХ-ДИП-исследованию проводится при отрицательных температурах, что способствует улучшению условий труда и соблюдению норм техники безопасности, так как значительно уменьшается летучесть растворителя и извлекаемых токсичных веществ.
Предложенная методика характеризуется экспрессностью и высокой воспроизводимостью результатов, что позволит в перспективе применять ее в мониторинге качества воды не только в отношении фенолов, но и других экологически опасных органических веществ. Важно также, что поставленная в работе задача решена с применением специально разработанного для пробоподготовки устройства – криоэкстрактора, серийный выпуск которого начат в 2021 г. в России.
Список литературы
Предельно допустимые концентрации (ПДК) химических веществ в воде водных объектов хозяйственно-питьевого и культурно-бытового водопользования: Гигиенические нормативы. ГН 2.1.5.1315-03. М.: Российский регистр потенциально опасных химических и биологических веществ МЗ РФ, 2003. 154 с.
Дмитриенко С.Г., Апяри В.В., Горбунова М.В., Толмачева В.В., Золотов Ю.А. Гомогенная жидкостная микроэкстракция органических соединений // Журн. аналит. химии. 2020. Т. 75. № 11. С. 963.
100-лет хроматографии / Отв. ред. Руденко Б.А. М.: Наука, 2003. 739 с.
Бехтерев В.Н. Способ извлечения органических веществ из водных сред экстракционным вымораживанием в поле центробежных сил. Патент РФ на изобретение № 2564999. Зарег. 11.09.2015, опубл. 10.10.15, бюл. № 28, приоритет 14.04.2014.
Bekhterev V.N. A method of recovery of organic substances from aqueous media by freeze-out extraction under the action of centrifugal force. Patent EPO №3357873. 2019. European Patent Bulletin. № 45. 06.11.2019. P. 940.
Бехтерев В.Н. Экстракционное вымораживание с центрифугированием – новая технология пробоподготовки в химическом анализе на примере органических оснований // Журн. аналит. химии. 2021. Т. 76. № 9. С. 859.
Koning S., Janssen H.-G., Brinkman U.A.Th. Modern methods of sample preparation for GC analysis // Chromatographia. 2009. V. 69. № 1. P. 33.
Raynie D.E. Modern extraction techniques // Anal. Chem. 2010. V. 82. № 12. P. 4911.
Ballesteros-Gomez A. Rubio S. Recent advances in environmental analysis // Anal. Chem. 2011. V. 83. № 12. P. 4579.
Цизин Г.И. Развитие методов концентрирования микрокомпонентов в России (1991–2010 гг.) // Журн. аналит. химии. 2011. Т. 66. № 11. С. 1135. (Tsysin G.I. Method of the preconcentration of trace components: Development in Russia (1991–2010) // J. Anal. Chem. 2011. V. 66. № 11. P. 1020.).
Федотов П.С., Малофеева Г.И., Савонина Е.Ю., Спиваков Б.Я. Твердофазная экстракция органических веществ: нетрадиционные методы и подходы // Журн. аналит. химии. 2019. Т. 74. № 9. С. 163.
Методика измерений массовой концентрации летучих фенолов в питьевых. поверхностных подземных пресных и сточных водах газохроматографическим методом. ПНД Ф 14.1:2:3:4.244-2007 (ФР.1.31.2007.03818). М.: ФБУ “ФЦАО”, 2011. 16 с.
Методика измерений массовой концентрации фенола в пробах питьевых. природных и сточных водах методом газожидкостной хроматографии. ПНД Ф 14.1:2:4.177-02 (ФР.1.31.2007.03818). М.: ФБУ “ФЦАО”, 2011. 18 с.
Кириченко В.Е., Первова М.Г., Пашкевич К.И., Назаров А.С. Определение фенолов в воде методами газовой хроматографии в виде ацетильных производных // Аналитика и контроль. 2001. Т. 5. № 1. С. 70.
Массовая концентрация фенолов в водах. Методика измерений методом высокоэффективной жидкостной хроматографии с применением твердофазной экстракции. Руководящий документ РД 52.18.750-2010. Обнинск: Министерство природных ресурсов и экологии. Росгидромет, 2011. 47 с.
Зайцев В.Н., Зуй М.Ф. Твердофазное микроэкстракционное концентрирование // Журн. аналит. химии. 2014. Т. 69. № 8. С. 1.
Бехтерев В.Н. Экстракционное вымораживание одноосновных карбоновых кислот из воды в ацетонитрил в условиях действия центробежных сил // Журн. физ. химии. 2016. Т. 90. № 10. С. 1558.
Bekhterev V.N. Extractive freezing-out in the analysis of organic compounds in the aqueous media // Mendeleev Communications. 2007. V. 17. № 4. P. 241.
Бехтерев В.Н. Выделение фенолов из воды экстракционным вымораживанием // Журн. аналит. химии. 2008. Т. 63. № 10. С. 1045.
Schneider G.M. Aqueous solutions at pressures up to 2 GPa: Gas–gas equilibria, closed loops, high-pressure immiscibility, salt effects and related phenomena // Phys. Chem. Chem. Phys. 2002. V. 4. P. 845.
Rowley H.H., Reed Wm.R. Solubility of water in diethyl ether at 25° // J. Am. Chem. Soc. 1951. V. 73. № 6. P. 2960.
Dearden J.C., Forbs W.F. Light absorption studies. Part XIV. The ultraviolet absorption spectrum of phenols // Can. J. Chem. 1959. V. 37. P. 1294.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии