

Журнал аналитической химии, 2023, T. 78, № 6, стр. 520-527
Особенности определения эфиров ксантофиллов в условиях обращенно-фазовой ВЭЖХ
В. И. Дейнека a, *, Т. Г. Буржинская a, И. П. Блинова a, Л. А. Дейнека a
a Белгородский государственный национальный исследовательский университет,
Институт фармации, химии и биологии
308015 Белгород, ул. Победы, 85, Россия
* E-mail: deineka@bsu.edu.ru
Поступила в редакцию 12.01.2023
После доработки 17.01.2023
Принята к публикации 18.01.2023
- EDN: DWNAHX
- DOI: 10.31857/S0044450223060026
Аннотация
Исследованы особенности хроматографического определения эфиров ксантофиллов на примере диэфиров лютеина цветков бархатцев в условиях обращенно-фазовой ВЭЖХ. Разработанный двухколоночный метод позволил установить, что в пробах с низкой растворимостью каротиноидов в использованном растворителе и на хроматографической колонке при применении подвижной фазы с низкой растворимостью каротиноидов возможно выпадение осадка диэфиров, нарушающее правильность хроматографического определения. Критическим фактором при этом является температура: хранение проб (растворов) в холодильнике не всегда целесообразно – возможно вымораживание основных компонентов. Показано, что использование подвижной фазы, содержащей от 0 до 10 об. % ацетонитрила в ацетоне при температуре не ниже 20°С допустимо для разделения диэфиров полностью транс-лютеина от цис-производных лютеина и производных зеаксантина на “мономерных” С18-фазах.
По типу функциональных групп каротиноиды разделяют на каротины, включающие углеводороды и их эпоксиды, и ксантофиллы, содержащие ОН-группы (или кетогруппы). Ксантофиллы в природных источниках чаще всего этерифицируются несколькими различными высшими жирными кислотами [1]. Это существенно осложняет разделение индивидуальных каротиноидов методом обращенно-фазовой ВЭЖХ [2], поэтому обычная практика – омыление эфиров ксантофиллов перед последующим определением [3, 4].
Среди экономически важных источников эфиров ксантофиллов можно выделить растения рода Tagetes (бархатцы) с оранжевой окраской лепестков цветков [5, 6]. Ксантофиллы (главным образом лютеин) этого растения используют в фармации для предупреждения возрастной макулярной дистрофии, в кормлении птиц, как пищевую добавку и как красители для пищевой промышленности [5]. Уникальность лепестков цветков бархатцев состоит в биосинтезе диэфиров лютеина с высокой концентрацией. Описано несколько примеров применения обращенно-фазовой ВЭЖХ [6–11] для контроля ксантофиллов в высушенных лепестках цветков бархатцев, хотя более обстоятельное исследование каротиноидов бархатцев представлено в работе [12]. Дополнительная информация о составе диэфиров лютеина получена с использованием вместо “мономерной” С18 “полимерной” С30 стационарной фазы [13, 14]. Однако целый ряд вопросов, связанных с хроматографическим определением диэфиров ксантофиллов, остается неисследованным. Это стало особенно важным после публикации работы [15], в которой установлено, что между результатами ВЭЖХ-определения каротиноидов на ряде коммерческих хроматографических колонок и количеством введенного образца может быть несоответствие, причины которого остались невыясненными.
Несоответствия, отмеченные в работе [15], стимулировали проведение настоящего исследования хроматографического поведения эфиров ксантофиллов в условиях обращенно-фазовой хроматографии с оценкой степени открытия (отношения количества найденного вещества к количеству вещества, введенного на колонку) каротиноидов по разработанному в данной работе двухколоночному способу.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использовали экстракты бархатцев (Tagetes erecta), физалиса декоративного (Physalis alkekengi) и плодов рябины (Sorbus aucuparia), выращенных в Белгороде в 2022 г. Лепестки бархатцев, пузыревидно-вздутую при плодах чашечку физалиса и плоды рябины высушивали без доступа прямого солнечного света. Экстракцию осуществляли растиранием растительного материала под слоем экстрагента (ацетона) в фарфоровой ступке до обесцвечивания последней порции экстрагента, все порции экстракта смешивали в мерной посуде и доводили до метки ацетоном.
Для определения концентрации диэфиров ксантофиллов использовали спектрофотометр Shimadzu UV-VIS с кварцевыми кюветами. После соответствующего разбавления фильтрата ацетоном его пропускали через насадочный фильтр с диаметром пор 0.2 мкм. Для расчета использовали коэффициент молярного поглощения 144 500 л · моль–1 · см–1 при 456 нм [16]. Установили, что линейная зависимость между концентрацией каротиноидов и оптической плотностью растворов сохраняется, по крайней мере, в диапазоне оптических плотностей от 0.1 до 1.1.
Для хроматографического разделения использовали экстракты, разбавленные ацетоном до концентрации не более 1.5 × 10–4 М по основному ксантофиллу. Такие экстракты могут храниться при температуре не ниже 20°С до 3 дней в темном месте. Хранения в холодильнике следует избегать.
Для разделения использовали хроматографические колонки 4.6 × 250 мм Kromasil 100-5C18, 4.6 × 250 мм Kromasil 150-5C18, 4.6 × 100 мм Kromasil 100-5C18 и защитную колонку Kromasil C18 с использованием хроматографа Agilent 1200 Infinity с диодно-матричным детектором. Хроматограммы регистрировали при 445 нм, хранили и обрабатывали в ПО Agilent ChemStation.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
При определении эфиров ксантофиллов в экстрактах лепестков бархатцев желательно разделить производные лютеина и зеаксантина, поскольку для профилактики возрастной макулярной дистрофии необходимы не только производные лютеина, но и зеаксантина [17]. Определение диэфиров зеаксантина (ЭЗ) на фоне диэфиров лютеина (ЭЛ) непростая задача, поскольку ЭЛ могут присутствовать не только в форме производных полностью транс-лютеина, но и в форме позднее элюирующейся на “мономерных” С18-стационарных фазах (как и ЭЗ) смеси цис-изомеров. Однако по нашему опыту в свежеприготовленных образцах содержание цис-изомеров заметно меньше содержания ЭЗ, что подтверждается электронными спектрами поглощения соответствующих хроматографических пиков. Следовательно, для приблизительной оценки уровня накопления производных зеаксантина можно использовать площадь пиков, элюирующихся сразу после пиков, соответствующих полностью транс-изомерам ЭЛ. При этом необходимо отделение производных зеаксантина от производных лютеина, в структуре которых один из радикалов относится к ненасыщенным кислотам [12]. Известно, что удерживание триацилглицеринов уменьшается при замене радикала пальмитиновой кислоты на олеиновую [18]. По аналогии эфир алкилоктадеценоат полностью транс-изомера лютеина будет элюироваться до соответствующего алкилпальмитата.
Для управления разделением сложных смесей при заданной “мономерной” стационарной С18-фазе можно использовать изменение типа органических компонентов подвижной фазы, их концентрации или температуры. В работе [19] изменением компонентов подвижной фазы удалось избежать наложения пиков диэфиров капсантина, имеющего в структуре одну карбонильную группу, и капсорубина с двумя карбонильными группами в молекуле.
Влияние типа компонентов подвижной фазы. Как экстрагент, так и подвижная фаза для разделения каротиноидов должны хорошо растворять эти соединения. В ацетонитриле и метаноле (компонент 1, К1), как в наиболее часто используемых компонентах подвижных фаз, растворимость β-каротина одинаковая и достаточно невысокая – 10 мг/л [16]. В связи с этим подвижная фаза должна содержать в качестве основного компонента (К2) растворитель с более высокой растворимостью каротиноидов в нем, и число таких растворителей из перечня обычно используемых в ВЭЖХ ограничено [16]. На рис. 1 представлены хроматограммы экстракта бархатцев в четырех типах элюентов, содержащих ацетонитрил (К1) и четыре различных компонента (К2), концентрации которых (на основе предварительных исследований) подбирали так, чтобы удерживание основных диэфиров было близким для всех четырех вариантов. Как видно, во всех хроматографических системах наблюдается один и тот же порядок элюирования диэфиров лютеина. Удерживание ЭЛ возрастает с ростом числа атомов углерода (ΣNС) в двух радикалах жирных кислот, поскольку возрастает сродство к ним стационарной C18-фазы. После каждого производного полностью транс-лютеина (например, 5 на рис. 1) появляется смесь (5а), содержащая полностью транс-зеаксантин и сумму цис-изомеров лютеина с аналогичной ΣNС. За ними, если это возможно, следует диэфир полностью-транс-лютеина (6б), в котором пальмитоильный радикал замещен на октадеценильный (предположительно, радикал олеиновой кислоты) с ΣN(С + 2).
Рис. 1.
Разделение каротиноидов экстракта цветков бархатцев в четырех типах элюентов: (а) – 70 об. % ацетона и 30 об. % ацетонитрила; (б) – 52 об. % пропанола-2 и 48 об. % ацетонитрила; (в) – 49 об. % этилацетата и 51 об. % ацетонитрила; (г) – 34 об. % метил-трет-бутилового эфира и 56 об. % ацетонитрила, 0.8 мл/мин. Колонка: 150 × 4.6 мм Kromasil 100‑5С18, 30°С. Вещества: 1 – лютеин, 2 и 3 – по два региоизомера миристата и пальмитата лютеина; диэфиры лютеина: 4 – лаурат-миристат, 5 – димиристат, 6 – миристат-пальмитат, 7 – дипальмитат, 8 – пальмитат-стеарат, 9 – дистеарат; буквой а обозначены соответствующие производные зеаксантина, буквой б – эфиры, содержащие радикалы октадеценовой кислоты.
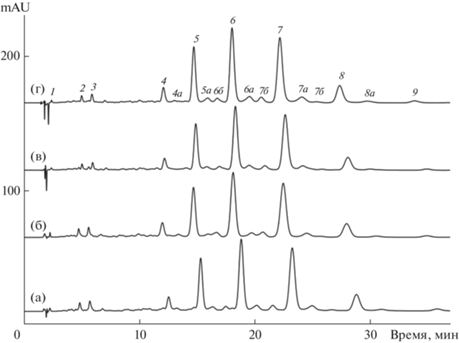
Одинаковый порядок элюирования в случае четырех различных добавок к ацетонитрилу в подвижной фазе неудивителен, поскольку производные лютеина и зеаксантина являются изомерами с одинаковым набором функциональных групп. Следовательно, для рассматриваемого случая ацетон, изопропанол, этилацетат и метил-трет-бутиловый эфир обладают примерно одинаковой селективностью.
Влияние концентрации органического модификатора подвижной фазы. С ростом концентрации К2 в подвижной фазе удерживание аналитов уменьшается тем сильнее, чем больше число дисперсионных взаимодействий (или число атомов углерода и водорода) между сорбатом и компонентами подвижной фазы. При этом степень разделения пар диэфиров типа 5а и 6б будет уменьшаться, что подтверждается экспериментально (рис. 2).
Рис. 2.
Разделение каротиноидов экстракта цветков бархатцев в четырех подвижных фазах состава ацетон–ацетонитрил, содержащих: (а) – 100 об. % ацетона; (б) – 95 об. % ацетона; (в) – 90 об. % ацетона; (г) – 85 об. % ацетона; (д) – 80 об. % ацетона, 0.8 мл/мин. Колонка: 250 × 4.6 мм Кромасил 100-5С18 с предколонкой Кромасил С18, 30°С. Эфиры лютеина: 1 – лаурат-миристат, 2 – димиристат, 3 – миристат-пальмитат, 4 – дипальмитат, 5 – пальмитат-стеарат, 6 – дистеарат.
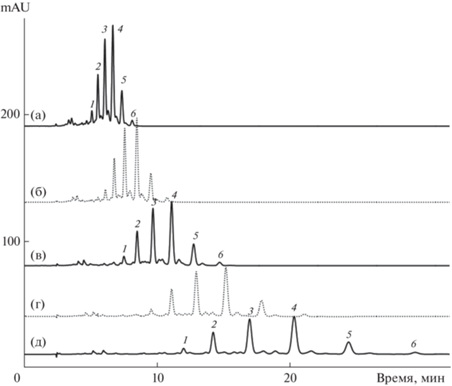
Данные рис. 2 свидетельствуют о том, что чем ниже в подвижной фазе концентрация К2, тем лучше разделяются компоненты смеси. Однако снижение концентрации К2 связано с другой проблемой – со снижением растворимости каротиноидов в подвижной фазе.
Влияние растворителя пробы на вид хроматограммы. В работе [20] показано, что при разделении антоцианов концентрация ацетонитрила в растворителе пробы может привести к артефактам, искажающим хроматограмму. Для проверки влияния растворителя пробы на разделение каротиноидов приготовили два образца с одинаковой концентрацией каротиноидов, но при использовании в первой пробе в качестве растворителя ацетона (П1), а во второй – смеси ацетона (70 об. %) с ацетонитрилом (П2). При регистрации в день приготовления хроматограммы обеих проб были практически идентичными, что указывало на возможность использования образцов в растворителе с большей растворяющей способностью, что удобно при пробоподготовке. Однако на второй день различия оказались значительными (рис. 3). На хроматограмме П2 площади пиков уменьшались (относительно площадей пиков для П1) с ростом удерживания диэфиров, т.е. со снижением их растворимости в растворителе пробы: от ~80% для лаурата-миристата и димиристата лютеина до ~50, ~40 и ~30% для дипальмитата, пальмитата-стеарата и дистеарата лютеина соответственно. Причина этого явления стала понятна после обнаружения на дне емкости, содержавшей вторую пробу, осадка.
Рис. 3.
Разделение каротиноидов двух образцов экстракта цветков бархатцев в элюенте состава 49 об. % этилацетата и 51 об. % ацетонитрила для проб П1 (А1) и П2 (А2). Колонка 150 × 4.6 мм Kromasil 100-5С18, 30°С, 0.8 мл/мин. Нумерация пиков – как на рис. 1.
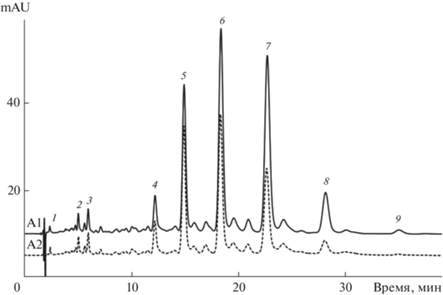
Следовательно, важнейшей причиной появления артефактов при хроматографировании каротиноидов может быть их недостаточная растворимость как в растворителе пробы, так и в подвижной фазе.
При недостаточной растворимости каротиноидов в растворителе пробы образуется осадок. При хранении растворов при пониженных температурах растворимость падает, т.е. к хранению растворов проб в холодильнике следует относиться с осторожностью.
Недостаточная растворимость каротиноидов в подвижной фазе, по нашему опыту, также может быть обнаружена по изменению значений давления в процессе хроматографирования: при спонтанной кристаллизации компонентов в процессе хроматографирования наблюдается постепенный рост давления со скачками. При исследовании растворимости антоцианов [21] наблюдали образование осадков даже в элюентах с высокой элюирующей способностью. Дело в том, что при хроматографировании происходит разделение суммы веществ (например, диэфиров) на индивидуальные компоненты, и вероятность кристаллизации возрастает, возрастает также температура плавления.
Двухколоночный способ контроля хроматографических процессов. Особенность хроматографического определения смеси веществ даже при наличии стандартных образцов состоит в том, что мы заранее не знаем, какой должна быть хроматограмма (по площадям пиков). Проблема легко решается, если воспользоваться следующим способом. Несколько раз (например, три раза) записываем хроматограмму при использовании колонки № 1, находим средние значения площадей пиков и их суммы. Затем присоединяем последовательно колонку № 2 и вновь трижды записываем хроматограммы. Обработка площадей хроматограмм в обоих случаях позволяет оценить потери или их отсутствие на колонке № 2 в сравнении с первой. Затем можно колонки поменять местами, оценив свойства первой из использованных колонок.
В ходе выполнения ряда исследований по предложенному способу установили, что при относительно слабой элюирующей силе подвижной фазы степень открытия каротиноидов может быть существенно меньше 100% (табл. 1). Однако при переходе к экстрагированию 100%-ным ацетоном потери каротиноидов в пределах погрешности эксперимента (порядка 1–1.5%) не удается обнаружить (табл. 2). Более того, некоторый рост площадей пиков в табл. 2 может быть связан с погрешностями определения площадей не полностью разделенных пиков (Rs < 1), которые более точно определяются на хроматограммах с лучшим разделением компонентов. Кроме того, как указывалось выше, свидетельством отсутствия артефактов является стабильная линия давления на входе в колонку. Следовательно, причиной снижения степени открываемости каротиноидов может быть образование осадка либо в пробе (при хранении при низкой температуре), либо на колонке в процессе хроматографирования. Можно добавить, что существует еще одна причина, приводящая к уменьшению открываемости аналитов в хроматографии – диффузия молекул аналита в галерейные поры [22]. Этот эффект, по всей видимости, также присутствует, но непрозрачность стенок колонки не позволяет его обнаружить. Однако по нашему опыту при твердофазной экстракции каротиноидов на патронах ДИАПАК С18 полная отмывка патрона от желтой окраски (при реэкстракции каротиноидов) весьма проблематична.
Таблица 1.
Площади пиков и степени открываемости эфиров ксантофиллов (n = 3) в элюенте состава 35 об. % метил-трет-бутилового эфира 65 об. % ацетонитрила
| Диэфир лютеина | Колонка или последовательно соединенные колоноки | α, % | |
|---|---|---|---|
| К1 | К1 + К2 | ||
| Димиристат | 719 ± 20 | 502 ± 11 | 70 |
| Миристат-пальмитат | 1688 ± 43 | 1099 ± 10 | 65 |
| Дипальмитат | 2241 ± 53 | 1471 ± 19 | 66 |
| Миристат-пальмитат | 790 ± 46 | 462 ± 38 | 58 |
| Дистеарат | 121 ± 19 | 48 ± 9 | 40 |
| Сумма площадей пиков | 5768 ± 200 | 3760 ± 25 | 65 |
Таблица 2.
Площади пиков диэфиров лютеина при разделении на колонке 150 × 4.6 мм Kromasil 100-5C18 (К1) и на двух последовательно соединенных колонках 150 × 4.6 мм Kromasil 100-5C18 (К1) и 250 × 4.6 мм Kromasil 100-5C18 (К2) в 100%-ном ацетоне при различных температурах (n = 3)
| Диэфир лютеина | К1 | К1 + К2, 20°С | ||
|---|---|---|---|---|
| 30°С | 25°С | 20°С | ||
| Димиристат | 1223 ± 12 | 1238 ± 4 | 1256 ± 10 | 1271 ± 15 |
| Миристат-пальмитат | 1858 ± 16 | 1887 ± 20 | 1907 ± 19 | 1907 ± 20 |
| Дипальмитат | 1672 ± 16 | 1697 ± 17 | 1719 ± 18 | 1738 ± 11 |
| Миристат-пальмитат | 503 ± 6 | 502 ± 7 | 513 ± 8 | 523 ± 5 |
| Дистеарат | 64 ± 2 | 60 ± 5 | 72± 4 | 71 ± 3 |
| Сумма площадей пиков | 5320 ± 50 | 5384 ± 42 | 5467 ± 35 | 5510 ± 52 |
Условия определения диэфиров лютеина и результаты определения каротиноидов цветков бархатцев. Экспериментально установлено, что при 19–20°С осадок не образуется в пробах с концентрацией диэфиров лютеина менее 0.0003 М, растворенных в ацетоне, но при хранении в бытовом холодильнике осадок появляется уже через двое суток при концентрации 2.8 × 10–4 М.
Установили, что элюент состава 10 об. % ацетонитрила и 90 об. % ацетона также может быть использован для разделения диэфиров ксантофиллов без проблем с выпадением осадков при разделении при температуре не ниже 20°С на колонке 250 × 4.6 мм Kromasil 100-5C18, защищенной предколонкой Kromasil C18, при скорости подвижной фазы 0.5 мл/мин или на двух последовательно соединенных колонках 100 × 4.6 мм Kromasil 100-5C18 и 250 × 4.6 мм Kromasil 100-5C18 в той же подвижной фазе при той же температуре; причем в этих условиях нет необходимости уменьшения скорости подачи подвижной фазы (ниже 1 мл/мин) (рис. 4).
Рис. 4.
Разделение каротиноидов экстракта цветков бархатцев в элюенте состава 10 об. % ацетонитрила и 90 об. % ацетона. (а): колонка 250 × 4.6 мм Kromasil 100-5С18 с предколонкой Kromasil С18, 20°С, 0.5 мл/мин. (б) и (в): колонки 100 × 4.6 мм Kromasil 100-5С18 и 250 × 4.6 мм Kromasil 100-5С18, 20°С, 1.0 мл/мин. Обозначение веществ 1–9 как на рис. 1, 10 – дилаурат лютеина. Дополнительные буквы обозначает производные полностью транс-лютеина с еще одним радикалом ненасыщенной кислоты.
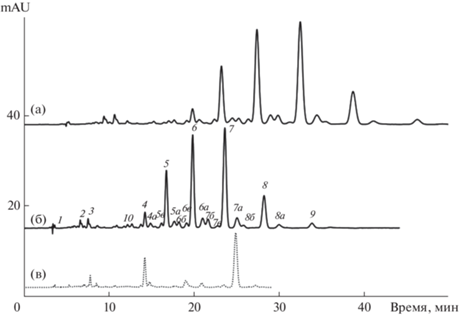
Тип радикалов жирных кислот в диэфирах лютеина обычно идентифицируют масс-спектрометрически [7, 12], но для диэфиров цветков бархатцев можно воспользоваться доступной из литературы [12] информацией об образовании эфиров в основном миристиновой и пальмитиновой кислотами с меньшим вкладом стеариновой и лауриновой кислот. Тогда достаточно использовать экстракт физалиса декоративного, содержащего в качестве основного компонента дипальмитат зеаксантина [23], для определения дипальмитата лютеина с последующим отнесением раньше и позднее элюирующихся соединений. Инкрементный подход [12, 24] позволяет в группе рядом элюирующихся соединений обнаружить дилаурат лютеина. На хроматограмме обнаруживается еще одна группа пиков с электронными спектрами поглощения, соответствующими полностью транс-лютеину, а по результатам контроля положения точек конвергенции [24] диэфиров, образованных с участием еще одной мононенасыщенной кислоты, пики этой группы веществ обозначены добавлением буквы в.
В табл. 3 представлены результаты определения молярной доли типов диэфиров лютеина по площадям пиков в пяти образцах (разного сорта) цветков бархатцев. Представленные данные свидетельствуют о высокой надежности определения жирнокислотного состава диэфиров в использованных условиях. На хроматограмме обнаруживаются также и другие соединения, среди которых сам лютеин (пик 1), содержание которого в экстракте находилось пределах 0.1–1.0%, моноэфиры лютеина (пики 2 и 3) – 2.0–2.6%; смесь диэфиров зеаксантина и цис-изомеров лютеина 7.7–9.8%, следовательно, на диэфиры полностью транс-лютеина приходилось 84.1–84.8%.
Таблица 3.
Условный жирнокислотный состав диэфиров лютеина (по площадям пиков на хроматограммах) пяти образцов высушенных лепестков бархатцев
| Образец | Концентрация каротиноидов, М | Доля диэфиров лютеина, мол. % | ||||||
|---|---|---|---|---|---|---|---|---|
| Л2 | ЛМ | М2 | МП | П2 | ПС | С2 | ||
| 1 | 2.8 × 10–4 | 1.9 | 5.1 | 16.5 | 28.7 | 33.1 | 12.5 | 2.2 |
| 1.4 × 10–4 | 2.0 | 5.1 | 16.6 | 28.6 | 33.0 | 12.5 | 2.3 | |
| 5.7 × 10–5 | 2.2 | 5.4 | 16.5 | 28.6 | 32.8 | 12.4 | 2.2 | |
| 3.1 × 10–5 | 2.0 | 4.9 | 16.5 | 28.6 | 33.0 | 12.7 | 2.2 | |
| 1.2 × 10–5 | 2.2 | 5.1 | 16.6 | 28.7 | 33.0 | 12.4 | 2.2 | |
| Среднее | 2.0 | 5.1 | 16.5 | 28.6 | 33.0 | 12.5 | 2.2 | |
| 2 | 3.4 × 10–5 | 0.5 | 2.8 | 11.2 | 24.5 | 39.3 | 17.74 | 4.0 |
| 3 | 2.7 × 10–5 | 0.6 | 4.2 | 15.6 | 28.9 | 34.2 | 13.51 | 3.0 |
| 4 | 3.6 × 10–5 | 0.7 | 5.4 | 16.3 | 27.3 | 32.3 | 14.33 | 3.6 |
| 5 | 4.4 × 10–5 | 0.9 | 4.9 | 17.6 | 30.2 | 33.4 | 11.4 | 1.7 |
Список литературы
Mariutti L.R.B., Mercadante A.Z. Carotenoid esters analysis and occurrence: What do we know so far? // Arch. Biochem. Biophys. 2018. V. 648. P. 36.
Mercadante A.Z., Rodrigues D.B., Petry F.C., Mariutti L.R.B. Carotenoid esters in foods – A review and practical directions on analysis and occurrence // Food Res. Int. 2017. V. 99. P. 830.
Sarkar C.R., Bhagawati B., Das L., Goswami B.C. An efficient condition of saponification of lutein ester from marigold flower // Ann. Biol. Res. 2012. V. 3. P. 1461.
Abdala A.F., Gallardo A.P., Olvera L.G., Silva E.M.E. Hydrolysis of carotenoid esters from Tagetes erecta by the action of lipases from Yarrowia lipolytica // Bioresour. Bioprocess. 2017. V. 4. 5.
Raut S., Thaneshwari T. Commercial application of lutein: Marigold flower pigment // Eco. Env. Cons. 2022. V. 28 (October Suppl. Issue). P. S315.
Piccaglia R., Marotti M., Grandi S. Lutein and lutein ester content in different types of Tagetes patula and T. erecta // Industr. Crops Prod. 1998. V. 8. P. 45.
Tsao R., Yang R., Young J.C., Zhu H., Manolis T. Separation of geometric isomers of native lutein diesters in marigold (Tagetes erecta L.) by high-performance liquid chromatography–mass spectrometry // J. Chromatogr. A. 2004. V. 1045. P. 65.
Sowbhagya H.B., Sampathu S.R., Krishnamurthy N. natural colorant from marigold – Chemistry and technology // Food Rev. Int. 2004. V. 20. P. 33-50.
Jiang X.-Y., Chen L.-S., Zhou C.-S. Lutein and lutein esters in marigold flowers by high performance chromatography // J. Cent. South Univ. Technol. 2005. V. 12. P. 306.
Hayashi T., Oka H., Ito Y., Goto T., Ozeki N., Itakura Y., Matsumoto H., Ohno H., Yoshida K., Miyazawa T., Nagase H. An HPLC method for the analysis of marigold color in food using lutein as an indicator // J. Liq. Chromatogr. Relat. Technol. 2005. V. 27. P. 335.
Vechpanich J., Shotipruk A. Recovery of free lutein from Tagetes erecta: determination of suitable saponification and crystallization conditions // Sep. Sci. Technol. 2010. V. 46. P. 265.
Лапшова М.С., Дейнека В.И., Дейнека Л.А., Блинова И.П., Третьяков М.Ю. Идентификация ксантофиллов в экстракте лепестков цветков бархатцев // Журн. аналит. химии. 2013. Т. 68. № 11. С. 1130.
Yung. J.C., Abdel-Aal E.-S.M., Rabalski I., Blackwell B.A. Identification of synthetic regioisomeric lutein esters and their quantification in a commercial lutein supplement // J. Agric. Food Chem. 2007. V. 55. P. 4965.
Дейнека В.И., Лапшова М.С., Захаренко Е.В., Дейнека Л.А. Особенности разделения двух классов эфиров высших насыщенных жирных кислот на “полимерной” обращенной фазе // Журн. физ. химии. 2013. Т. 87. № 11. С. 1943.
Epler K.S., Sander L.C., Ziegler R.G., Wise S.A., Craft N.E. Evaluation of reversed-phase liquid chromatographic columns for recovery and selectivity of selected carotenoids // J. Chromatogr. 1992. V. 595. P. 89.
Craft N.E., Soares J.H., Jr. Relative solubility, stability, and absorptivity of lutein and β-carotene in organic solvents // J. Agric. Food Chem. 1992. V. 40. P. 431.
Mrowicka M., Mrowicki J., Kucharska E., Majsterek I. Lutein and zeaxanthin and their roles in age-related macular degeneration – Neurodegenerative disease // Nutrients. 2022. V. 14. Article 827
Туртыгин А.В., Дейнека В.И., Дейнека Л.А. Определение состава триацилглицеринов в маслах семян граната методами обращенно-фазовой ВЭЖХ и спектрофотометрии // Журн. аналит. химии. 2013. Т. 68. № 6. С. 619.
Дейнека В.И., Буржинская Т.Г., Дейнека Л.А. Проблемы идентификации ксантофиллов лепестков Lilium pumilum L. при применении метода обращенно-фазовой ВЭЖХ со спектрофотометрическим и масс-спектрометрическим детектированием // Аналитика и контроль. 2019. № 4. С. 501.
Дейнека В.И., Сидоров А.Н., Дейнека Л.А., Тыняная И.И. Пробоподготовка при ВЭЖХ определении антоцианов и бетацианинов. Эффект растворителя образца // Сорбционные и хроматографические процессы. 2016. Т. 16. Вып. 3. С. 384.
Дейнека В.И., Дейнека Л.А., Сидоров А.Н., Костенко М.О., Блинова И.П. Оценка растворимости антоцианов с использованием насадок для твердофазной экстракции // Журн. физ. химии. 2016. Т. 90. № 4. С. 622.
Дейнека В.И., Дейнека Л.А., Сидоров А.Н., Саенко И.И., Костенко О.М. Оценка свойств сорбентов концентрирующих патронов для твердофазной очистки: роль “галерейных” пор // Сорбционные и хроматографические процессы. 2016. Т. 16. Вып. 5. С. 624.
Weller P., Breithaupt D.E. Identification and quantification of zeaxanthin esters in plants using liquid chromatography-mass spectrometry // J. Agric. Food Chem. 2003. V. 51. P. 7044.
Дейнека В.И., Дейнека Л.А. Инкрементный подход в анализе каротиноидов методом ОФ ВЭЖХ. Разделение диэфиров ксантофиллов // Сорбционные и хроматографические процессы. 2006. Т. 6. Вып. 3. С. 366.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии