

Журнал аналитической химии, 2023, T. 78, № 7, стр. 637-647
Новые сорбенты для определения аминокислот в почвенных экстрактах методом гидрофильной жидкостной хроматографии с масс-спектрометрическим детектированием
Н. Ю. Чикурова a, А. В. Горбовская a, А. Н. Ставрианиди a, b, Е. С. Фёдорова b, А. О. Шемякина a, А. К. Буряк b, А. С. Ужель a, А. В. Чернобровкина a, *, О. А. Шпигун a
a Московский государственный университет имени М.В. Ломоносова, химический факультет
Москва, Ленинские горы,1, стр. 3, 119991 Россия
b Институт физической химии и электрохимии им. А.Н. Фрумкина Российской академии наук
Ленинский просп., 31, корп. 4,
Москва, 119071 Россия
* E-mail: chernobrovkina@analyt.chem.msu.ru
Поступила в редакцию 09.12.2022
После доработки 09.02.2023
Принята к публикации 16.02.2023
- EDN: VRRQNX
- DOI: 10.31857/S0044450223070046
Аннотация
Разработаны сорбенты на основе различных матриц – силикагеля и сополимера стирола и дивинилбензола – для определения аминокислот методом гидрофильной хромато-масс-спектрометрии. Выбран оптимальный вариант структуры функционального слоя в двух сериях полученных неподвижных фаз, обеспечивающий лучшую гидрофилизацию для каждой матрицы. Изучены механизмы удерживания, выбраны условия масс-спектрометрического детектирования, разделения и определения 16 аминокислот. Проведена оценка применимости полученных сорбентов и способа определения аминокислот для анализа почвенных экстрактов.
Аминокислоты играют важную роль в азотном обмене почвы и растений, а также в процессе почвообразования [1]. Они составляют группу неспецифических соединений почвы, количество свободных аминокислот в различных типах почв неодинаково. Определение веществ данного класса в почвах уже несколько лет представляет интерес для агрохимиков.
Традиционными методами определения аминокислот являются обращенно-фазовая хроматография на сорбентах на основе модифицированного октадецильными группами силикагеля с предколоночной дериватизацией и катионообменная высокоэффективная жидкостная хроматография (ВЭЖХ) на сульфированном полистирол-дивинилбензоле (ПС-ДВБ) с постколоночной дериватизацией [2]. В последние годы определение соединений данного класса получило развитие благодаря возможности селективного разделения полярных недериватизированных аминокислот напрямую методом гидрофильной хроматографии (ГИХ) [3]. В случае если непременным условием является высокая чувствительность метода, то его реализация возможна путем сочетания жидкостного хроматографа с масс-спектрометрическим (МС) детектором [4–6]. В то же время успешное сочетание метода гидрофильной хроматографии с масс-спектрометрией возможно благодаря летучести и низкой вязкости подвижной фазы, содержащей большое количество органического растворителя. Среди коммерческих фаз для ГИХ-МС-определения полярных соединений, в том числе аминокислот, представлено большое разнообразие фаз на основе силикагеля: диольные [7], амидные [8, 9], цвиттер-ионные [10] и с привитыми макромолекулами [10].
Самые распространенные коммерчески доступные фазы для гидрофильной хроматографии – это сорбенты на основе силикагеля. Однако их применение для ВЭЖХ с МС-детектированием затруднено ввиду низкой стабильности матрицы и смывания привитых функциональных групп, приводящего к загрязнению детектора. Именно поэтому актуальным является поиск новых способов модифицирования силикагеля для увеличения его стабильности или использование альтернативных матриц для создания сорбентов, которые можно применять в ГИХ-МС.
В качестве инертной матрицы, механически стабильной и устойчивой во всем диапазоне pH, перспективен ПС-ДВБ с высокой степенью сшивки, широко используемый в качестве матрицы для анионообменников [11–14]. При этом требуется выбор подходящего способа гидрофилизации и одновременного экранирования поверхности данной гидрофобной матрицы для обеспечения приемлемых факторов удерживания гидрофильных соединений, селективности и эффективности колонок с использованием элюентов, пригодных для МС-детектирования [15]. К ранее предложенным способам гидрофилизации можно отнести разветвление функциональных слоев, повышение гидрофильности функциональных центров и их удаление от поверхности матрицы за счет гидрофильных спейсеров [16, 17]. Наибольшей гидрофилизации удается достичь за счет объединения всех перечисленных выше подходов и создания сложных функциональных слоев, например ковалентно привитых конденсационных полимеров, получаемых in situ из вторичного амина и диэпоксида [18].
Цель настоящего исследования – разработка новых неподвижных фаз для ГИХ-МС, выбор условий определения аминокислот в данном режиме на полученных сорбентах и оценка их применимости к анализу реальных объектов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приборы и материалы. Аминокислоты: D,L-фенилаланин, D,L-пролин, D,L-серин, L-изолейцин, L-лейцин, L-метионин, L-гистидин, L-лизин х. ч. (Serva, Германия); L-аспарагин, аланин, глицин, аспарагиновая кислота х. ч. (Merck, Германия); L-глутамин, L-валин, L-треонин х. ч. (Reanal, Венгрия).
Почва: два образца предоставлены Аналитическим центром МГУ. Адрес и место отбора проб: Истринский район, д. Борзые, ул. Невская (первая проба); хутор Шулька, Иткуловский сельсовет (вторая проба). Органическое вещество почвы составляло 31.5 и 29.5% соответственно.
Прочие реактивы: ацетонитрил, “HPLC gradient grade”; кислота уксусная ледяная, 99.5%; кислота ортофосфорная, 85% х. ч., хлорид калия ч. д. а. (Panreac, Испания). Этанол ч. д. а.; ацетат аммония ч. (Лабтех, Россия); ацетон ч. д. а. (ХимМед, Россия); 1,4-диоксан ч. д. а. (Компонент-Реактив, Россия).
Для изучения хроматографических свойств сорбентов использовали: жидкостной хроматограф Vanquish Flex с флуоресцентным детектором FLD и диодно-матричным детектором DAD (Thermo Scientific, США); систему ВЭЖХ Dionex Ultimate 3000 (Thermo Scientific, США), состоящую из двухканального насоса высокого давления, автоматической системы ввода пробы, термостата колонок и детектора на диодной матрице. Хроматограммы региcтрировали с помощью программного пакета Chromeleon 7.0.
Для проведения ГИХ-МС-анализа на колонке С-М использовали жидкостной хромато-масс-спектрометр (Shimadzu, Япония), состоящий из следующих модулей: квадрупольный масс-спектрометр LCMS-2020 c ионизацией аналитов электрораспылением (ESI); два ВЭЖХ-насоса LC-20AD; дегазатор DGU-20A; контроллер CBM-20A; автосeмплер SIL-20AC и термостат CTO-20AC. Программное обеспечение для сбора данных и управления хроматографом – LabSolutions ver. 5.55. В качестве подвижной фазы использовали смесь 10 мМ аммонийно-формиатного буферного раствора и ацетонитрила (80 : 20, по объему). Скорость потока – 0.8 мл/мин.
Для проведения ГИХ-МС-анализа на колонке П-ДМА использовали систему, состоящую из жидкостного хроматографа Agilent 1260 (Agilent, США) и гибридного времяпролетного масс-спектрометра Bruker Maxis Impact (Bruker Scientific, США). Объем вводимой пробы 5 мкл. В качестве подвижной фазы использовали смесь 10 мМ формиата аммония и ацетонитрила (85.5 : 14.5, по объему) с добавлением 0.05% муравьиной кислоты. Скорость потока – 0.8 мл/мин. Температура термостата колонки 30°C. Параметры МС-детектирования были следующими: электрораспылительная ионизация (ЭРИ); режим регистрации положительных ионов; диапазон сканирования 50–500 m/z; температура источника 200°C; потоки газа (N2) для осушения и распыления 7 л/мин и 1.5 л/мин соответственно; напряжение на капилляре 4.5 кВ. Ширина окна m/z для измерения площадей пиков по выбранным ионам ±0.01 Да.
Система ГX-МС Agilent GC (8890)-MS (5977B) (Agilent Technologies, США) состояла из моноквадрупольного МС-детектора с источником электронной ионизации, термостата колонки, охлаждаемого узла ввода пробы, термодесорбера (TDU 2), автоматической роботизированной системы ввода пробы (Gerstel, Германия) и линии подачи газов (He, 1 мл/мин). Для изучения структуры сорбентов использовали колонку ГХ: две соединенные последовательно колонки HP-5MS 15 м × 0.25 мм, 0.25 мкм (Agilent Technologies, США), а также стеклянные термодесорбционные трубки (5 × 0.5 см), кварцевую стекловату ч. д. а. (TRAJAN, Австралия).
Для упаковки сорбентов использовали стальные колонки размером 100 × 3 мм и 100 × 4 мм, которые заполняли при помощи насоса Knauer K-1900 (Knauer, Германия).
Синтез сорбентов и заполнение хроматографических колонок. Неподвижные фазы на основе силикагеля с привитыми аминопропильными радикалами Диасфер-110-Амин (диаметр сферических частиц 5 мкм, средний диаметр пор 11 нм, удельная поверхность 200 м2/г; ЗАО “БиоХимМакСТ”, Россия) синтезировали согласно рекомендациям [19, 20]. К матрице последовательно добавляли кетон (ацетон), соответствующий изоцианид и карбоновую кислоту (гликолевую). Аббревиатура (табл. 1) указывает на изоцианид, использованный в синтезе сорбентов по реакции Уги [19, 21] (М – 2-морфолиноэтилизоцианид, Т – п-толуолсульфонилметилизоцианид).
Таблица 1.
Обозначения неподвижных фаз и факторы удерживания уридина согласно тесту Танака
| Матрица | Фаза | Функциональный слой | lg P* | kU | ||
|---|---|---|---|---|---|---|
| Силикагель | C-М | Изоцианид | Морфолинэтил | 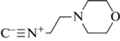 |
0.19 | 4.0 |
| C-Т | п-Толуолсульфонилметил | 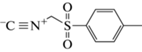 |
1.15 | 2.9 | ||
| Полистирол-дивинилбензол (ПС-ДВБ) | П-ДМА | Амин | Диметиламин | 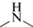 |
–0.17 | 1.0 |
| П-МЭА | Метилэтаноламин | 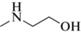 |
–1.15 | 0.8 | ||
Для синтеза сорбентов на полимерной основе использовали микросферические частицы ПС-ДВБ (степень сшивки 50%, размер частиц 5.5 ± ± 0.5 мкм, средний диаметр пор 4 нм, площадь поверхности 650 м2/г). Модифицирование ПС-ДВБ проводили в соответствии с методикой, описанной в работе [22], но в оптимизированных условиях для стадий ацилирования и восстановительного аминирования. Синтез конденсационных слоев инициировали путем выдерживания смеси 1,4-бутандиолдиглицидилового эфира (1.37 ммоль) и диметиламина (ДМА) (1.43 ммоль) в 10 мл дистиллированной воды в ультразвуковой ванне в течение 10 мин. Затем переливали смесь в колбу емк. 100 мл, добавляли 10 мл дистиллированной воды и суспензию, состоящую из 1 г аминированного ПС-ДВБ, 5 мл дистиллированной воды и 5 мл этанола. Синтез вели в течение 4 ч при 60°С и получали фазу П-ДМА (табл. 1). Синтез фазы П-МЭА с метилэтаноламином (МЭА) в структуре имел следующие отличия: время инициализации синтеза конденсационных слоев увеличили на 1 ч, количества реагентов для синтеза – в три раза, температуру синтеза – до 80°C.
Колонки заполняли при давлении 300–400 бар. Для приготовления суспензии сорбентов на основе силикагеля использовали смесь 10 мл 0.1 М раствора KCl, 8 мл этанола и 2 мл 1,4-диоксана, на основе ПС-ДВБ – 20 мл воды. В качестве подвижной фазы для упаковки сорбентов на основе силикагеля использовали дегазированный ацетонитрил, для полимерных – 0.015 М раствор карбоната натрия. Размеры колонок для сорбентов на основе силикагеля – 100 × 3 мм, на основе ПС-ДВБ – 100 × 4 мм.
Изучение структуры сорбентов. Изучение термодесорбции высушенных образцов сорбентов на основе силикагеля методом ГХ-МС доказало закрепление фрагментов на поверхности матрицы. При нагревании до 250°С сорбентов С-М и С-Т регистрировали сигналы остатков изоцианидов. На масс-хроматограмме продуктов термического разложения С-М наблюдали интенсивный пик, соответствующий 2-морфолиноэтанамину, основные сигналы в масс-спектре: m/z 100 (C5H10NO+), m/z 70 (C4H8N+), m/z 56 (C3H6N+), сигнал молекулярного иона малоинтенсивен. На масс-хроматограмме С-Т наблюдали интенсивный пик, соответствующий 4-метилтиофенолу, основные сигналы в масс-спектре: m/z 124 (молекулярный ион С7H8S+•), m/z 91 $\left( {{{{\text{C}}}_{7}}{\text{H}}_{7}^{ + }} \right),$ m/z 79 $\left( {{{{\text{C}}}_{{\text{6}}}}{\text{H}}_{7}^{ + }} \right),$ m/z 77 $\left( {{{{\text{C}}}_{{\text{6}}}}{\text{H}}_{5}^{ + }} \right).$
Анализ реальных объектов. К 2 г почвы добавляли 20 мл воды, встряхивали в течение часа, после отстаивания образец фильтровали через фильтр “белая лента”. Образцы подвергали очистке путем твердофазной экстракции на картридже Chromabond® C18 ec f. Картридж кондиционировали 10 мл ацетонитрила и 5 мл воды. К полученным водным экстрактам добавляли ацетонитрил для достижения соотношения водная часть–ацетонитрил согласно составу подвижной фазы.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для решения задачи определения аминокислот в режиме ГИХ-МС предложено использовать неподвижные фазы на основе двух различных типов матриц – силикагеля и ПС-ДВБ – и подходящие для них способы гидрофилизации и экранирования поверхности. Модифицирование 3-аминопропилсиликагеля осуществляли по многокомпонентной реакции Уги, позволяющей формировать гидрофильные амидные функциональные группы и получать селективные и эффективные фазы [19, 20]. С целью улучшения их разделяющей способности для проведения синтеза применяли новые изоцианиды, различающиеся структурой и гидрофильностью (величины параметра Ханша lg P и структурные формулы приведены в табл. 1). Гидрофилизацию и экранирование гидрофобной матрицы аминированного ПС-ДВБ проводили путем ковалентного закрепления положительно заряженных конденсационных полимеров, получаемых in situ [18]. Таким образом синтезировали два сорбента на основе силикагеля по реакции Уги с разными изоцианидами в функциональных слоях, а также два сорбента на основе ПС-ДВБ с конденсационными полимерными слоями, сформированными с помощью аминов различной гидрофильности (табл. 1). Изучали свойства полученных сорбентов, а также механизмы удерживания аминокислот. Для оценки возможности применения разработанных фаз в режиме ГИХ-МС выбрали оптимальные структуры функциональных слоев для сорбентов на основе разных матриц.
Механизмы удерживания аминокислот. Для выбора условий разделения аминокислот в режиме ГИХ полезна оценка вклада различных типов возможных для них взаимодействий с сорбентами разной структуры.
Согласно теории метода гидрофильной хроматографии [23], близость коэффициента корреляции к единице в уравнении (1) свидетельствует о реализации адсорбционного механизма удерживания, а в уравнении (2) соответственно – о преобладании распределительного механизма:
(2)
$\lg k{\kern 1pt} ' = {{a}_{1}}w\left( {{{{\text{H}}}_{{\text{2}}}}{\text{O}}} \right) + {{b}_{1}}.$Для сорбента С-М установили, что при содержании водной составляющей в элюенте от 15 до 35 об. % при концентрации фосфатного буферного раствора в подвижной фазе 0.35 мМ вклад в удерживание аминокислот вносят как распределительный, так и адсорбционный механизмы, поскольку коэффициенты корреляции для двух зависимостей близки к единице (табл. 2). Также показано, что удерживание аспарагина на фазе П-ДМА при содержании 10–40 об. % воды в элюенте преимущественно регулируется адсорбционными взаимодействиями с функциональными группами сорбента.
Таблица 2.
Уравнения и коэффициенты корреляции логарифмической и билогарифмической зависимостей факторов удерживания аминокислот на сорбентах С-М, П-ДМА от доли водной части в подвижной фазе
| Сорбент | Вещество | lg k' = aw + b | lg k' = algw + b | ||||
|---|---|---|---|---|---|---|---|
| a | b | r | a | b | r | ||
| С-М | Phe | –3.52 | 1.29 | 0.991 | –1.93 | –0.79 | 0.999 |
| Met | –3.88 | 1.55 | 0.994 | –2.13 | –0.74 | 0.999 | |
| Tyr | –4.28 | 1.71 | 0.991 | –2.35 | –0.82 | 0.999 | |
| Pro | –4.35 | 1.85 | 0.992 | –2.39 | –0.72 | 0.999 | |
| Asn | –4.92 | 2.29 | 0.998 | –2.68 | –0.61 | 0.998 | |
| Ser | –5.39 | 2.55 | 0.997 | –2.94 | –0.62 | 0.999 | |
| Ala | –5.71 | 2.59 | 0.994 | –3.12 | –0.78 | 0.999 | |
| П-ДМА | Asn | –0.54 | 1.87 | 0.968 | –2.78 | 4.24 | 0.999 |
В условиях разделения аминокислоты находятся в форме цвиттер-ионов, и при pH 6.5 фосфатного буферного раствора вблизи их изоэлектрических точек они практически электронейтральны. Представляло интерес оценить, можно ли в таком случае ожидать электростатических взаимодействий диссоциировавших карбоксильных групп с анионообменными центрами сорбентов и определить факторы влияния на удерживание этих аналитов для сорбентов различной структуры. Для оценки вклада электростатических взаимодействий в удерживание аминокислот изучили зависимости их факторов удерживания от концентрации буферного раствора, которую варьировали от 1 до 10 мМ (рис. 1).
Рис. 1.
Влияние концентрации буферного раствора на факторы удерживания аминокислот. Условия: сорбент С-М (слева), подвижная фаза фосфатный буферный раствор с pH 6.5–CH3CN (20 : 80, по объему); сорбент П-ДМА (справа), подвижная фаза фосфатный буферный раствор с pH 6.5–CH3CN (25 : 75, по объему). Диапазон концентраций буферного раствора в водной части элюента 1–10 мМ. Скорость потока – 1 мл/мин; УФ-детектирование при 210 нм.
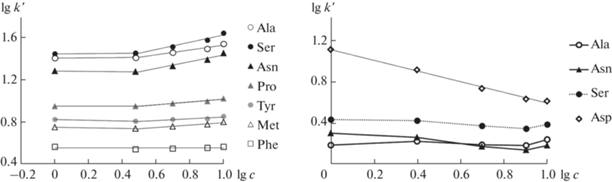
Билогарифмическая зависимость, полученная для сорбента С-М на основе силикагеля, представлена двумя диапазонами (рис. 1). При концентрации буферного раствора 1–3 мМ факторы удерживания аминокислот не зависят от концентрации элюирующего иона, тогда как при ее повышении до 10 мМ наблюдается небольшое их возрастание (доля ацетонитрила в подвижной фазе составляла 80 об. %). Вероятно, это связано с увеличением толщины приповерхностного водного слоя, в который предпочтительнее переходят гидратированные фосфат-ионы, являющиеся космотропами, что, как следствие, приводит к увеличению удерживания полярных аналитов. Интересно, что данный эффект отсутствует для большинства аминокислот на сорбенте П-ДМА с гидрофобной матрицей. В этом случае только для аспарагиновой кислоты установлено преобладание электростатических взаимодействий (рис. 1) с наклоном соответствующей зависимости (3), равным –0.51 (r = 0.998):
где [c]m – концентрация элюирующего иона в подвижной фазе (доля ацетонитрила – 75 об. %). Таким образом, повышение концентрации буферного раствора в составе элюента приводит к существенному снижению удерживания отрицательно заряженной в условиях анализа аспарагиновой кислоты (pIAsp = 3.0).
Выбор условий разделения аминокислот. При разделении аминокислот в режиме ГИХ использовали УФ-детектирование при λп = 210 нм, а также 15 и 20%-ный нелетучий фосфатный буферный раствор с рН 6.5 в составе элюента для фаз на основе ПС-ДВБ и силикагеля соответственно (рис. 2) [19, 20]. Оценивали селективность и факторы удерживания аминокислот на четырех разработанных сорбентах. Согласно тесту Танака [24], введение гидрофильного изоцианида в функциональный слой сорбента на основе силикагеля привело к получению более гидрофильного сорбента С-М по сравнению с С-Т (табл. 1) и обеспечило наибольшие времена удерживания определяемых компонентов. В то же время использование более гидрофильного метилэтаноламина для создания конденсационных полимерных слоев на ПС-ДВБ не позволило увеличить гидрофильность сорбента по сравнению с ДМА. Это может быть связано с меньшей ионообменной емкостью сорбента П-МЭА, о чем свидетельствует меньший фактор удерживания хлорида при использовании в качестве элюента 15 мМ раствора KOH (для сорбента П-ДМА – 1.5, для П-МЭА – 1.2). Аналогично большие времена удерживания аминокислот согласуются с увеличением kU по тесту Танака (табл. 1).
Рис. 2.
Хроматограммы модельных водно-ацетонитрильных (15 : 85, по объему) растворов аминокислот при их разделении на сорбентах С-М (а), С-Т (б), П-ДМА (в), П-МЭА (г). (а, б) Phe, Met, Tyr (5 мг/л), Pro (20 мг/л), Asn (15 мг/л), Ser, Ala (50 мг/л). (в) Pro (35 мг/л), Val (35 мг/л), Asn, Gln (20 мг/л), Ala, Ser, Asp (50 мг/л). (г) Leu (10 мг/л), Val (10 мг/л), Tyr, Thr (20 мг/л), Ala, Ser (50 мг/л). Условия разделения: сорбенты С-М (а) и С-Т (б), колонка 100 × 3 мм, подвижная фаза CH3CN–1 мМ фосфатный буферный раствор с pH 6.5 (80 : 20, по объему), скорость потока – 1 мл/мин, УФ-детектирование при 210 нм; сорбенты П-ДМА (в), П-МЭА (г), колонка 100 × 4 мм, подвижная фаза CH3CN–5 мМ фосфатный буферный раствор с pH 6.5 (85 : 15, по объему), скорость потока – 1 мл/мин, УФ-детектирование при 210 нм.
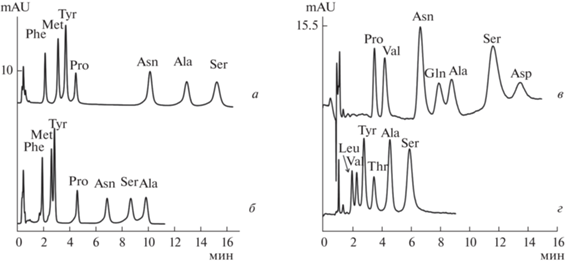
Для выбора состава подвижной фазы, совместимой с МС-детектированием, провели разделение аминокислот с использованием аммонийно-ацетатного (10–20 мМ, pH 5.8), а также аммонийно-формиатного (10–20 мМ, pH 4.7) буферных растворов и рефрактометрического детектирования. При переходе на летучие буферные растворы наблюдали уменьшение факторов удерживания аминокислот на всех сорбентах с сохранением селективности. Показано снижение эффективности в пределах 25% на сорбентах на основе разных матриц, одинаковое для двух буферных систем. Тем не менее эффективность по аминокислотам достигала 25 000 тт/м на сорбенте С-М на основе силикагеля и 15 000 тт/м на фазе П-ДМА на основе ПС-ДВБ.
Таким образом, в двух сериях выбраны сорбенты, обеспечивающие наибольшие факторы удерживания аминокислот: полученный по реакции Уги с использованием 2-морфолиноэтилизоцианида сорбент С-М на основе силикагеля и фаза П-ДМА с диметиламином в конденсационном полимерном слое на матрице ПС-ДВБ. Выбрали составы подвижной фазы: 14.5 и 20% 10 мМ аммонийно-формиатного буферного раствора (pH 4.7) и 85.5 и 80% ацетонитрила для разделения аминокислот с точки зрения экспрессности анализа и симметрии пиков для фаз С-М и П-ДМА соответственно.
Выбор масс-спектрометрических параметров для детектирования аминокислот. В условиях электрораспылительной ионизации эффективность распыления и испарения капель увеличивается при низких скоростях потока подвижной фазы. При этом ухудшается форма и, как следствие, разрешение пиков аминокислот. Для достижения компромисса между чувствительностью МС-детектирования и разделением аминокислот в качестве оптимальной скорости потока выбрали 0.8 мл/мин.
В состав подвижной фазы в условиях ГИХ при использовании МС-детектора помимо воды и ацетонитрила могут входить легколетучие компоненты буферных растворов, например муравьиная кислота. При этом интенсивность сигналов [M + H]+ ионов увеличивается за счет создания кислой среды и подавления диссоциации аналитов. Большие концентрации буферных растворов в подвижной фазе негативно сказываются на ионизации. Интенсивность ионов для всех аминокислот в 2–3 раза выше при элюировании подвижной фазой состава вода–ацетонитрил с добавлением 0.1% муравьиной кислоты (50 : 50, по объему) по сравнению с фазой 10 мМ аммонийно-формиатный буферный раствор (pH 4.7)–ацетонитрил (50 : 50, по объему). Повышение объемной доли ацетонитрила до 80% положительно сказалось на интенсивности сигналов в масс-спектрах.
На интенсивность сигнала при МС-детектировании также влияют настройки масс-спектрометра. Варьировали разность потенциалов (DL voltage) между выходом из линии десольватации и входом в квадрупольный массив линз (QArray) от 0 до 70 В. Оптимальное значение этого параметра выбирали по наибольшей высоте пика для каждого компонента. Также варьировали напряжение постоянного тока в квадрупольном массиве линз (QArray voltage) от 0 до 30 В. При этом интенсивность аддуктных ионов с ацетонитрилом уменьшалась при увеличении DL и QArray потенциалов. Оптимальные значения DL и QArray потенциалов – 30 и 10 В соответственно. Дальнейшее их увеличение приводило к снижению интенсивности целевых [M+H]+ ионов.
Следует отметить, что величина фонового сигнала по полному ионному току на МС-хроматограммах в режиме сканирования при элюировании через колонку С-М подвижной фазой состава аммонийно-формиатный буферный раствор–ацетонитрил (20 : 80, по объему) со скоростью 0.8 мл/мин в 3.5 раза превышала уровень фона, полученный без колонки в тех же условиях. Наиболее интенсивные сигналы: m/z 120, 138, 161 и 179, вероятно, соответствуют отщепляемому в циклической форме триаминовому фрагменту (m/z 138, [M + H]+) функциональной группы сорбента. В таком случае сигнал с m/z 120 образуется при отщеплении от него одной молекулы воды, а сигналы с m/z 161 и 179 являются ацетонитрильными аддуктами этих ионов. На колонках с сорбентом на основе ПС-ДВБ дополнительных сигналов и увеличения уровня фона не наблюдали, что свидетельствует о высокой стабильности закрепления функционального слоя на полимерной матрице.
При разделении и определении аминокислот с МС-детектированием основной задачей является разделение пиков изомерных соединений изолейцина и лейцина, лизина и глутамина. Также необходимо отделить от пика лизина следующий за ним пик серина, который может давать сигнал c m/z 147 [M + ACN + H]+, совпадающий по молекулярной массе с регистрируемыми сигналами [M + H]+ ионов глутамина и лизина. Отмечено, что после промывки сорбента водой достигается полное разделение этих трех веществ. Хроматограмма модельной смеси аминокислот на сорбенте С-М представлена на рис. 3а.
Рис. 3.
Хроматограммы модельных водно-ацетонитрильных (15 : 85, по объему) растворов аминокислот при их разделении на сорбентах С-М (а) и П-ДМА (б). (а) Phe (10 мг/л), Leu, ILeu (15 мг/л), Met (10 мг/л), Tyr (30 мг/л), Arg, Val, Pro (10 мг/л), Thr, Gly, Glu (15 мг/л), Asn (20 мг/л), His, Lys (10 мг/л), Ser (20 мг/л), Ala (15 мг/л). (б) Thr, Val, Phe, Leu, Pro, Met, Val, Gln, Asn, Ser, Ala (10 мг/л), Tyr (20 мг/л). Условия: сорбент С-М, колонка 100 × 3 мм, подвижная фаза CH3CN–10 мМ аммонийно-формиатный буферный раствор с pH 4.7 (80 : 20, по объему); сорбент П-ДМА, колонка 100 × 4 мм, подвижная фаза CH3CN– аммонийно-формиатный буферный раствор с pH 4.7 (85.5 : 14.5, по объему); скорость потока 0.8 мл/мин; МС-детектирование.
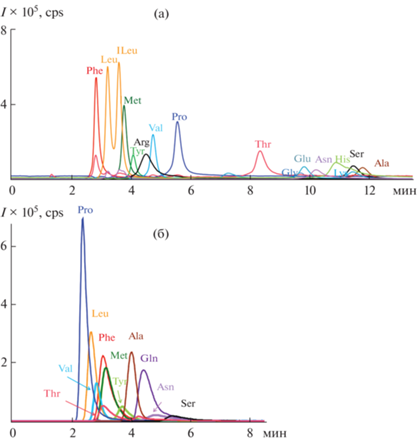
Разделение аминокислот на колонке П-ДМА проводили в выбранных условиях с регистрацией масс-хроматограмм в режиме мониторинга выбранных ионов на масс-спектрометре высокого разрешения (Δm/z = 0.01 Да). Хроматограмма модельной смеси аминокислот на сорбенте П-ДМА с диметиламином в конденсационном полимерном слое представлена на рис. 3б.
Определение аминокислот в модельных экстрактах из почв. Для оценки применимости разработанных сорбентов к анализу реальных объектов приготовили модельные растворы вытяжек из почвы с добавлением определяемых аминокислот. Правильность определения проверяли методом введено–найдено с добавками аминокислот на двух уровнях содержаний вблизи нижней границы диапазона линейности. Найденные количества согласуются с введенными (табл. 3, 4). Хроматограммы приведены на рис. 4.
Таблица 3.
Результаты определения аминокислот в модельном экстракте первого образца почвы (сорбент С-М)
| Вещество | сmin, мг/л | clim, мг/л | Диапазон линейности, мг/л | Уравнение, y = (A ± Δ)x нг/мл |
R2 | Процент восстановления ± sr, % | |
|---|---|---|---|---|---|---|---|
| 0.25 мг/л | 1 мг/л | ||||||
| Phe | 0.03 | 0.1 | 0.1–10 | y = (570 ± 30)x | 0.9985 | 101 ± 5 | 95 ± 3 |
| Leu | 0.03 | 0.1 | 0.1–10 | y = (330 ± 30)x | 0.9983 | 90 ± 10 | 97 ± 10 |
| Ile | 0.03 | 0.1 | 0.1–10 | y = (400 ± 20)x | 0.9979 | 87 ± 6 | 85 ± 2 |
| Met | 0.03 | 0.1 | 0.1–10 | y = (710 ± 70)x | 0.9985 | 87 ± 5 | 88 ± 8 |
| Tyr | 0.5 | 2 | 2–30 | y = (40 ± 2)x | 0.9987 | <clim | 94 ± 9 |
| Arg | 0.03 | 0.1 | 0.1–10 | y = (530 ± 50)x | 0.9991 | <clim | 36 ± 4 |
| Val | 0.03 | 0.2 | 0.2–10 | y = (220 ± 10)x | 0.9995 | 87 ± 9 | 92 ± 4 |
| Pro | 0.03 | 0.1 | 0.1–10 | y = (450 ± 50)x | 0.9975 | 95 ± 2 | 100 ± 10 |
| Thr | 0.03 | 0.2 | 0.2–10 | y = (250 ± 30)x | 0.9989 | 100 ± 10 | 92 ± 8 |
| Gly | 0.7 | 2 | 2–30 | y = (10.0 ± 0.5)x | 0.9991 | <clim | 92 ± 5 |
| Gln | 0.3 | 0.8 | 0.8–10 | y = (83 ± 6)x | 0.9979 | <clim | 88 ± 8 |
| Asn | 0.6 | 2 | 2–30 | y = (25 ± 2)x | 0.9995 | 90 ± 10 | 86 ± 2 |
| His | 0.2 | 0.5 | 0.5–10 | y = (180 ± 20)x | 0.9969 | <clim | 88 ± 9 |
| Lys | 0.2 | 0.5 | 0.5–10 | y = (150 ± 9)x | 0.9991 | <clim | 100 ± 5 |
| Ser | 0.7 | 2 | 2–30 | y = (15 ± 1)x | 0.9973 | <clim | 90 ± 7 |
| Ala | 0.5 | 2 | 2–30 | y = (40 ± 3)x | 0.9981 | <clim | 87 ± 6 |
Таблица 4.
Результаты определения аминокислот в модельном экстракте второго образца почвы (сорбент П-ДМА)
| Вещество | сmin, мг/л | clim, мг/л | Диапазон линейности, мг/л | Уравнение, y = (A ± Δ)x нг/мл |
R2 | Процент восстановления ± sr, % | |
|---|---|---|---|---|---|---|---|
| 0.5 мг/л | 1 мг/л | ||||||
| Phe | 0.08 | 0.3 | 0.3–5 | y = (1020 ± 80)x | 0.9932 | 102 ± 8 | 93 ± 2 |
| Leu | 0.4 | 1 | 1–10 | y = (290 ± 20)x | 0.9916 | 90 ± 10 | 90 ± 3 |
| Met | 0.3 | 0.8 | 0.8–10 | y = (300 ± 30)x | 0.9957 | 85 ± 8 | 97 ± 4 |
| Tyr | 0.4 | 1 | 1–10 | y = (330 ± 20)x | 0.9974 | 104 ± 9 | 96 ± 5 |
| Val | 0.3 | 0.8 | 0.8–10 | y = (270 ± 10)x | 0.9921 | 93 ± 7 | 101 ± 9 |
| Pro | 0.03 | 0.1 | 0.1–5 | y = (1250 ± 70)x | 0.9984 | 45 ± 7 | 42 ± 8 |
| Thr | 0.3 | 0.8 | 0.8–10 | y = (320 ± 20)x | 0.9972 | 93 ± 10 | 91 ± 4 |
| Gln | 0.8 | 3 | 3–10 | y = (230 ± 30)x | 0.9893 | 120 ± 8 | 120 ± 10 |
| Asn | 1 | 3 | 3–10 | y = (62 ± 7)x | 0.9869 | 121 ± 2 | 119 ± 4 |
| Ser | 1 | 3 | 3–10 | y = (26 ± 3)x | 0.9818 | 115 ± 4 | 123 ± 4 |
| Ala | 0.08 | 0.3 | 0.3–5 | y = (450 ± 20)x | 0.9981 | 90 ± 7 | 93 ± 2 |
Рис. 4.
Хроматограммы холостого образца (а), а также хроматограммы вытяжек из почвы с добавками аминокислот на двух уровнях содержаний 0.5 мг/л (б) и 1 мг/л (в). Условия: сорбент П-ДМА, колонка 100 × 4 мм, подвижная фаза CH3CN–аммонийно-формиатный буферный раствор с pH 4.7 (85.5 : 14.5, по объему); скорость потока 0.8 мл/мин; МС-детектирование.
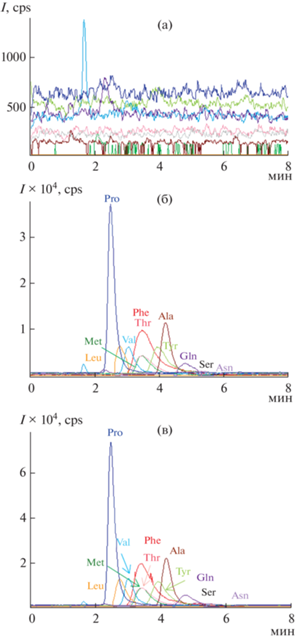
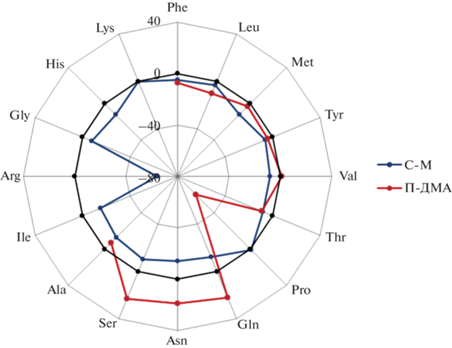
В случае сорбента С-М (табл. 3) значимый матричный эффект (∼70%) наблюдали только для аргинина. Несмотря на высокий фоновый сигнал, пределы обнаружения были ниже, чем на сорбенте П-ДМА, даже с учетом использования разных МС-детекторов, что может быть связано с большей эффективностью сорбента С-М на основе силикагеля. При разделении и определении аминокислот в образцах с добавками на П-ДМА наблюдали незначительные положительные матричные эффекты для серина, аспарагина и глутамина и значительный отрицательный матричный эффект для пролина (∼60%) вследствие его слабого удерживания на полимерном сорбенте (табл. 4). Такого рода матричные эффекты можно объяснить как селективным связыванием аминокислот компонентами почвы, так и подавлением или усилением ионизации в присутствии других компонентов почв, если их пики перекрываются с пиками аналитов на хроматограммах. Других существенных различий в матричных эффектах для разных сорбентов не наблюдали (рис. 5).
Рис. 5.
Средние матричные эффекты (%) для аминокислот на сорбентах С-М и П-ДМА (уровень добавки – 1 мг/л).
* * *
Выбраны условия, обеспечивающие определение 16 аминокислот в режиме ГИХ-МС на сорбенте на основе силикагеля и 13 веществ данного класса на фазе на основе полимера ПС-ДВБ. Установлена меньшая величина фонового сигнала при использовании сорбентов на основе ПС-ДВБ по сравнению с модифицированными фазами на основе силикагеля, что свидетельствует о перспективности использования ПС-ДВБ в качестве матрицы сорбентов для ГИХ-МС. При этом эффективность фаз, полученных при модифицировании 3-аминопропилсиликагеля по реакции Уги, выше, чем таковых на основе полимерной матрицы. Показано, что разработанные сорбенты подходят для прямого ГИХ-МС-определения большого количества аминокислот в различных видах почв без дериватизации.
Работа выполнена при поддержке Российского научного фонда, грант № 20‑13‑00140, в рамках Программы развития Междисциплинарной научно-образовательной школы Московского университета “Будущее планеты и глобальные изменения окружающей среды”, с использованием оборудования ЦКП МГУ “Технологии получения новых наноструктурированных материалов и их комплексное исследование”, приобретенного МГУ по программе обновления приборной базы в рамках национального проекта “Наука” и в рамках Программы развития МГУ.
Список литературы
Dell'mour M., Jaitz L., Oburger E., Puschenreiter M., Koellensperger G., Hann S. Hydrophilic interaction LC combined with electrospray MS for highly sensitive analysis of underivatized amino acids in rhizosphere research // J. Sep. Sci. 2010. V. 33. P. 911.
Weiss J. Handbook of Ion Chromatography. 4th Ed. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA, 2016. 1576 p.
Themelis T., Gotti R., Gatti R. A novel hydrophilic interaction liquid chromatography method for the determination of underivatized amino acids in alimentary supplements // J. Pharm. Biomed. Anal. 2017. V. 145. P. 751.
Qiu J., Craven C., Wawryk N., Carroll K, Li X. Integration of solid phase extraction with HILIC-MS/MS for analysis of free amino acids in source water // J. Environ. Sci. 2022. V. 117. P. 190.
Violi J.P., Bishop D.P., Padula M.P., Westerhausen M.T., Rodgers K.J. Acetonitrile adduct analysis of underivatised amino acids offers improved sensitivity for hydrophilic interaction liquid chromatography tandem mass-spectrometry // J. Chromatogr. A. 2021. V. 1655. Article 462530.
Lozowicka B., Kaczynski P., Iwaniuk P. Analysis of 22 free amino acids in honey from Eastern Europe and Central Asia using LC-MS/MS technique without derivatization step // J. Food Compost. Anal. 2021. V. 98. Article 103837.
Kohler I., Derks R.J.E., Giera M. The rise of hydrophilic interaction chromatography in untargeted clinical metabolomics // LC GC Eur. 2016. V. 29. P. 60.
Schriewer A, Heilen K.J, Hayen H, Jiang W. Direct analysis of amino acids by HILIC–ESI-MS // The Application Notebook. 2017. V. 30. P. 386.
Thermo Scientific. Application note 73151. Underivatized amino acid analysis in wine by HILIC separation and mass detection. URL: https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/an-73151-hilic-ms-amino-acids-wine-an73151-en.pdf (07.12.22).
Agilent. Application note. Methods for the Analysis of Underivatized Amino Acids by LC/MS. URL: https://www.agilent.com/cs/library/applications/ 5991-8582EN_HILIC_Underivatized_Amino_Acid_application.pdf (07.12.22).
Uzhel A.S., Gorbovskaya A.V., Zatirakha A.V., Smolenkov A.D., Shpigun O.A. Manipulating selectivity of covalently-bonded hyperbranched anion exchangers toward organic acids. Part I: Influence of primary amine substitutes in the internal part of the functional layer // J. Chromatogr. A. 2018. V. 1589. P. 65.
Uzhel A.S., Gorbovskaya A.V., Zatirakha A.V., Smolenkov A.D., Shpigun O.A. Manipulating selectivity of covalently-bonded hyperbranched anion exchangers toward organic acids. Part II: Effect of mono- and dicarboxylic amino acids in the internal part of the functional layer // J. Chromatogr. A. 2019. V. 1596. P. 117. https://doi.org/10.1016/j.chroma.2019.03.006
Uzhel A.S., Zatirakha A.V., Smolenkov A.D., Shpigun O.A. Manipulating selectivity of covalently-bonded hyperbranched anion exchangers toward organic acids. Part III: Effect of diamine structure in the external part of the functional layer // J. Chromatogr. A. 2019. V. 1602. P. 310. https://doi.org/10.1016/j.chroma.2019.05.052
Uzhel A.S., Gorbovskaya A.V., Zatirakha A.V., Smolenkov A.D., Shpigun O.A. Manipulating selectivity of covalently-bonded hyperbranched anion exchangers toward organic acids. Part IV: General algorithm based on the variation of external part of the functional layer // J. Chromatogr. A. 2020. V. 1634. Article 461648. https://doi.org/10.1016/j.chroma.2020.461648
Baygildiev T., Zatirakha A., Rodin I., Braun A., Stavrianidi A., Koryagina N., Rybalchenko I., Shpigun O. Rapid IC-MS/MS determination of methylphosphonic acid in urine of rats exposed to organophosphorous nerve agents // J. Chromatogr. B. 2017. V. 1058. P. 32.
Liu J., Wang Y., Cheng H., Wang N., Wu S., Zhang P., Zhu Y. High-capacity anion exchangers based on poly (glycidylmethacrylate-divinylbenzene) microspheres for ion chromatography // Talanta. 2016. V. 159. P. 272.
Shchukina O.I., Zatirakha A.V., Smolenkov A.D., Nesterenko P.N., Shpigun O.A. Anion exchangers with branched functional ion exchange layers of different hydrophilicity for ion chromatography // J. Chr-omatogr. A. 2015. V. 1408. P. 78.
Zhang K., Lou C., Zhu Y., Zhi M., Zeng X., Shou D. Covalently grafted anion exchangers with linear epoxy-amine functionalities for high-performance ion chromatography // Talanta. 2019. V. 194. P. 485.
Chikurova N.Yu., Shemiakina A.O., Shpigun O.A., Chernobrovkina A.V. Multicomponent Ugi reaction as a tool for fast and easy preparation of stationary phases for hydrophilic interaction liquid chromatography. Part I: The influence of attachment and spacing of the functional ligand obtained via the Ugi reaction // J. Chromatogr. A. 2022. V. 1666. Article 462804.
Chikurova N.Yu., Shemyakina A.O., Bryskina D.E., Nuriev V.N., Komarov A.A., Statkus M.A., Stavrianidi A.N., Chernobrovkina A.V. A Novel Adsorbent for Hydrophilic Chromatography Based on Silica Modified by the Ugi Reaction // J. Anal. Chem. 2021. V. 76. P. 1083.
Dömling A., Ugi I. Multicomponent reactions with isocyanides // Angew. Chem. 2000. V. 39. P. 3168.
Zatirakha A.V., Smolenkov A.D., Pirogov A.V., Nesterenko P.N., Shpigun O.A. Preparation and characterization of anion exchangers with dihydroxy-containing alkyl substitutes in the quaternary ammonium functional groups // J. Chromatogr. A. 2014. V. 1323. P. 104.
Jin G., Guo Z., Zhang F., Xue X., Jin Y., Liang X. Study of the retention equation in hydrophilic interaction liquid chromatography // Talanta. 2008. V. 76. P. 522.
Kawachi Y., Ikegami T., Takubo H., Ikegami Y., Miyamoto M., Tanaka N. Chromatographic characterization of hydrophilic interaction liquid chromatography stationary phases: Hydrophilicity, charge effects, structural selectivity, and separation efficiency // J. Chromat-ogr. A. 2011. V. 1218. P. 5903.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии