

Астрономический журнал, 2022, T. 99, № 9, стр. 767-783
Формирование двухкольцевых полициклических ароматических углеводородов при рекомбинации бензил и пропаргил радикалов в условиях околозвездных оболочек звезд асимптотической ветви гигантов
В. С. Красноухов 1, 2, *, П. С. Пивоваров 2, М. В. Загидуллин 1, В. Н. Азязов 1, А. М. Мебель 3, А. Н. Морозов 3
1 Самарский филиал Физического института им П.Н. Лебедева РАН
443011 Самара, Россия
2 Самарский университет
443086 Самара, Россия
3 Кафедра химии и биохимии, Международный университет Флориды
33199 Флорида, Майами, США
* E-mail: vkrasnoukhov@fian.smr.ru
Поступила в редакцию 08.04.2022
После доработки 26.05.2022
Принята к публикации 21.06.2022
- EDN: CPHSTQ
- DOI: 10.31857/S0004629922090079
Аннотация
Механизмы и кинетика образования двухкольцевых полициклических ароматических углеводородов при рекомбинации радикалов бензила (С7Н7) и пропаргила (С3Н3) в условиях околозвездных оболочек богатых углеродом звезд асимптотической ветви гигантов, а также их горения уточнены на основе высокоточных методов квантовой химии и развитой теории переходного состояния. На основе построенных диаграмм поверхностей потенциальных энергий и расчетных зависимостей кинетических констант процессов от температуры и давления выявлены реакционные пути и их относительные вклады в состав конечных продуктов. Показано, что в условиях околозвездных оболочек звезд асимптотической ветви гигантов стабилизации начальных комплексов С10Н10, в отличии от пламени горения, не происходит, что способствует росту выхода двуциклических продуктов реакции.
1. ВВЕДЕНИЕ
Полициклические ароматические углеводороды (ПАУ) – органические молекулы, состоящие из конденсированных бензольных колец, вместе с их (де)гидрированными [1], алкилированными [2, 3], протонированными [4, 5] и ионизированными [6] производными считаются присутствующими повсюду в межзвездном пространствe и в околозвездных оболочках. При этом до 20% углеродного баланса в нашей Галактике приписывается как раз молекулам этого класса [1, 7–9]. Также была выдвинута гипотеза, что они представляют собой недостающее звено между малыми молекулами углерода и углеродсодержащими наночастицами [10, 11]. Некоторые из наблюдаемых диффузионных межзвездных полос [1, 2, 4, 5, 12, 13], которые представляют собой дискретные полосы поглощения, наложенные на кривую межзвездного поглощения в диапазоне от ~ 400 нм (видимая область) до 1.2 мкм (ближняя инфракрасная область), а также неидентифицированные полосы инфракрасного излучения в диапазоне 3–14 мкм [10, 14, 15], могут быть приписаны ПАУ. Наиболее прямым свидетельством существования ПАУ в межзвездном пространстве является их обнаружение в углеродистых хондритах, таких как Murchison, Allende и Orgueil [16–19], где изотопный анализ 13C/12C и D/H подтверждает их околозвездное происхождение [18], а также в кометах [20] и марсианских метеоритах [21]. Недавно ароматическая молекула бензонитрил (C6H5CN) была идентифицирована в молекулярном облаке TMC-1 [22].
Предполагается, что ПАУ образуются в результате роста из небольших ароматических молекул, таких как бензол и толуол, например, в оболочках богатых углеродом звезд асимптотической ветви гигантов (АВГ) и планетарных туманностях [23, 24], через процессы роста молекулярной массы. Механизм “отрыв водорода – присоединение углерода (HACA)” [25–28], заимствованный из кинетической теории образования и роста ПАУ на Земле, при горении углеводородного топлива [26, 29–34], был первоначально привлечен для предсказания содержания ПАУ в оболочках звезд [25, 27]. В последнее время [9] на основе экспериментальных и расчетных данных был предложен целый ряд новых реакционных путей, ведущих к укрупнению ПАУ в космических условиях. В отличие от НАСА, где к фенилу последовательно присоединяются две молекулы ацетилена, прежде чем образуется двухкольцевой ПАУ, в новом механизме [23] “отрыв водорода – присоединение винилацетилена” (HAVA) для этого достаточно одного столкновения с молекулой винилацетилена С4Н4. В обзоре Кайзера и Хансена [9], кроме HACA и HAVA, детально описываются известные к настоящему времени механизмы роста ПАУ “добавление фенила–дегидроциклизация” [35] (PAC), “радикал–радикальные реакции” [36] (RRR), “добавление метилидина–циклизация–ароматизация” [37] (MACA). Поиски новых реакционных путей, ведущих к росту молекулярной массы ПАУ, продолжают вестись во многих лабораториях мира. В работе Френклаха и Фейгельсона [25] отмечается, что результаты по моделированию выхода ПАУ в оболочке звезд АВГ весьма чувствительны к большому числу химических и астрофизических параметров, значения которых в большинстве случаев только грубо оцениваются. Эта проблема решается, в том числе, за счет разработки кинетических схем, учитывающих наиболее важные и значительные реакционные пути, ведущие к формированию и росту ПАУ, а также нахождения точных значений кинетических констант вовлеченных элементарных процессов.
Особую роль в процессах роста ПАУ могут играть метилзамещенные и, в более общем случае, алкилированные ПАУ, молекулой-прототипом которых является толуол. В высокотемпературных средах, например, в околозвездных оболочках или при горении, они могут образовываться в результате процессов метилирования/алкилирования, т.е. в результате реакций преодоления CH3/алкил-радикала на H, которые требуют значительных барьеров [38–42]. Бензил радикал образуется из толуола путем отрыва атома Н от метильной группы.
В данной работе рассматривается один из возможных путей образования ПАУ в реакции рекомбинации бензил (C7H7) и пропаргил (C3H3) радикалов, являющейся потенциально важной стадией образования второго ароматического кольца. По аналогии с реакцией C3H5 + C3H3, ведущей к образованию бензола, нафталин может образовываться в реакции C7H7 + C3H3 и последующих реакциях, как предложено Колкетом и Сири [43], а затем Мариновым и соавт. [44]. Механизм реакции С7Н7 + С3Н3 ранее изучался в работе Мацуги и Миёси методами квантовой химии и теоретической химической кинетики [45]. Принимая во внимание последние достижения в развитии теории переходного состояния, механизм и кинетика этой важной реакции заслуживают нового рассмотрения, направленного на уточнение значений констант скоростей реакционных каналов, ведущих к различным продуктам, и их относительных выходов в зависимости от температуры и давления. В данном исследовании константы скорости реакции радикалов бензилa и пропаргилa, зависящие от температуры и давления, рассчитывались методом Райса–Рамспергера–Касселя–Маркуса и основного кинетического уравнения (РРКМ-ОУ), тогда как константы скорости в пределе высокого давления для безбарьерных входных и выходных каналов, соответственно, оценивались с использованием теории переходного состояния с варьируемой координатой реакции (VRC-TST) и теории фазового пространства. Уточненные константы будут полезны при построении более надежных кинетических моделей образования нафталина и роста ПАУ как в астрохимии, так и в химии горения.
2. МЕТОДЫ РАСЧЕТА
2.1. Расчет поверхности потенциальной энергии
Были применены ab initio расчеты для исследования поверхности потенциальной энергии (ППЭ) реакции бензил + пропаргил. На первом этапе были оптимизированы геометрии реагентов, продуктов, всех локальных минимумов C10H10 и переходных состояний с использованием метода теории функционала плотности (ТФП) B3LYP [46–48] с базисным набором 6-311G**. Частоты колебаний и энергия нулевых колебаний (ZPE) рассчитывались с использованием того же уровня теории. Окончательное уточнение одноточечных энергий было выполнено в рамках подхода модельной химии [49–51] G3(MP2,CC), где общая энергия рассчитывается как
Механизм реакции включает несколько бирадикальных соединений (синглетов с открытыми оболочками), для которых оптимизация геометрии и расчеты частот колебаний выполнены с использованием неограниченного подхода UB3LYP/6-311G**, а полные энергии уточнены в рамках композитного триплет-синглетно-щелевого метода [45, 52]:
2.2. Кинетические расчеты
Рассчитанные относительные энергии реагентов, продуктов, промежуточных и переходных состояний на ППЭ реакции и их молекулярные параметры в дальнейшем использовали в статистических расчетах констант скорости реакции и коэффициентов ветвления продуктов. В частности, константы скорости, зависящие от энергии и углового моментa (E,J-разрешенные), оценивались в рамках теории РРКМ [58]. Модель гармонического осциллятора – жесткого роторa (RRHO) использовалась для вычисления количества состояний для переходных состояний и плотности состояний для соответствующих локальных минимумов. В расчетах переходного состояния применялась туннельная поправка Эккарта [59]. Функции распределения внутренних роторoв рассматривались в рамках приближения заторможенного ротора, где потенциалы внутреннего вращения были взяты из расчетов B3LYP/6-311G** Мацуги и Миёси [45]. Константа скорости, разрешенная по E,J для начальной безбарьерной ассоциации бензил и пропаргил радикалов, была рассчитана с использованием теории переходного состояния с варьируемой координатой реакции (VRC-TST) [60–62]. Для многочисленных безбарьерных реакций диссоциации, приводящих к различным продуктам C10H9 + H и C6H5 + C4H5, теория фазового пространства [63] была использована для оценки E,J-разрешенных констант скорости их обратных реакций бимолекулярной ассоциации в пределе высокого давления (ВД). Показатели степени и префакторы для потенциалов в расчетах теории фазового пространства были подобраны так, чтобы константы скорости при ВД соответствовали константам скорости наиболее схожих аналогичных реакций-прототипов, оцененных в работe Клиппенштейна и др. [64], а также в нашей предыдущей публикации [65], в рамках наиболее точных VRC-ТСТ расчетов. Выбор реакции-прототипа для каждого конкретного случая обсуждается ниже.
Зависимые от T и p (температуры и давления) феноменологические константы скорости были рассчитаны с использованием подхода одномерного основного кинетического уравнения [66] (ОУ), реализованного в пакете MESS [67]. Параметры Леннардa-Джонсa, (ε/см–1, σ/Å) = (390, 4.46), и столкновительной передачи энергии, n = = 0.62, α300 = 424 см–1, для расчетов ОУ были взяты из предыдущего исследования систем [68] C9Hx/Ar и использовались в “экспоненциально убывающей” [69] модели столкновительной передачи энергии для температурной зависимости параметра разброса α деактивирующей части функции передачи энергии α(T) = α300(T/300 K)n.
2.3. VRC-TST расчеты
Теория VRC-TST [60–62] была применена для исследования входных каналов реакции бензил + + пропаргил. В этом методе используется такое приближение, что внутримолекулярные степени свободы взаимодействующих фрагментов не активны в реакции рекомбинации. Поэтому реактивный поток определяется межмолекулярными модами взаимодействующих фрагментов, тогда как внутрифрагментарные моды сохраняются. Реакционный поток оценивается путем вариационной оптимизации числа состояний в классическом фазовом пространстве, определяемом поверхностью раздела между взаимодействующими фрагментами [61]. Взаимодействующие комплексы со случайной ориентацией на поверхности раздела были получены с использованием структур, не изменяющих свои геометрические свойства при взаимодействии с молекулами (“жесткие” структуры), бензила и пропаргила, рассчитанные для минимального энергетического пути (МЭП) на разделяющей поверхности. Использование в расчетах “жестких” структур подтверждается приближением “сохраняющихся” мод. Оптимизация структур МЭП для входных каналов реакции бензил + пропаргил проводилaсь на уровне теории CASSCF [70] (12e,12o)/DZP [71, 72] с уравновешенным усреднением состояния волновой функции по двум нижним дублетам. МЭП сканировался путем фиксирования соответствующего расстояния C–C, в то время как другие степени свободы были оптимизированы. Активное пространство (12e,12o) включало π-системы как пропаргила, так и бензила, а также электроны, участвующие в разрыве/образовании связи. Для сходимости интегралов фазового пространства для каждой точки, разделяющей поверхности с помощью одноточечных расчетов на уровне CASPT2(12e,12o)/cc-pVDZ, были оценены энергии около 5000 конформаций “жестких” бензил + + пропаргил комплексов. Вычисленная энергия была дополнительно уточнена с помощью специальных одномерных поправок на полный базисный набор (ПБН) следующим образом:
где Eжесткий – энергия одиночной точки CASPT2 (12e,12o)/cc-pVDZ структуры на разделяющей поверхности, состоящей из взаимодействующих “жестких” фрагментов. Поправка ПБН рассчитывалась с использованием энергий CASPT2(12e, 12o)/cc-pVnZ [53, 54] (n = D, T, Q) “жестких” МЭП-структур:Поправки вычислялись в явном виде только для структур МЭП, а их зависимость от расстояния, соответствующего образующейся связи С‒С, интерполировалась с помощью сплайнов.
3. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИE
3.1. Поверхность потенциальной энергии
Рисунки 1–5 иллюстрируют построенные диаграммы потенциальной энергии, показывающие относительные энергии локальных минимумов, переходных состояний и продуктов по отношению к реагентам C7H7 + C3H3, а также их трехмерную геометрию. Вычисленные здесь энергии приведены вместе со значениями, полученными в предыдущем исследовании Мацуги и Миёси [45] на уровне CBS-QB3 для структур с закрытой оболочкой и с использованием композитного триплет-синглетно-щелевого метода для синглетов с открытой оболочкой. Видно, что относительные энергии, полученные в настоящей работе, весьма близки к тем, о которых сообщают Мацуги и Миёси [45], причем различия не превышают 3 ккал/моль, а в большинстве случаев находятся в пределах 1–2 ккал/моль.
Рис. 1.
Диаграмма потенциальной энергии для начальных каналов реакции C7H7 + C3H3. Все относительные энергии указаны в ккал/моль по отношению к реагентам. Значения в скобках представлены для сравнения с работой Мацуги и Миёси [37].

Рис. 2.
Диаграмма потенциальной энергии каналов реакций изомеризации и разложения начального интермедиата w2. Все относительные энергии указаны в ккал/моль по отношению к реагентам C7H7 + C3H3. Значения в скобках представлены для сравнения с работой Мацуги и Миёси [37].
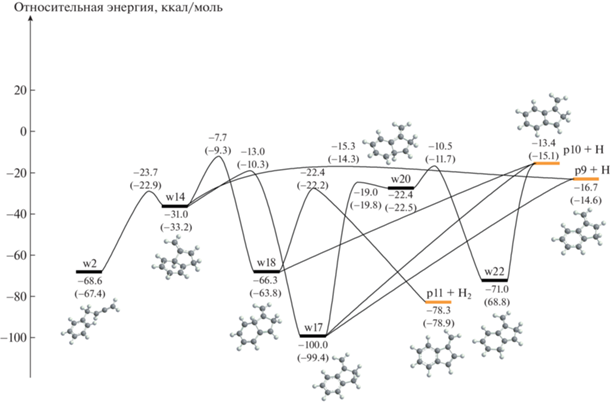
Рис. 3.
Диаграмма потенциальной энергии каналов реакций изомеризации и распада начального интермедиата w1. Все относительные энергии указаны в ккал/моль по отношению к реагентам C7H7 + C3H3. Значения в скобках представлены для сравнения с работой Мацуги и Миёси [37].

Рис. 4.
Диаграмма потенциальной энергии каналов реакций изомеризации и разложения интермедиата w17 на H-нафталинил радикалы и нафталин. Все относительные энергии указаны в ккал/моль по отношению к реагентам C7H7 + C3H3. Значения в скобках представлены для сравнения с работой Мацуги и Миёси [37].

Рис. 5.
Диаграмма потенциальной энергии реакции C7H7 + C3H3 в триплетном электронном состоянии. Все относительные энергии указаны в ккал/моль по отношению к реагентам.

Во входном канале, в зависимости от того, присоединяется ли пропаргил к внекольцевой группе СН2 бензила своим СН2- или СН-концом, могут образовываться два разных начальных комплекса: 3-бутинилбензол (w1) или 2,3-бутадиенилбензол (w2), соответственно, см. рис. 1. Эта часть диаграммы потенциальной энергии показывает наиболее короткие реакционные пути, в которых за первоначальным образованием связи сразу следует разрыв другой связи C–H или C–C, приводящий к продуктам p1–p4. Все эти каналы являются эндотермическими. Наименее эндотермическим из них является образование 1-фенил-2,3-бутадиенил радикала (р1) + Н, лежащего на 11.8 ккал/моль выше реагентов. Другие продукты c потерей H включают 1-бензилпропаргил (p2) и 1-бензилалленил (p3) с более высокой относительной энергией 19.6 и 17.4 ккал/моль соответственно. Продукт фенил (p4) + i-C4H5, образующийся при разрыве внешней связи C–C рядом с кольцом, имеет самую высокую относительную энергию 21.8 ккал/моль. Хотя продукты р1–р4 не являются энергетически выгодными, ведущие к ним каналы предпочтительны с точки зрения энтропии, что увеличивает вероятность их образования при высоких температурах. Данное заключение проверяется, в частности, путем расчета полных энтропий продуктов p3 и p4 при 1000 К (176.57 и 213.29 Кал/Моль/К соответственно) в сравнении с, например, энтропией переходного состояния w14 → p9 + H (163.17 Кал/Моль/К). Количественно подтверждается, что энтропии реакций безбарьерного распада, как правило, выше, чем энтропии активации реакций, идущих через переходные состояния.
Далее мы рассматриваем энергетически выгодные каналы, ведущие к метиленинданил радикалам (p5, p9 и p10) + H и метиленинденам (p6 и p11) + H2. Пути к p9, p10 и p11, начинающиеся с w2, показаны на рис. 2. Боковая цепь в w2 может замкнуться в пятичленное кольцо с группой CH2 вне кольца, образуя w14, с которого может оторваться атом водорода с переходом к p9, 1‑метилен-2-инданил радикалу, лежащему на 16.7 ккал/моль ниже исходных реагентов. Высшее по энергии переходное состояние по пути w2 → w14 → p9 + H находится на 15.3 ккал/моль ниже реагентов. Альтернативно, w14 может изомеризоваться в w17 за счет 1,3-H миграции в пятичленном кольце, а w17 может потерять атом H из двух разных положений с образованием p9 или p10 (3-метилен-1-инданил, 13.4 ккал/моль ниже энергии реагентов) без переходных состояний. 1,2-H сдвиг в w14 приводит к w18, и последний может потерять либо H с образованием p10, либо H2 с образованием 1-метилениндена. Хотя продукт p11 + H2 в целом является сильно экзотермическим (на 78.3 ккал/моль), переходное состояние с отрывом H2 требует высоких энтропийных затрат и находится на 22.4 ккал/моль ниже реагентов, но на 55.9 ккал/моль выше продукта. Интермедиат w17 также может изомеризоваться в w22 через w20 за счет последовательных 1,2-H миграций внутри шестичленного кольца, при этом w22 в конечном итоге диссоциирует в p10 + H. По-видимому, исход реакции должен контролироваться переходными состояниями, соединяющими w14 и p9 + H, w17 и w18, соответственно, находящимися на 15.3, 13.0 и 7.7 ккал/моль ниже по энергии исходных реагентов C7H7 + C3H3.
Замыканию пятичленного кольца в другом начальном интермедиате w1, w1 → w4 → w8 предшествует 1,2-H сдвиг от боковой цепи к шестичленному кольцу (рис. 3). Бициклический интермедиат w8 может далее изомеризоваться посредством трех различных 1,2-H миграций с образованием w10, w11 или w12, среди которых путь к w10 явно предпочтительнее из-за его значительно более низкой высоты барьера. Все три промежуточных соединения w10–w12 могут отсоединять атом водорода без перевала с образованием 2-метилен-1-инданила p5, лежащего на 20.4 ккал/моль ниже реагентов. Кроме того, w10 может отстрелить молекулярный водород с образованием 2-метилениндена, но этот шаг реакции имеет высокий выходной барьер.
На рис. 4 показаны пути дальнейших превращений интермедиата w17 (образованного из w2, рис. 2), ведущих в нафталин (р14) или в его непосредственных предшественников. Внекольцевая группа CH2 в w17 может внедряться в пятичленное кольцо, образуя w23. Последний затем может подвергаться 1,2-H миграции из групп CH2 в новом шестичленном кольце к соседнему “голому” (безводородному) атому углерода, что приводит к орто- и параизомерам дигидронафталина w24 и w25 соответственно. Как w24, так и w25 могут терять атом H, не встречая на своем пути перевалов при движении вверх по “ложбине”, образуя 1H-нафталинил радикал p12. Промежуточное соединение w24 также может разлагаться до 2H-нафталинила p13 + H, тогда как w25 может отсоединить H2 с образованием нафталина p14, проходя через перевал. Хотя пути от w17 к продуктам p12–p14 довольно выгодны с энергетической точки зрения, они гораздо более требовательны к энтропии, чем простые потери H от этого промежуточного состояния, производящие p9 или p10.
Наконец, мы также исследовали возможность протекания реакции C7H7 + C3H3 в триплетном электронном состоянии (рис. 5). Здесь два радикала могут рекомбинировать через барьер в 15.0 ккал/моль с образованием начального интермедиата w1T, в котором пропаргил присоединен к СН2-группе бензила СН2-концом. w1T находится на 17.4 ккал/моль ниже реагентов по энергии и далее может подвергаться замыканию шестичленного цикла, приводящему к промежуточному соединению w2T, расположеннoму на уровне –11.3 ккал/моль, через переходное состояние, расположенное на 9.9 ккал/моль выше C7H7 + C3H3. Таким образом, циклоприсоединение C7H7 + + C3H3 с образованием w2T является ступенчатым, а не одношаговым, как в случае недавно изученного нами циклоприсоединения бензил + + бензил в триплетном состоянии [73]. Несмотря на тщательный поиск, нам не удалось найти переходное состояние для одношагового циклоприсоединения C3H3 к C7H7. Промежуточное соединение w2T теряет атом H в месте соединения двух колец с образованием продукта C10H9 p15, лежащего на 6.1 ккал/моль выше реагентов через переходное состояние, расположенное на 14.3 ккал/моль выше реагентов. Затем p15 может служить предшественником образования нафталина, который может быть образован миграцией H от CH2 к соседнему свободному атому C с последующей потерей H из другой группы CH2; реакция p15 → нафталин + H является экзотермической на 3.5 ккал/моль и ожидается, что она будет быстрой в условиях горения. Мы также попытались найти путь от w2T к 1H-нафталенилу p12 через миграцию H с последующей потерей атома водорода, но все попытки найти переходное состояние для миграции H в w2T сходятся к переходному состоянию для потери H, соединяющему w2T с р15.
Обратимся теперь к выбору прототипных реакций радикальной рекомбинации, подобных обратным реакциям для безбарьерного разложения промежуточных соединений C10H10 при разрыве одинарной связи. Константы скорости в пределе ВД для этих прототипных реакций, рассчитанные в ранних [65, 66] работах с использованием подхода VRC-TST, используются здесь в наших расчетах РРКМ-ОУ. В частности, из-за сходства радикалов i-C4H5 и аллила константа скорости (C6H5) p4 + H была подогнана в рамках теории фазового пространства к константе скорости C6H5 + аллил [58], деленной на 2, с учетом различия в симметрийном факторе для пути реакции. Константа скорости p1 + H принималась равной константе скорости H + аллила [59], также разделенную на 2. Для p2 + H → w1/w2 в качестве прототипов были соответственно выбраны [59] реакции H + CH3CHCCH → CH3CH2CCH и H + + CH3CHCCH → CH3CHCCH2. Константа скорости H + CH3CHCCH → CH3CHCCH2 также использовалась для p3 + H → w2. Для реакций, обратных образованию изомеров метиленинданил радикалов, в том числе p5 + H → w10/w11/w12, p9 + H → w17 и p10 → w17/w18, мы использовали константу скорости C5H5 + H [59], деленную на коэффициент 5 для каждого канала, исходя из того, что присоединение H происходит к ароматическому π-радикалу.
3.2. Константы скорости
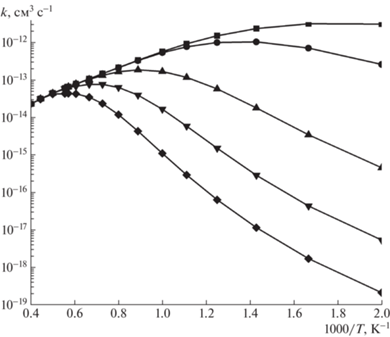
На рис. 6 показана общая константа скорости реакции C7H7 + C3H3 в пределе ВД, рассчитанная здесь с использованием теории VRC-TST, в сравнении с константой скорости, полученной Мацуги и Миёси [45] в рамках микроканонического вариационного TST (VTST) метода. Отчетливые различия видны как в значениях, так и в температурной зависимости рассчитанных констант скорости. Константа скорости, рассчитанная в подходе VTST, показывает отрицательную температурную зависимость во всем диапазоне температур 500–2000 К, рассматриваемом Мацуги и Миёси [45], уменьшаясь от 5.8 × 10–11 до 9.1 × × 10–12 см3 с–1. Константа скорости VRC-TST показывает более сложную температурную зависимость, отрицательную при низких температурах и положительную при более высоких температурах, с минимальным значением 2.8 × 10–11 см3 с–1, достигаемым около 800 К. Такое поведение типично для реакций между двумя резонансно стабилизированными радикалами. Константы скорости VRC-TST и VTST близки при 900 K, но VTST значение превышает текущий VRC-TST результат в 1.93 раза при 500 K. Разница еще больше при высоких температурах, где константы скорости VRC-TST выше, чем VTST в 3.9 и 6.1 раза при 2000 и 2500 К соответственно. Отметим, что для 2500 К мы экстраполировали выражение, выведенное Мацуги и Миёси [45] в температурном интервалe 500–2000 К. Спад общей константы скорости, рассчитанной при различных конечных давлениях, определяется балансом между столкновительной стабилизацией исходных интермедиатов w1 и w2, их разложением обратно на реагенты и диссоциацией на бимолекулярные продукты путем отщепления H или i-C4H5. В диапазоне температур, где преобладает столкновительная стабилизация, до 800, 1000, 1125 и 1375 К при давлениях 30 Торр, 1, 10 и 100 атм, соответственно, константы скорости при конечных давлениях по величине очень близки к пределу ВД. При более высоких температурах распад w1 и w2 обратно в C7H7 + C3H3 становится значительным и наблюдается резкое уменьшение полной константы скорости в диапазонах 800–1750 К (30 Торр), 1000–2000 К (1 атм), 1125–2250 К (10 атм) и 1375–2500 К (100 атм). При высоких температурах вступают в действие каналы, ведущие к образованию бимолекулярных продуктов, и константы полной скорости при конечном давлении показывают положительную температурную зависимость, при 2500 К их значения сливаются. Следует отметить, что мы повторили наши расчеты РРКМ-ОУ с параметрами Леннарда-Джонса и передачи энергии, использованными Мацуги и Миёси [45], но значения констант скорости изменились относительно мало, с наибольшими различиями до 13–52%, которые наблюдаются вблизи температур, при которых отрицательная температурная зависимость переходит в положительную. В других диапазонах температур различия обычно не превышают нескольких процентов.
Рис. 6.
Суммарная константа скорости реакции C7H7 + C3H3, рассчитанная на пределе ВД ($\blacksquare $) и при конечных давлениях: ($ \bullet $) – 1 атм, ($\blacktriangle $) – 30 Торр, ($\diamondsuit $) – 10 атм, (×) – 100 атм, (+) – результаты работы Мацуги и Миёси [37] для ВД.

Анализ расчетных констант скоростей показывает, что прямая реакция преимущественно образует стабилизированные столкновениями комплексы w1 и w2. В частности, на w1 и w2 приходится более 90% общего выхода продукта до 1375, 1650, 1800 и 2000 К при 30 Торр, 1, 10 и 100 атм соответственно. Соотношение выходов w1/w2 остается между 3.3 при 500 К и 1.4 при самых высоких температурах, когда оба промежуточных соединения все еще существуют в виде стабильных химических соединений; это соотношение регулируется константами скорости в пределе ВД для присоединения пропаргила к бензилу по концам CH2 и CH соответственно. При высоких температурах начинают преобладать бимолекулярные продукты, включающие p3 + H, C6H5 (p4) + + i‑C4H5, p2 + H, p9 + H, p1 + H, p5 + H и p10 + H. Основной вклад вносят p3, p4 и p2, за которыми следует p9, выход которого при 1 атм достигает максимума 2.4% при 2000 K. Зависящие от давления константы скорости отдельных каналов продуктов реакции C7H7 + C3H3 показаны на рис. 7(a) для давления 1 атм. Как видно на рис. 7(б), w1 и w2 в основном разлагаются обратно на реагенты с константами скоростей, практически не зависящими от давления, за исключением низких температур, где их значения слишком малы и поэтому незначительны. Константы скорости мономолекулярного разложения w1 и w2 до бензила + пропаргила быстро растут с температурой и становятся выше 102 с–1 при 1000 и 1125 К и 105 с–1 при 1250 и 1500 К соответственно. Это указывает на то, что при температурах, соответствующих горению, вероятно, устанавливается равновесие между C7H7 + C3H3 и w1/w2. Метиленинданильные продукты p9 и p10 известны как вероятные предшественники нафталина [46, 61]. Маловероятно, что обратные реакции (p1–p4) + Н дадут существенный вклад в формирование нафталина.
Рис. 7.
Константы скорости для основных каналов продуктов для C7H7 + C3H3 при давлении 1 атм: ($\square $) – w1, (◊) – w2, (+) – p1, ($ \bullet $) – p2, ($\blacksquare $) – p3, (×) – p4, (⬟) – p5, ($\blacktriangle $) – p9, ($\blacktriangledown $) – p10, а также общие константы для 1 атм ($\rlap{--} $) и предела высокого давления ($ - {\kern 1pt} - $) (а); на панели (б) сравнение констант скоростей мономолекулярного распада w1/w2 ($\blacksquare $ для w1, $\diamondsuit $ для w2) c работой Мацуги и Миёси [37] (+ для w1, × для w2) при 1 атм.

Мацуги и Миёси [45] предложили следующий упрощенный механизм образования метиленинданил радикалов р9 и р10, которые затем переходят к образованию нафталина р14 и его метиленинденового изомера р11:
(R1)
${{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{{\text{7}}}} + {{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{3}}}} \leftrightarrows {\text{w1}};$(R2)
${{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{{\text{7}}}} + {{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{3}}}} \leftrightarrows {\text{w2}};$(R3)
${{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{{\text{7}}}} + {{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{3}}}} \to ({\text{p}}9 + {\text{p}}10) + {\text{H}};$Наши расчеты дают более высокие относительные выходы p3 и p4 и более низкие выходы p9 и p10. Разница возникает из-за разной обработки выходных безбарьерных каналов с использованием VTST в работе Мацуги и Миёси и в рамках теории фазового пространства (соответствующей подходящим результатам VRC-TST) в настоящей работе. Ввиду этого различия и нынешних результатов мы предлагаем несколько расширенный вариант этой упрощенной модели, который должен включать, помимо (R1)–(R4), обратные реакции (R3) и (R4), а также:
(R5)
${{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{7}} + {{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{3}}}} \leftrightarrows {\text{p3}} + {\text{H}};$(R6)
${{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{{\text{7}}}} + {{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{3}}}} \leftrightarrows {\text{p4}} + {\text{i - }}{{{\text{C}}}_{{\text{4}}}}{{{\text{H}}}_{5}};$(R7)
${\text{p3}} + {\text{H}} \leftrightarrows {\text{p4}} + {\text{i - }}{{{\text{C}}}_{{\text{4}}}}{{{\text{H}}}_{5}};$(R9)
${\text{p}}4 + {\text{i - }}{{{\text{C}}}_{{\text{4}}}}{{{\text{H}}}_{{\text{5}}}} \to ({\text{p9}} + {\text{p10}}) + {\text{H}}.$Модифицированные выражения Аррениуса для констант скоростей этих реакций, рассчитанные при различных давлениях, представлены в табл. 1. Кроме того, еще более упрощенный механизм, предложенный Мацуги и Миёси [45] для высокотемпературных условий, (T/K) > 100 × × log10(p/атм) + 1400 в диапазоне давлений 0.01–100 атм, включая вторичные реакции с образованием нафталина и метилениндена,
(R3)
${{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{{\text{7}}}} + {{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{3}}}} \to ({\text{p9}} + {\text{p10}}) + {\text{H}};$Таблица 1.
Параметры модифицированных выражений Аррениуса $k = A{{T}^{n}}{\text{exp}}\left( { - {{E}_{{\text{a}}}}{\text{/}}RT} \right)$ или k = ${{A}_{1}}{{T}^{{n1}}} \times $ $ \times \;{\text{exp(}}{\kern 1pt} - {\kern 1pt} E_{{\text{a}}}^{1}{\text{/}}RT{\text{)\;\;}}$ + ${{A}_{2}}{{T}^{{n2}}}{\text{exp(}}{\kern 1pt} - {\kern 1pt} E_{{\text{a}}}^{2}{\text{/}}RT{\text{)}}$ для рассмотренных реакций. Предэкспоненциальные множители A приведены в см3 моль–1 c–1 для бимолекулярных реакций и в c–1 для мономолекулярных реакций, Ea даны в кал моль–1
| Реакция | p | A1 | n1 | Ea1 | A2 | n2 | Ea2 | T, K |
|---|---|---|---|---|---|---|---|---|
| C7H7 + C3H3 → → w1 | 30 Торр | –8.93E+94 | –24.809 | 33 636 | 5.70E+73 | –18.313 | 24 736 | 500–1500 |
| 1 атм | 1.28E+107 | –27.068 | 59 203 | 1.83E+44 | –9.709 | 11 303 | 500–1800 | |
| 10 атм | 6.70E+93 | –22.922 | 56 043 | 7.70E+36 | –7.3731 | 8868.3 | 500–2000 | |
| 100 атм | 2.50E+73 | –16.9 | 46 525 | 2.79E+25 | –3.8193 | 4617.2 | 500–2000 | |
| w1 → C7H7 + + C3H3 | 30 Торр | –7.25E+62 | –14.372 | 81 402 | 1.00E+52 | –10.936 | 77 705 | 500–1500 |
| 1 атм | 2.48E+108 | –26.473 | 1.24E+05 | 1.43E+50 | –10.383 | 79 688 | 500–1800 | |
| 10 атм | 1.49E+94 | –22.094 | 1.19E+05 | 1.35E+43 | –8.1597 | 77 364 | 500–2000 | |
| 100 атм | 1.59E+76 | –16.78 | 1.11E+05 | 4.50E+33 | –5.2046 | 73 915 | 500–2000 | |
| C7H7 + C3H3 → → w2 | 30 Торр | 2.10E+113 | –29.31 | 59 212 | 4.09E+49 | –11.593 | 13 080 | 500–1650 |
| 1 атм | 54 680 | 2.3973 | –3260.8 | –1.23E+35 | –5.7493 | 28 537 | 500–1800 | |
| 10 атм | 1.75E+83 | –19.915 | 52 172 | 2.89E+29 | –5.2333 | 6272.4 | 500–2000 | |
| 100 атм | 8.04E+75 | –17.585 | 52 248 | 1.60E+24 | –3.5836 | 4435.7 | 500–2250 | |
| w2 → C7H7 + + C3H3 | 30 Торр | 3.62E+115 | –28.957 | 1.26E+05 | 8.63E+54 | –12.101 | 82 528 | 500–1650 |
| 1 атм | 1.84E+12 | 1.2236 | 67 123 | –3.61E+48 | –8.5394 | 104 550 | 500–1800 | |
| 10 атм | 8.24E+84 | –19.458 | 1.18E+05 | 2.17E+36 | –6.2187 | 76 321 | 500–2000 | |
| 100 атм | 4.28E+75 | –16.618 | 1.16E+05 | 3.51E+31 | –4.7109 | 74 677 | 500–2250 | |
| C7H7 + C3H3 → → (p9 + p10) + H | 1.04 × 10–6 Торр | 2.20E+57 | –14.205 | 1.56E+04 | 8.30E+22 | –3.6679 | 3582.8 | 500–2500 |
| 1.04 × 10–4 Торр | 1.91E+69 | –17.453 | 2.52E+04 | 1.33E+30 | –5.5803 | 11 740 | 500–2500 | |
| 1.04 × 10–1 Торр | 4.69E+38 | –7.1394 | 35 358 | 3.70E+41 | –7.2932 | 52 967 | 500–2500 | |
| 30 Торр | 1.12E+62 | –14.179 | 43 583 | 7.19E+91 | –25.372 | 38 601 | 500–2500 | |
| 1 атм | 5.24E+15 | –1.4309 | 15 522 | 6.51E+74 | –17.459 | 62 385 | 500–2500 | |
| 10 атм | 1.79E+08 | 0.70467 | 13 997 | 1.19E+72 | –16.48 | 68 323 | 500–2500 | |
| 100 атм | 3.67E–04 | 4.1014 | 10 673 | 1.32E+62 | –13.566 | 68 593 | 500–2500 | |
| C7H7 + C3H3 → → (p9 + p10) + H |
30 Торр | 1.12E+62 | –14.179 | 43 583 | 7.19E+91 | –25.372 | 38 601 | 500–2500 |
| 1 атм | 5.24E+15 | –1.4309 | 15 522 | 6.51E+74 | –17.459 | 62 385 | 500–2500 | |
| 10 атм | 1.79E+08 | 0.70467 | 13 997 | 1.19E+72 | –16.48 | 68 323 | 500–2500 | |
| 100 атм | 3.67E–04 | 4.1014 | 10 673 | 1.32E+62 | –13.566 | 68 593 | 500–2500 | |
| (p9 + p10) + + H → C7H7 + + C3H3 | 30 Торр | 9.57E+72 | –16.131 | 63 017 | 4.49E+99 | –26.38 | 56 615 | 500–2500 |
| 1 атм | 3.70E+25 | –3.0819 | 34 269 | 4.37E+85 | –19.382 | 81 767 | 500–2500 | |
| 10 атм | 2.04E+18 | –1.0132 | 32 802 | 2.24E+82 | –18.25 | 87 277 | 500–2500 | |
| 100 атм | 3.67E+06 | 2.4061 | 29 493 | 4.32E+72 | –15.402 | 87 779 | 500–2500 | |
| w2 → (p9 + + p10) + H | 30 Торр | 1.08E+96 | –24.173 | 1.07E+05 | 4.70E+34 | –7.092 | 62 181 | 500–1650 |
| 1 атм | 1.01E–01 | 4.0166 | 49 114 | –6.53E+21 | –2.0438 | 74 872 | 500–1800 | |
| 10 атм | 6.15E+68 | –15.828 | 98 771 | 1.07E+22 | –3.0821 | 58 080 | 500–2000 | |
| 100 атм | 3.59E+79 | –18.512 | 1.15E+05 | 2.36E+21 | –2.728 | 59 312 | 500–2250 | |
| (p9 + p10) + + H → w2 | 30 Торр | 7.42E+101 | –25.663 | 57 623 | 9.05E+39 | –8.4865 | 11 661 | 500–1650 |
| 1 атм | 293.33 | 3.1861 | –2094.1 | –2.23E+24 | –2.5979 | 23 429 | 500–1800 | |
| 10 атм | 3.71E+76 | –17.826 | 51 535 | 7.31E+24 | –3.7105 | 6682.9 | 500–2000 | |
| 100 атм | 1.96E+82 | –19.103 | 64 302 | 5.72E+21 | –2.619 | 6824.1 | 500–2250 | |
| C7H7 + C3H3 → → p3 + H | 30 Торр | 4.29E+39 | –6.9536 | 44 315 | 3.91E+22 | –8.249 | –2982.8 | 500–2500 |
| 1 атм | –9.38E+62 | –14.021 | 56 419 | 1.57E+38 | –6.4631 | 45 769 | 500–2500 | |
| 10 атм | 1.37E+10 | 1.2209 | 32 036 | 6.47E+64 | –13.527 | 76 788 | 500–2500 | |
| 100 атм | 8.0561 | 3.9003 | 29 818 | 1.42E+63 | –12.84 | 84 002 | 500–2500 | |
| p3 + H → → C7H7 + C3H3 |
30 Торр | 1.02E+46 | –8.8226 | 27 411 | 1.99E+29 | –9.855 | –17 535 | 500–2500 |
| 1 атм | –3.14E+65 | –14.719 | 37 615 | 7.00E+44 | –8.4131 | 28 799 | 500–2500 | |
| 10 атм | 1.85E+16 | –0.58032 | 14 970 | 1.19E+71 | –15.366 | 59 776 | 500–2500 | |
| 100 атм | 1.23E+07 | 2.0833 | 12 777 | 1.65E+69 | –14.625 | 66 806 | 500–2500 | |
| C7H7 + C3H3 → → p4 + i-C4H5 |
30 Торр | 1.13E+42 | –7.8662 | 44 222 | 500–2500 | |||
| 1 атм | –7.66E+67 | –15.615 | 58 386 | 2.05E+43 | –8.1257 | 47 768 | 500–2500 | |
| 10 атм | 2.11E+16 | –0.76155 | 34 494 | 3.42E+70 | –15.353 | 79 532 | 500–2500 | |
| 100 атм | 1.16E+06 | 2.2237 | 31 805 | 7.31E+67 | –14.395 | 85 953 | 500–2500 | |
| p4 + i-C4H5 → → C7H7 + C3H3 |
30 Торр | 1.56E+41 | –7.6696 | 23 178 | 500–2500 | |||
| 1 атм | –1.77E+64 | –14.549 | 36 106 | 5.19E+43 | –8.2906 | 27 236 | 500–2500 | |
| 10 атм | 1.90E+15 | –0.51828 | 13 302 | 1.06E+70 | –15.254 | 58 733 | 500–2500 | |
| 100 атм | 1.11E+05 | 2.46 | 10 627 | 1.37E+67 | –14.237 | 64 941 | 500–2500 | |
| p3 + H → → p4 + i-C4H5 |
30 Торр | 5.40E+46 | –9.1633 | 36 993 | 3.48E+15 | –0.35084 | 18 675 | 500–2500 |
| 1 атм | 7.16E+49 | –9.7203 | 45 944 | 4.37E+67 | –16.955 | 38 315 | 500–2500 | |
| 10 атм | 7.62E+12 | 0.36256 | 22 293 | 6.10E+63 | –13.402 | 62 795 | 500–2500 | |
| 100 атм | 1.18E+06 | 2.3236 | 21 196 | 5.05E+64 | –13.424 | 71 627 | 500–2500 | |
| p4 + i-C4H5 → → p3 + H |
30 Торр | 1.62E+37 | –6.4235 | 31 525 | 39 131 | 2.7294 | 12 375 | 500–2500 |
| 1 атм | 1.29E+43 | –7.7902 | 42 184 | 1.09E+56 | –13.506 | 32 720 | 500–2500 | |
| 10 атм | 6.73E+05 | 2.3692 | 18 206 | 7.58E+56 | –11.427 | 58 916 | 500–2500 | |
| 100 атм | 7.29E–02 | 4.3798 | 17 056 | 9.29E+57 | –11.496 | 67 869 | 500–2500 | |
| p3 + H → (p9 + + p10) + H | 30 Торр | 1.51E+43 | –8.7983 | 22 729 | 500–2500 | |||
| 1 атм | –8.93E+64 | –15.313 | 34 996 | 2.28E+44 | –9.0396 | 26 066 | 500–2500 | |
| 10 атм | 1.07E+15 | –0.98967 | 11 791 | 9.44E+70 | –16.088 | 58 027 | 500–2500 | |
| 100 атм | 2.36E+05 | 1.8134 | 9411.6 | 3.95E+68 | –15.207 | 64 895 | 500–2500 | |
| p4 + i-C4H5 → → (p9 + p10) + H |
30 Торр | 2.82E+42 | –8.7796 | 21 469 | 500–2500 | |||
| 1 атм | –2.87E+63 | –15.055 | 33 471 | 3.33E+43 | –8.9921 | 24 737 | 500–2500 | |
| 10 атм | 2.84E+14 | –1.0312 | 10 475 | 1.00E+70 | –15.991 | 57 083 | 500–2500 | |
| 100 атм | 5520.2 | 2.0835 | 7593.3 | 4.62E+66 | –14.857 | 63 140 | 500–2500 |
Сравнение констант скоростей различных реакций, рассчитанных в данной работе, в работе Мебеля и др. [68] и Мацуги и Миёси [45] показало, что для (R3) наша константа скорости ниже значений Мацуги и Миёси при низких температурах от 8 раз при 500 К, 2 раз при 1000 К, до 40–50% около 1400–1500 К. Однако константы скорости практически совпадают при температурах выше 1600 К, т.е. в температурном режиме, наиболее подходящем этому механизму. Константы скорости для (R1) и (R2) как в прямом, так и в обратном направлениях в целом оказались весьма схожи. Например, при 1000 К константы скоростей в прямом направлении различаются на 40–45%, а в обратном на 22–34%. Однако есть два основных отличия: во-первых, коэффициент ветвления w1/w2, контролируемый константами скорости входного канала, заметно выше в настоящих расчетах, и, во-вторых, константы скорости прямой реакции, рассчитанные Мацуги и Миёси, быстро падают до очень малых значений при температурax выше 1500 К. Наши результаты показывают, что прямые константы скоростей для образования w1 и w2 относительно мало уменьшаются с температурой и остаются высокими (выше 10–12 см3 с–1) до 1800 К. Выше этой температуры наши расчеты показывают, что w1 и w2 становятся нестабильными и превращаются в продукты их разложения, в основном C7H7 + C3H3. При сравнении констант скоростей мономолекулярного распада метиленинданил радикалов p9 и p10 на метиленинден (p11) и нафталин (p14), полученными Мебелем и др. в 2016 г. [68] и Мацуги и Миёси [45], обнаружились существенные различия. Наши константы скорости образования р11 в целом более чем на порядок превышают значения Мацуги и Миёси, тогда как для образования р14 различия меньше и уменьшаются с 8.5 раза при 800 К до 1.4 раза при 2000 К. Кроме того, наши расчеты предсказывали гораздо более высокий выход метилениндена по сравнению с нафталином, тогда как Мацуги и Миёси предсказывали обратное. Различия в константах скорости бимолекулярной реакции Н-стимулированной изомеризации метилениндена в нафталин (R12) менее выражены. Константы скорости, рассчитанные в 2016 г., выше при низких температурах от 4.7 раза при 500 К до 1.8 при 1000 К. При более высоких температурах значения Мацуги и Миёси немного выше, но различия находятся в пределах 30%. Подводя итог, можно сказать, что, хотя настоящие расчеты кинетики реакции C7H7 + C3H3 качественно подтверждают механизм, предложенный Мацуги и Миёси, количественные различия в рассчитанных константах скорости требуют их включения в обновленный подробный кинетический механизм образования двуциклических продуктов, включая нафталин.
Наконец, мы обращаемся к возможной роли триплетного электронного состояния в реакции бензил + пропаргил. На рис. 8 сравниваются константы скорости образования возможных предшественников нафталина p9 и p10 в реакции через синглетную C10H10 ППЭ и p15 через триплетную поверхность. Очевидно, что преобладает образование р9 и что роль триплетного пути является незначительной, поскольку рассчитанная константа скорости С7Н7 + С3Н3 → р15 + Н на 3‒4 порядка ниже, чем для образования р9 и примерно на 2 порядка ниже константы скорости образования p10. Несмотря на то что реакция в триплетном состоянии непосредственно приводит к образованию нафталинового каркаса, реакция тормозится относительно высоким барьером и наличием интермедиатов, разложение которых обратно на реагенты конкурирует с образованием продуктов.

3.3 Кинетика реакции C7H7 + C3H3 в условиях оболочек звезд АВГ
Рассмотрим зависимости констант скоростей реакции бензил + пропаргил от давления и температуры присущих околозвездным оболочкам звезд АВГ. На рис. 9 приводятся температурные зависимости суммарной константы скорости реакций ведущих к двуциклическим продуктам p9 + p10 при давлениях 1.04 × 10–6 Торр, 1.04 × × 10‒4 Торр, 1.04 × 10–1 Торр, а также более высоких давлений 30 Торр и 1 атм. Показано, что при высоких температурах, стремящихся к 2500 К, константы скоростей образования данных метиленинданил радикалов для присущих давлений в оболочках звезд АВГ [25, 27] находятся на уровне около 2.31 × 10–14 см3 с–1. Однако при уменьшении температуры и давления скорость образования (p9 + p10) увеличивается на порядки, что указывает на очевидное ослабление стабилизации начальных комплексов w1 и w2 и рост выхода двуциклических продуктов реакции. Таким образом, в оболочке звезд АВГ выход двуциклических соединений в реакции C7H7 + C3H3 растет с удалением от центра звезды, где температура и давление монотонно падают.
Рис. 9.
Константы скорости реакции C7H7 + C3H3 → (p9 + p10) + H, рассчитанные при различных давлениях. Обозначения для представленных давлений: ($\blacksquare $) – 1.04×10–6 Торр, ($ \bullet $) – 1.04×10–4 Торр, ($\blacktriangle $) – 1.04×10–1 Торр, ($\blacktriangledown $) – 30 Торр, ($\diamondsuit $) – 1 атм.
Важную роль в процессах роста ПАУ могут играть метилзамещенные и, в более общем случае, алкилированные ПАУ, молекулой-прототипом которых является толуол, как на некоторых космических объектах [9, 34, 37, 73, 74], так и в горении [75, 76]. Отмечается [74], что СН радикал в оболочках звезд АВГ формируется в рекомбинационном процессе
где М – третья частица, или в элементарном процессе имеющем два канала продуктов реакции. В дальнейших столкновениях атомарного водорода с СН или СН2 производится метил радикал СН3. Существует ряд других реакционных путей, в продуктах которых наличествует СН3. Метан, присутствующий в оболочках звезд АВГ, реагируя с такими активными компонентами как Cl, O, H, OH и т.д., также вносит вклад в баланс СН3.Бензил радикал C7H7 в оболочках звезд АВГ образуется в реакциях толуола, например,
(R13)
${{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{{\text{8}}}} + {\text{Н}} \leftrightarrow {{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{7}} + {{{\text{Н}}}_{{\text{2}}}}.$В свою очередь C7H8 формируется из бензола в реакциях
(R14)
${{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{6}}}} + {\text{C}}{{{\text{H}}}_{{\text{2}}}} \to {{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{{\text{8}}}},$(R15)
${{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{6}}}} + {\text{C}}{{{\text{H}}}_{{\text{3}}}} \to {{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{{\text{8}}}} + {\text{H}}$(R16)
${{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{5}}}} + {\text{C}}{{{\text{H}}}_{{\text{3}}}} \to {{{\text{C}}}_{{\text{7}}}}{{{\text{H}}}_{8}}.$В недавнем обзоре [75] приведены разнообразные реакционные пути, ведущие к C7H7 и C7H8 в пламенах. Пути образования бензола и фенил радикала в оболочках звезд АВГ подробно описаны, например, в работе Шершнеф и др. [27]. Второй компонент изучаемой реакции пропаргил радикал C3H3 присутствует в оболочках звезд АВГ в концентрациях (см., например, рис. 6–8 из работы Шершнеф [77]) на 4–5 порядков выше, чем для фенил радикала, в скелет которого достраивается второе ароматическое кольцо в механизме НАСА.
Хотя пока не имеется данных о содержании толуола в оболочках звезд АВГ, во многих пламенах отношение концентраций толуол/бензол варьируется в пределах 0.1–0.5 (см. табл. 2 из работы Руве и др. [76]). Исходя из схожести углеводородных процессов в горении и оболочках богатых углеродом звезд АВГ можно предположить, что в некоторых из этих звездных объектов содержание толуола будет сравнимо с бензолом и, исходя из этого, реакцию C7H7 + C3H3 целесообразно включать в кинетическую модель, описывающую рост молекулярной массы ПАУ в оболочках звезд АВГ. Определение вклада данной реакции в кинетику ПАУ не является целью данной работы. Отметим только его очевидное преимущество перед механизмом НАСА, где второе кольцо добавляется к бензолу в последовательности: отрыв водорода – присоединение ацетилена – отрыв водорода – присоединение ацетилена. Тогда как из толуола последовательность существенно короче: отрыв водорода – присоединение пропаргила. Это обстоятельство может иметь решающее значение в конкуренции двух реакционных путей, ведущих к двухкольцевым ПАУ.
4. ЗАКЛЮЧЕНИЕ
В текущем исследовании были пересмотрены механизм и кинетика реакции C7H7 + C3H3 с использованием развитых методов теории переходного состояния для оценки критических констант скорости безбарьерных входных и выходных каналов реакции. Расчеты подтверждают механизм реакции, предложенный в более ранней работе Мацуги и Миёси [45], но предлагают уточненные количественные данные. Расчеты кинетических констант показали три различных температурных режима, границы которых зависят от давления. При более низких температурах в реакции преобладает столкновительная стабилизация двух начальных промежуточных соединений, 3-бутинилбензола w1 и 2,3-бутадиенилбензола w2, где образование w1 является предпочтительным из-за более высокой константы скорости соответствующего входного канала. В промежуточном температурном интервале w1 и w2 эффективно разлагаются обратно до реагентов, что приводит к резкому падению зависимой от давления полной константы скорости реакции. В высокотемпературном режиме w1 и w2 перестают быть стабильными, и реакция протекает в прямом направлении, без стабилизации интермедиатов С10Н10 и с образованием бимолекулярных продуктов. К таковым относятся “быстрые” продукты, образующиеся при непосредственном разрыве связи C–H в w1/w2 с образованием изомеров C10H9 p1–p3, состоящих из бензольного кольца с боковой цепью, вместе с атомом водорода, или при разрыве связи C–C с образованием фенил радикала C6H5 (p4) + i-C4H5. Более продолжительные пути ведут к предшественникам нафталина, метиленинданил радикалам p9 и p10 + H, и включают замыкание боковой цепи C4 на пятичленное кольцо перед потерей атома H. Наиболее вероятные бимолекулярные продукты включают p3 + H и p4 + i-C4H5, за которыми следуют p9 + H, p2 + H, p1 + H и p10 + H. Последующие изомеризация и разложение p9 и p10 ведут к образованию бензофульвена и нафталина, где первый также может перегруппировываться во второй посредством изомеризации с помощью присоединения атома водорода. Важно отметить, что в условиях оболочек звезд АВГ, где давление газа намного меньше 1 Торр, процесс стабилизации начальных комплексов w1 и w2 сильно замедлен, что способствует росту выхода двуциклических ПАУ в продуктах реакции.
По сравнению с предыдущими результатами Мацуги и Миёси [45], настоящие расчеты показывают более сложную температурную зависимость общей константы скорости в пределе высокого давления, а также более высокие относительные выходы p3 и p4 по сравнению с p9. В связи с этим мы предлагаем обновить кинетическую схему образования нафталина, инициированного реакцией бензил + пропаргил, за счет включения каналов С7Н7 + С3Н3 → р3 + Н/р4 + + i‑С4Н5 и вторичных каналов р3 + Н/р4 + + i‑C4H5, которые могут производить p9 + H. Количественные изменения констант скорости, основанные на настоящих расчетах и представленные в табл. 1, предлагаются для уточненных кинетических моделей образования нафталина и роста ПАУ в околозвездных оболочках звезд АВГ и в пламенах горения.
Список литературы
M. G. Rawlings, A. J. Adamson, C. C. M. Marshall, and P. J. Sarre, Monthly Not. Roy. Astron. Soc. 485, 3398 (2019).
R. Ruiterkamp, T. Halasinski, F. Salama, B. H. Foing, L. J. Allamandola, W. Schmidt, and P. Ehrenfreund, Astron. and Astrophys. 390, 1153 (2002).
M. Tsuge, C.-Y. Tseng, and Y.-P. Lee, Physical Chemistry Chemical Physics 20, 5344 (2018).
N. L. J. Cox, J. Cami, A. Farhang, J. Smoker, A. Monreal-Ibero, R. Lallement, P. J. Sarre, C. C. M. Marshall, K. T. Smith, and C. J. Evans, Astron. and Astrophys. 606, A76 (2017).
R. Ruiterkamp, N. L. J. Cox, M. Spaans, L. Kaper, B. H. Foing, F. Salama, and P. Ehrenfreund, Astron. and Astrophys. 432, 515 (2005).
C. Boersma, J. Bregman, and L. J. Allamandola, Astrophys. J. 858, 67 (2018).
A. G. Tielens, Rev. of Mod. Phys., 85, 1021 (2013).
M. Kalpana, E. Babu, D. Mani, R. Tripathi, and N. Bhandari, Plan. and Space Sci. 198, 105177 (2021).
R. I. Kaiser and N. Hansen, The Journal of Physical Chemistry A 125, 3826 (2021).
A. Leger and J. L. Puget, Astron. and Astrophys. 137, L5 (1984).
R. I. Kaiser, D. S. N. Parker, and A. M. Mebel, Annual Reviews of Physical Chemistry 66, 43 (2015).
W. W. Duley, Faraday Discussions 133, 415 (2006).
N. L. J. Cox, J. Cami, L. Kaper, P. Ehrenfreund, B. H. Foing, B. B. Ochsendorf, S. H. M. van Hooff, and F. Salama, Astron. and Astrophys. 569, A117 (2014).
M. Tsuge, M. Bahou, Y.-J. Wu, L. Allamandola, and Y.‑P. Lee, Astrophys. J. 825, 96 (2016).
H.-S. Kim, D. R. Wagner, and R. J. Saykally, Phys. Rev. Lett. 86, 5691 (2001).
R. Zenobi, J.-M. Philippoz, R. N. Zare, and P. R. Buseck, Science 246, 1026 (1989).
Y. Wang, Y. Huang, C. M. O. D. Alexander, M. Fogel, and G. Cody, Geochimica et Cosmochimica Acta 69, 3711 (2005).
A. G. Tielens, Ann. Rev. Astron. and Astrophys. 46, 289 (2008).
A. Bergantini and R. I. Kaiser, Chem 1, 822 (2016).
S. A. Sanford, J. Aléon, C. M. Alexander, T. Araki, S. Bajt, G. A. Baratta, J. Borg, J. P. Bradley, D. E. Brownlee, J. R. Brucato, and M. J. Burchell, Science 314, 1720 (2006).
D. S. McKay, E. K. Gibson Jr., K. L. Thomas-Keprta, H. Vali, C. S. Romanek, S. J. Clemett, X. D. Chillier, C. R. Maechling, and R. N. Zare, Science 273, 924 (1996).
B. A. McGuire, A. M. Burkhardt, S. Kalenskii, C. N. Shin-gledecker, A. J. Remijan, E. Herbst, and M. C. McCarthy, Science 359, 202 (2018).
D. S. N. Parker, F. Zhang, Y. S. Kim, R. I. Kaiser, A. Lan-dera, V. V. Kislov, A. M. Mebel, and A. G. Tielens, Proceedings of the National Academy of Science 109, 53 (2012).
A. M. Mebel, A. Landera, R. I. Kaiser, The Journal of Physical Chemistry A 121, 901 (2017).
M. Frenklach and E. D. Feigelson, Astrophys. J. 341, 372 (1989).
M. Frenklach, Physical Chemistry Chemical Physics 4, 2028 (2002).
I. Cherchneff, J. R. Barker, and A. G. Tielens, Astrophys. J. 401, 269 (1992).
I. Cherchneff, EAS Publications Series 46, 177 (2011).
M. Frenklach, D. W. Clary, W. C. Gardiner Jr., and S. E. Stein, Symposium (International) on Combustion 20, 887 (1985).
M. Frenklach, Proceedings of the Combustion Institute 22, 1075 (1988).
V. V. Kislov, N. I. Islamova, A. M. Kolker, S. H. Lin, and A. M. Mebel, Journal of Chemical Theory and Computation 1, 908 (2005).
A. M. Mebel, Y. Georgievskii, A. W. Jasper, and S. J. Klip-penstein, Proceedings of the Combustion Institute 36, 919 (2017).
M. Frenklach, R. I. Singh, and A. M. Mebel, Proceedings of the Combustion Institute 37, 969 (2019).
L. Zhao, R. I. Kaiser, W. Lu, B. Xu, M. Ahmed, A. N. Mo-rozov, A. M. Mebel, A. H. Howlader, and S. F. Wnuk, Nature Communications 10, 3689 (2019).
B. Shukla, A. Susa, A. Miyoshi, and M. Koshi, The Journal of Physical Chemistry A 112, 2362 (2008).
V. S. Krasnoukhov, D. P. Porfiriev, I. P. Zavershinskiy, V. N. Azyazov, and A. M. Mebel, The Journal of Physical Chemistry A 121, 9191 (2017).
S. Doddipatla, G. R. Galimova, H. Wei, A. M. Thomas, C. He, Z. Yang, A. N. Morozov, C. N. Shingledecker, A. M. Mebel, and R. I. Kaiser, Science Advances 7, eabd4044 (2021).
P. M. Holt and J. A. Kerr, International Journal of Chemical Kinetics 9, 185 (1977).
D. Robaugh and W. Tsang, The Journal of Physical Chemistry 90, 4159 (1986).
I. V. Tokmakov and M. C. Lin, International Journal of Chemical Kinetics 33, 633 (2001).
J. Park and M. C. Lin, International Journal of Chemical Kinetics 33, 803 (2001).
V. V. Kislov and A. M. Mebel, The Journal of Physical Chemistry A 111, 3922 (2007).
M. B. Colket and D. J. Seery, Proceedings of the Combustion Institute 25, 883 (1994).
N. M. Marinov, W. J. Pitz, C. K. Westbrook, A. E. Lutz, A. M. Vincitore, and S. M. Senkan, Proceedings of the Combustion Institute 27, 605 (1998).
A. Matsugi and A. Miyoshi, International Journal of Chemical Kinetics 44, 206 (2011).
C. T. Lee, W. T. Yang, and R. G. Parr, Phys. Rev. B 37, 785 (1988).
A. D. Becke, The Journal of Chemical Physics 98, 5648 (1993).
R. Krishnan, J. S. Binkley, R. Seeger, and J. A. Pople, The Journal of Chemical Physics 72, 650 (1980).
L. A. Curtiss, K. Raghavachari, P. C. Redfern, V. Rassolov, and J. A. Pople, The Journal of Chemical Physics 109, 7764 (1998).
L. A. Curtiss, K. Raghavachari, P. C. Redfern, A. G. Ba-boul, and J. A. Pople, Chemical Physics Letters 314, 101 (1994).
A. G. Baboul, L. A. Curtiss, P. C. Redfern, and K. Raghavachari, The Journal of Chemical Physics 110, 7650 (1999).
J. A. Miller, S. J. Klippenstein, Y. Georgievskii, L. B. Har-ding, W. D. Allen, and A. C. Simmonett, The Journal of Physical Chemistry A 114, 4881 (2010).
P. Celani and H.-J. Werner, The Journal of Chemical Physics 112, 5546 (2000).
T. Shiozaki, G. Werner, P. Celani, and H.-J. Werner, The Journal of Chemical Physics 135, 081106 (2011).
T. H. Dunning, The Journal of Chemical Physics 90, 1007 (1989).
M. J. Frisch et al., Gaussian 09 (Gaussian, Inc., Wallingford CT, 2009).
H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, and M. Schutz, Wiley Interdisciplinary Reviews: Computational Molecular Science 2, 242 (2012).
R. A. Marcus, The Journal of Chemical Physics 20, 359 (1952).
W. H. Miller, Journal of the American Chemical Society 101, 6810 (1979).
S. J. Klippenstein, The Journal of Chemical Physics 96, 367 (1992).
Y. Georgievskii, J. A. Miller, and S. J. Klippenstein, Physical Chemistry Chemical Physics 9, 4259 (2007).
Y. Georgievskii and S. J. Klippenstein, The Journal of Chemical Physics 118, 5442 (2003).
S. J. Klippenstein and J. I. Cline, The Journal of Chemical Physics 103, 5451 (1995).
L. B. Harding, S. J. Klippenstein, and Y. Georgievskii, The Journal of Physical Chemistry A 111, 3789 (2007).
A. N. Morozov and A. M. Mebel, The Journal of Physical Chemistry A 123, 1720 (2019).
Y. Georgievskii, J. A. Miller, M. P. Burke, and S. J. Klippenstein, The Journal of Physical Chemistry A 117, 12146 (2013).
Y. Georgievskii and S. J. Klippenstein, https://tcg.cse.anl.gov/papr (2015).
A. M. Mebel, Y. Georgievskii, A. W. Jasper, and S. J. Klip-penstein, Faraday Discussions 195, 637 (2016).
J. Troe, The Journal of Chemical Physics 66, 4745 (1977).
P. E. M. Siegbahn, J. Almlof, A. Heiberg, and B. O. Roos, The Journal of Chemical Physics 74, 2384 (1981).
T. H. Dunning Jr., The Journal of Chemical Physics 53, 2823 (1970).
T. H. Dunning and P. J. Hay, Methods of Electronic Structure Theory (Springer, Boston, MA, 3, 1, 1977).
R. I. Kaiser, L. Zhao, W. Lu, M. Ahmed, V. S. Krasnoukhov, V. N. Azyazov, and A. M. Mebel, Nature Communications 13, 786 (2022).
G. Santoro, L. Martínez, K. Lauwaet, M. Accolla, G. Tajuelo-Castilla, P. Merino, J. M. Sobrado, R. J. Peláez, V. J. Herrero, I. Tanarro, and Á. Mayoral, Astrophys. J. 895, 97 (2020).
C. W. Zhou, A. Farooq, L. Yang, and A. M. Mebel, Progress in Energy and Combustion Science 90, 100983 (2022).
L. Ruwe, K. Moshammer, N. Hansen, and K. Kohse-Höinghaus, Combustion and Flame 175, 34 (2017).
I. Cherchneff, Astron. and Astrophys. 545, A12 (2012).
Дополнительные материалы отсутствуют.
Инструменты
Астрономический журнал