

Биоорганическая химия, 2019, T. 45, № 2, стр. 145-154
Оптимизация синтеза [MeArg1, Nle10]апелина-12 и изучение его стабильности в плазме крови человека методом протонного магнитного резонанса
М. В. Сидорова , М. Е. Палькеева , А. А. Азьмуко , М. В. Овчинников , А. С. Молокоедов , В. Н. Бушуев , О. И. Писаренко
ФГБУ “Национальный медицинский исследовательский центр кардиологии” Минздрава РФ
121552 Москва, 3-я Черепковская ул., 15а, Россия
Поступила в редакцию 19.03.2018
После доработки 09.06.2018
Принята к публикации 10.04.2018
Аннотация
Разработан способ твердофазного синтеза MeArg1, Nle10-аналога апелина-12 (метилина) с использованием Fmoc-методологии в сочетании с временной защитой гуанидиновой функции остатков аргинина протонированием (солеобразованием) на стадии образования амидной связи. С помощью спектроскопии протонного магнитного резонанса проведена сравнительная оценка протеолитической стабильности апелина-12 и его структурного аналога в плазме крови человека; показано, что время полураспада аналога в плазме примерно в три раза больше, чем у природного пептида.
ВВЕДЕНИЕ
Разработка подходов к снижению повреждения сердца, вызванного ишемическим и реперфузионным стрессом, является актуальной задачей экспериментальной кардиологии. В последние годы в различных лабораториях осуществляется направленный поиск новых соединений, способных воздействовать на адаптационные механизмы в миокардиальных клетках в условиях изменившегося кислородного и энергетического обеспечения и таким образом уменьшать повреждение сердца. При ишемии и последующей реперфузии в миокарде увеличивается выработка ряда присущих организму факторов – адипокинов, цитокинов и вазоактивных пептидов, инициирующих механизмы запрограммированного клеточного выживания, которые запускаются каскадами реперфузионных киназ. К таким соединениям относится адипокин апелин, являющийся лигандом сопряженного с G-белками APJ-рецептора и представляющий собой 77-членный полипептид.
Апелин активно экспрессируется различными тканями, включая эндотелиальные и гладкомышечные клетки коронарных сосудов и кардиомиоциты. Активация системы апелин/APJ-рецептор оказывает вазодилатирующее действие на сосуды, стимулирует транспорт ионов Ca2+ в кардиомиоциты, улучшая сократимость миокарда, снижает толерантность к инсулину при ожирении, препятствует образованию атеросклеротических бляшек [1]. Показано, что С-концевые фрагменты апелина (апелин-36, апелин-17, апелин-13 и апелин-12) способны улучшать восстановление функции сердца и снижать необратимые повреждение кардиомиоцитов при реперфузии после длительной ишемии [2]. Пептид апелин-12 (H-Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Met-Pro-Phe-OH, A12), соответствующий С-концевому фрагменту апелина у животных различных видов и человека, является минимальной активной частью молекулы исходного апелина-77. Он обладает высоким сродством к APJ-рецептору и оказывает мощное защитное действие при экспериментальной ишемии и реперфузии сердца у крыс [3, 4].
Эти данные указывают на возможность использования пептидов в качестве потенциальных кардиопротекторных лекарственных средств, однако чувствительность пептидов к протеолитической деградации в организме человека является серьезным препятствием к их практическому применению. Во многих лабораториях ведется поиск модифицированных апелиновых пептидов, обладающих селективным действием и метаболической устойчивостью [5, 6].
Нами ранее были синтезированы оригинальные пептиды на основе природного апелина-12 [7, 8]. С целью увеличения протеолитической стабильности в структуры этих соединений были внесены следующие модификации: N-концевой остаток аргинина был защищен от действия аминопептидаз Nα-метильной группой; подверженный окислению остаток метионина был заменен на норлейцин; С-концевой остаток фенилаланина защищали от действия карбоксипептидаз амидированием. Полученные аналоги обладали кардиопротекторной активностью при моделировании ИБС и хронической сердечной недостаточности на лабораторных животных. Было выяснено, что наибольшей защитной эффективностью обладает пептид химической структуры H-MeArg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Nle-Pro-Phe-OH, названный метилином. Преимуществом метилина по сравнению с природными кардиопротекторами является комбинированный характер его действия, который включает метаболическую коррекцию, антиоксидантную защиту и стабилизацию мембран кардиомиоцитов, поддержку внутриклеточного ионного гомеостаза и вазодилатационные свойства [9, 10]. Антиишемические и инотропные (увеличение силы сердечных сокращений) свойства этого пептида могут найти клиническое применение в кардиологической практике.
Данная работа является продолжением наших исследований в области дизайна, синтеза и изучения пептидных кардиопротекторов и посвящена разработке оптимизированной методики синтеза и оценке протеолитической стабильности наиболее перспективного в плане практического применения MeArg1, Nle10-аналога апелина-12 – метилина.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Оптимизация схемы синтеза метилина. При твердофазном синтезе (ТФС) пептидов, содержащих в последовательности несколько остатков аргинина, часто возникают проблемы, связанные с защитами гуанидиновой функции этой аминокислоты. Как правило, в ТФС в сочетании с Nα-Fmoc-защитой для блокирования гуанидиновой функции аргинина применяют группы тозильного типа (Mtr, Pmc или Pbf). При отщеплении Mtr- или Pmc-защит требуются достаточно жесткие условия и длительные обработки, часто сопровождающиеся образованием побочных продуктов например, реакция сульфатирования гидроксильных групп остатков серина и/или треонина (с выходом побочного продукта до 50%) [11]. Эта побочная реакция характерна и при синтезе апелиновых пептидов.
Ранее мы показали [8], что при получении апелина-12 содержание побочного О-сульфата в “сыром” продукте ТФС может колебаться от 6 до 48% в зависимости от состава смеси, использованной для заключительного деблокирования и отщепления пептида от полимерного носителя. При синтезе метилина мы также столкнулись с образованием соответствующего побочного продукта – О-сульфата метилина. Было установлено, что степень сульфатирования остатка Ser снижается в присутствии воды как скэвенджера в составе деблокирующей смеси, однако полностью подавить эту реакцию не удается. При ТФС апелиновых пептидов в граммовых количествах данная проблема встает более остро, поскольку при укрупнении масштаба синтеза, как правило, повышается интенсивность побочных процессов и, как следствие этого, усложняется очистка пептидов.
С целью исключения возможности образования трудноотделимых побочных продуктов, описанных выше, мы предприняли попытку ТФС метилина с использованием введения остатка аргинина с незащищенной гуанидиновой функцией, хотя примеры реализации такого подхода в твердофазном синтезе пептидов немногочисленны. Принципиальная возможность использования в синтезе производных NG-незамещенного аргинина обусловлена тем, что основность гуанидиновой функции (рКа ≈ 12.5) намного выше основности α-аминогруппы эфиров аминокислот; эта разница в основности позволяет в ходе пептидного синтеза селективно блокировать гуанидиновую группу протонированием, что и обеспечивает ее временную защиту на стадии создания амидной связи [12]. Сообщалось об успешном ТФС аналогов рилизинг-фактора лютеинизирующего гормона (LH-RH) с применением тактики минимальной защиты, когда аргинин защищали солеобразованием [13].
В нашей лаборатории с применением этой тактики были осуществлены успешные синтезы брадикинина и октапептидного фрагмента 584–591 гликопротеина gp41 ВИЧ1 ступенчатым наращиванием пептидной цепи [14], β-амилоида-(1–42) [15] фрагментной конденсацией на твердой фазе с использованием аргинина без защиты гуанидиновой функции. Для протонирования гуанидиновой группы аргинина использовали хлоргидрат [13] или бромгидрат пиридина [14, 15]; для присоединения соответствующих производных Boc- или Fmoc-Arg(Н+)-ОН применяли карбодиимидный метод [13–15] или регент Кастро – BOP [14]. В работе [14] было показано, что конденсация Nα-Fmoc-аргинина с пептидилполимерами в присутствии N,N-диизопропилкарбодиимида или BOP не сопровождается рацемизацией.
Синтез метилина проводили с С-конца с применением Fmoc-методологии. Для блокирования функциональных групп боковых цепей аминокислот использовали следующие защиты: But – для гидроксильной функции серина, Trt – для имидазольной функции гистидина и Boc – для ε‑аминогруппы лизина, гуанидиновые группы аргининов не защищали. Для исключения из синтетического цикла проблемной стадии пролинсодержащего дипептидилполимера (H-Pro-Phe-P), на первом этапе ТФС фенилаланил-полимер конденсировали с дипептидным блоком Fmoc-Nle-Pro-OH, далее пептидную цепь наращивали по одной аминокислоте. Для присоединения N-концевого остатка аргинина использовали его NαBoc,Nα-метильное производное (Boc-MeArg), остаток аргинина в третье положение пептида вводили в виде Fmoc-производного.

Схема 1 . Получение Boc-MeArg-OH.
Синтез Boc-MeArg-OH осуществляли, опираясь на описанную методику [16], по схеме 1 , исходя из Вос-L-глутамина (I). На первой стадии под действием уксусного ангидрида в пиридине происходит дегидратация γ-карбоксамидной функции Вос-глутамина с образованием соответствующего нитрила (II). Затем проводили селективное метилирование α-аминогруппы соединения (II) с использованием иодистого метила и гидрида натрия в тетрагидрофуране с образованием кристаллической N-Вос-2-метиламино-4-циано-L-масляной кислоты (III). Гидрогенолизом продукта (III) на платиновом катализаторе в изопропиловом спирте получали NαBoc,NαMe-орнитин (IV), в который вводили гуанидиновую группу действием S-метилизотиомочевины в присутствии триэтиламина с образованием целевого NαBoc,NαMe-аргинина (V). Соединение (V) очищали на катионообменнике в пиридин-ацетатном буфере до 95%-й чистоты. Полученный продукт был идентичен коммерческому образцу. Структуру полупродуктов и целевого соединения (V) подтверждали данными спектроскопии 1Н-ЯМР, а их чистоту анализировали с помощью ВЭЖХ на обращенной фазе.
Для разработки оптимальной схемы синтеза были проведены тестовые ТФС метилина в автоматическом и ручном режиме, в которых меняли масштаб синтеза (от 0.5 до 1.5 ммоль), реагенты для протонирования гуанидиновой функции аргинина и способ создания амидной связи. Для протонирования гуанидиновой группы применяли либо бромгидрат пиридина [14, 15], либо 1-гидроксибензотриазол (HOBT). При этом оказалось, что HOBT эффективно блокирует боковую функцию аргинина, а получаемые при этом соли Nα-защищенного аргинина существенно лучше, чем соответствующие хлор- или бромгидраты, растворяются в применяемых для ТФС растворителях.
Для создания амидной связи использовали карбодиимидный метод, соли фосфония/урония – реагент Кастро гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония (ВОР) [17] или тетрафторборат N,N,N',N'-тетраметил-О -(бензотриазол-1-ил)урония (TBTU) [18].
Важным нюансом конденсации производных NG-незащищенного аргинина с использованием BOP или TBTU является возможность проведения реакции без применения органического основания. Обычно при использовании ВОР и TBTU в ТФС для активации карбоксильной группы применяют 2–3 эквивалента органического основания (диизопропилэтиламина или N-метилморфолина), необходимого для связывания кислых противоионов – гексафторфосфата или тетрафторбората. Известно, что избытки оснований способны привести к рацемизации присоединяемой аминокислоты. В случае активации карбоксильной группы Fmoc-Arg-OH или Fmoc-MeArg-OH в присутствии ВОР или TBTU их гуанидиновая группа (с рКа ≈ 12.5) играет роль органического основания, являясь акцептором кислых противоионов и при этом происходит ее блокирование.
Дополнительным преимуществом этого способа конденсации является тот факт, что активированное производное аргинина получается in situ в условиях непрерывного технологического процесса, т.е. не требуется введения дополнительной стадии приготовления активированных производных аргинина.
В наших экспериментах лучшие результаты на стадии введения в пептидную цепь остатков аргинина были получены при 2-кратном проведении соответствующих конденсаций с использованием ВОР или TBTU (см. табл. 1). В работе [14] было показано, что в ходе ТФС на стадии деблокирования NG-незамещенного Arg-содержащего пептидилполимера пиперидиновым реагентом происходит депротонирование гуанидиновой функции. Для предотвращения возможного ацилирования гуанидина при следующей стадии конденсации, мы применяли 5-минутную промывку аминосвободного Arg-пептидилполимера 5% раствором HOBT в DMF или NMP непосредственно перед введением следующей аминокислоты. Оказалось, что эта дополнительная обработка исчерпывающе защищает гуанидиновую группу остатков аргинина протонированием и не препятствует ацилированию α-аминогруппы Arg-пептида, сидящего на полимере. Кроме того, в отличие от бромгидрата пиридина, 1-гидроксибензотриазол хорошо растворим в органических растворителях, и его растворы в вышеприведенных растворителях устойчивы при хранении.
Таблица 1.
Сравнительная оценка синтезов метилина Н-MeArg1-Pro-Arg3-Leu-Ser-His-Lys-Gly-Pro-Nle-Pro-Phe-ОН с постоянной и временной защитой остатков аргинина
| Способ защиты функциональных групп аргинина | Конденси-рующий реагент | Состав деблокирующей смеси и время реакции | Содержание метилина в сыром продукте ТФС, % | Способ очистки и чистота (%) по ВЭЖХ | Выход* метилина, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| МеArg1 | Arg3 | ||||||||
| Nα | NG | Nα | NG | ИОХ | ИОХ + + ВЭЖХ | ||||
| Схема с постоянной защитой гуанидиновых групп остатков Arg1 и MeArg3 | |||||||||
| Fmoc | Mtr | Fmoc | Pmc | TBTU/NMM | TFA/тиоанизол/ вода/TIBS 16 ч | 60–65 | 90 | 97 | 49–60 |
| Схема с временной защитой гуанидиновых групп остатков Arg1 и MeArg3 протонированием (солеобразованием) | |||||||||
| Boc | H+ | Fmoc | H+ | ВОР | TFA/вода/TIBS 1.5–2.0 ч | 68–75 | 98 | – | 58–65 |
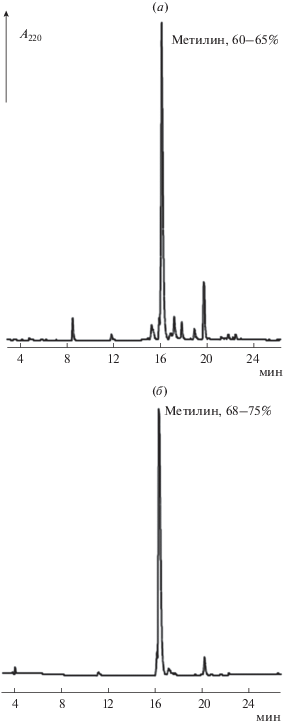
В табл. 1 дана сравнительная оценка результатов синтеза метилина по традиционной схеме ТФС с постоянными защитами гуанидиновой функции остатков аргинина (как описано в работе [8]) и с временной защитой аргининов протонированием, а на рис. 1 представлены профили аналитической ВЭЖХ соответствующих “сырых” продуктов ТФС. С точки зрения выхода целевого пептида обе схемы синтеза дают сопоставимые результаты, однако состав “сырого” продукта, полученного с использованием незащищенного аргинина, благодаря отсутствию трудноотделимых примесей позволяет получить пептид с чистотой более 98% в одну стадию с помощью ионообменной хроматографии (ср. рис. 1а и б). Чистота пептида подтверждена данными ВЭЖХ, а структура пептида – данными масс-спектрометрии и 1Н-ЯМР-спектроскопии, представленными в табл. 2.
Таблица 2.
Химические сдвиги (δ, м.д.) сигналов протонов метилина в DMSO-d6 (300 K)
| Протоны | МеArg1 | Pro2 | Arg3 | Leu4 | Ser5 | His6 | Lys7 | Gly8 | Pro9 | Nle10 | Pro11 | Phe12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NαH | 8.92, 8.78 | – | 8.31 | 7.78 | 8.1 | 8.3 | 8.14 | 8.19 | – | 7.96 | – | 7.98 |
| CαH | 4.22 | 4.46 | 4.21 | 4.39 | 4.285 | 4.59 | 4.29 | 3.98, 3.86 | 4.36 | 4.385 | 4.355 | 4.38 |
| CβH | 1.78, 1.52 | 2.14, 1.78 | 1.77, 1.52 | 1.54, 1.41 | 3.54, 3.758 | 3.10, 2.99 | 1.67, 1.52 | 2.01, 1.08 | 1.41 | 2.02, 1.80 | 3.01, 2.91 | |
| CγH | 1.87 | 1.83 | 1.83 | |||||||||
| CδH | 3.45, 3.72 | 0.85, 0.82 | 3.58, 3.46 | 3.58, 3.46 | ||||||||
| NαMe | 2.52 |
Рис. 1.
Аналитическая ВЭЖХ “сырых” продуктов ТФС метилина с постоянной (а) и временной (б) защитой остатков аргинина. Колонка Kromasil 100 C18, 5 мкм, 4.6 × 250 мм. Пептиды элюировали со скоростью 1 мл/мин градиентом буфера Б в буфере А от 10 до 70% за 30 мин, буфер А – 0.05 M KH2PO4, pH 3.0, буфер Б – 70% ацетонитрил в буфере А, детекция при 220 нм.
Таким образом, разработанная методика получения метилина с временным блокированием гуанидиновых функций остатков аргинина на стадии его включения в пептид с использованием традиционных для ТФС реагентов образования амидной связи позволяет существенно упростить пост-синтетические процедуры и последующую очистку сырого продукта.
Изучение протеолитической деградации метилина с помощью спектроскопии протонного магнитного резонанса. Следующим этапом работы было сравнительное изучение протеолитической стабильности апелина-12 и метилина в плазме крови человека. Пептиды в биологических системах подвергаются быстрому ферментативному гидролизу, что существенно ограничивает их практическое применение. Изучение протеолитической стабильности пептидов и путей их деградации дает важную информацию, необходимую для целенаправленного создания протеолитически устойчивых соединений, в частности, путем защиты участков пептидной цепи с преимущественно гидролизуемыми амидными связями. Задача количественного определения введенных в организм пептидов представляется достаточно сложной, требует разработки специальных подходов и до сих пор во многом остается нерешенной.
В настоящее время для изучения протеолитической деградации пептидов in vivo и in vitro применяют различные методы. Одним из подходов к исследованию метаболизма пептидов является использование в этом анализе аналогов изучаемых пептидов, содержащих изотопные метки. Так, биодеградацию семакса, селанка, [Leu]энкефалина и гексапептидного фрагмента фактора дифференцировки лейкоцитов (HLDF-6-амида) исследовали с помощью равномерно меченных тритием аналогов этих пептидов, а для анализа меченых соединений применяли ВЭЖХ [19, 20].
Наиболее часто для изучения биодеградации пептидов используется комбинация ВЭЖХ с масс-спектрометрией [21]. Авторы работы [22] разработали чувствительный метод определения присутствующих в плазме крови человека апелинов-12, -13, -17, -36, и [Pyr1]апелина-13, основанный на катионообменной экстракции апелиновых пептидов из плазмы крови с последующей мультиплексной жидкостной хроматографией/ тандемной масс-спектрометрией (LC/MS/MS). В этой работе для количественной оценки каждой из апелиновых форм использовались внутренние стандарты апелинов-12, -13, -17, -36 и [Pyr1]апелина-13, содержащие стабильные изотопы 13С и 15N.
Для анализа ферментативных превращений пептидов в различных биологических системах используется также ЯМР-спектроскопия [23–26]. Высокая степень индивидуальности спектров ЯМР [27] пептидов дает возможность непосредственно в ходе ферментативного гидролиза определить как качественный, так и количественный состав образующихся продуктов распада. При этом, что особенно важно, для идентификации структуры продуктов гидролиза не требуется их выделения из биологической смеси [23–26]. Метод позволяет, используя один образец пептида, непрерывно следить за кинетикой его гидролиза. Одним из ограничений метода является его относительно низкая чувствительность. Поэтому приходится работать в области концентраций пептидов, заметно превышающих фармакологические. Тем не менее, метод непрерывной регистрации гидролиза пептидов удобен при изучении взаимосвязи структуры и ферментативной стабильности в рядах пептидных аналогов. Ранее мы успешно использовали 1Н-ЯМР для сравнительной оценки протеолитической стабильности ряда пептидных ингибиторов киназы легких цепей миозина [28, 29].
Фрагменты спектров ЯМР природного апелина-12 в плазме крови человека, полученные через различные промежутки времени после его добавления в плазму крови человека, даны на рис. 2. Сравнение этих спектров показывает, что амплитуда сигналов пептида при 4.37 и 3.35 м.д., относящихся к протонам СαН- и СδН2-групп остатков Arg1 в составе пептида, со временем уменьшается; при этом в спектрах появляются новые, более узкие, сигналы при 3.88 и 3.35 м.д., амплитуда которых со временем увеличивается. Эти сигналы отнесены, соответственно, к протонам СαН- и СδН2-групп свободного аргинина, что было подтверждено неизменностью химических сдвигов этих сигналов при добавлении аминокислоты – аргинина в раствор. Сигналов других свободных аминокислот, в частности, C-концевого Phe12 на начальном этапе деградации обнаружено не было.
Рис. 2.
Фрагменты спектров 1Н-ЯМР апелина-12 в плазме крови человека (после вычитания спектра плазмы), полученные через 20 (а), 100 (б), 210 (в), 400 (г), 600 (д) и 800 мин (е) после добавления пептида в плазму. Сигналы протонов остатков аминокислот – продуктов протеолитической деградации помечены символом *.

Следовательно, первоначальной стадией деградации апелина-12 является гидролиз связи Arg1–Pro2N-концевой части пептида c образованием свободного аргинина. Время полудеградации N-концевой части апелина-12 в плазме крови оказалось равным приблизительно 2 ч 45 мин. Значительное перекрывание сигналов протонов СδН2-групп остатков Arg1 (3.35 м.д.) и Arg3 (3.32 м.д.) в составе апелина-12, а также и СδН2-протонов отщепившегося свободного аргинина при 3.35 м.д. затрудняет получение информации о пути распада и времени полужизни этого пептида в плазме. В дальнейшем, после трехчасовой инкубации апелина-12 в плазме, кроме сигналов протонов свободного аргинина, в спектрах появляются сигналы и других свободных аминокислот: δ-СН3-группы свободного Leu4 при 1.05, 1.06 м.д., СδН2-пролина при 3.45, 3.52 м.д., а также протонов ароматического кольца С-концевого Phe12.
Кинетическая картина (рис. 3) изменения амплитуды сигналов протонов (С2,6Н, С4Н, С3,5Н) ароматического кольца С-концевого остатка Phe12 и, в первую очередь, амплитуды сигналов протонов С3,5Н отщепившегося фенилаланина при 7.53 м.д. позволила оценить время полужизни С-концевого остатка Phe12 в апелине-12 в плазме крови, которое оказалось близким к 8 ч. Следовательно, скорость отщепления С-концевого остатка Phe12 в плазме, по крайней мере, в три раза меньше скорости гидролиза связи Arg1–Pro2 в N‑концевой части апелина-12. Таким образом, определяющей фактором в деградации апелина-12 в плазме крови является последовательное отщепления N-концевых аминокислотных остатков, начиная с отщепления Arg1 .
Рис. 3.
Фрагменты спектров 1Н-ЯМР апелина-12 в плазме крови человека (после вычитания спектра плазмы), полученные через 300 (а), 500 (б), 700 (в), 850 (г), 1000 (д) и 1300 мин (е) после добавления пептида в плазму. Сигналы протонов остатка фенилаланинина – продукта протеолитической деградации помечены символом *.

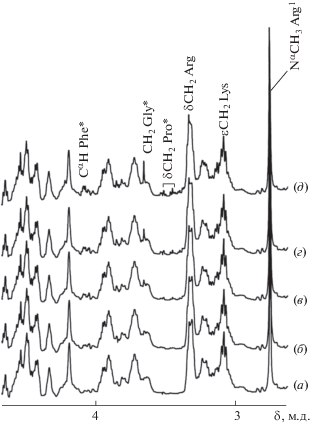
В отличие от апелина-12 в спектрах метилина не наблюдается каких-либо существенных изменений амплитуды сигналов протонов аминокислотных остатков N-концевой части пептида, по крайней мере в первые 4 ч его инкубации в плазме (рис. 4). В частности, не изменяется амплитуда сигналов протонов СδН2-групп (3.34 м.д.) остатков МеArg1, Arg3, а также протонов метильной группы MeArg1 при 2.63 м.д., т.е. связь МеArg1– Pro2 в метилине не гидролизуется. Ранее также было показано, что метилирование N-концевых остатков в пептидах в значительной степени защищает их от действия аминопептидаз [26, 27]. Время полужизни метилина в плазме крови определяется только скоростью отщепления С-концевого остатка Phe12, свидетельством чего является, как и в случае апелина-12, появление в спектрах метилина, например, сигналов протонов ароматического кольца свободного фенилаланина-С3,5Н при 7.53 м.д. Скорость отщепления остатка Phe12 в метилине практически такая же, как в апелина-12 и близка к 8 ч.
Рис. 4.
Фрагменты спектров 1Н-ЯМР метилина в плазме крови человека (после вычитания спектра плазмы), полученные через 20 (а), 300 (б), 500 (в), 700 (г) и 1000 мин (д) после добавления пептида в плазму. Сигналы протонов остатков аминокислот – продуктов протеолитической деградации помечены символом *.
Разумеется, значения времен полужизни пептидов, полученные в экспериментах in vitro, не соответствуют времени их реальной сохранности в кровотоке, т.к. в этих экспериментах концентрации пептидов существенно выше, а активность ферментов плазмы крови ниже, чем в физиологических условиях. Кроме того, следует отметить значительное уширение сигналов рассмотренных пептидов в плазме крови по сравнению с их раствором в D2O без заметного изменения их химических сдвигов. Этот факт можно объяснить обратимым связыванием пептидов с компонентами плазмы крови, что может приводить к увеличению времени их распада. Однако проведенные сравнительные эксперименты позволяют построить ряд относительной устойчивости пептидов к протеолитической деградации.
ЗАКЛЮЧЕНИЕ
Возможность использования модифицированных пептидов апелина в качестве потенциальных лекарственных средств для терапии сердечно-сосудистых заболеваний диктует необходимость разработки оптимизированных способов синтеза этих соединений и изучения их стабильности в биологических системах. Представленная схема получения метилина исключает возможность образования побочных продуктов сульфатирования гидроксильной группы остатка серина, что обеспечивает упрощение пост-синтетических процедур и очистки пептида. Исследование протеолитической стабильности метилина c помощью 1H-ЯМР выявило существенное возрастание времени полужизни пептида в плазме человека по сравнению с природным апелином-12. Такие свойства могут повысить эффективность внутривенного введения метилина больным с различной патологией сердца, а также обосновывают возможность его использования в составе комплекса мер, направленных на защиту миокарда, при проведении операций на открытом сердце. Эти результаты являются принципиально важными для организации экспериментального производства метилина и его запланированных клинических испытаний в ФГБУ “НМИЦ кардиологии” Минздрава России.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использованы производные L-аминокислот (Fluka, NovaBiochem и Bachem Швейцария), BOP, HOBt, TBTU, NMM (Fluka, Швейцария), TIBS (Sigma-Aldrich, США). Для синтеза применяли DMF, N-метилпирролидон (NMP), дихлорметан, 4-метилпиперидин и TFA (Fluka, Швейцария). Для синтеза и очистки пептидов использовали ацетонитрил и изопропиловый спирт (Panreac, Испания). Аналитическую ВЭЖХ пептидов проводили на приборе Knauer (ФРГ) на колонке Kromasil (Швеция) 4.6 × 250 мм, 5 мкм; пептиды элюировали со скоростью 1 мл/мин градиентом буфера Б в буфере А: от 10 до 70% за 30 мин, буфер А – 0.05 M KH2PO4, pH 3.0, буфер Б – 70% ацетонитрила в буфере А, детекция при 220 нм. Препаративную ВЭЖХ пептидов проводили на приборе Knauer (ФРГ) на колонке Диасорб С16 130Т (25 × 250 мм), размер частиц сорбента 10 мкм. В качестве элюентов использовали буфер А – 0.01 M NH4OAc, pH 4.5, буфер Б – 70% ацетонитрила в буфере А. Пептиды элюировали градиентом 0.5% в минуту буфера Б в буфере А от 100% буфера А со скоростью 10 мл/мин, детекция при 226 нм.
Ионообменную хроматографию проводили на колонке с СМ-сефадексом (Sigma-Aldrich, США) 25 × 700 мм, пептиды элюировали линейным градиентом концентрации пиридин-ацетатного буфера с рН 4.5 от 0.05 до 2 М.
ЯМР-спектры сняты на спектрометре WH-500 Bruker 500 МГц (ФРГ) в DMSO-d6 при 300 K, концентрация пептидов составляла 2–3 мг/мл, химические сдвиги измерялись относительно тетраметилсилана и масс-спектрометрии (масс-спектры регистрировали на приборе VISION 2000, Termobioanalysis corp., Finnigan, США).

Схема 2 . Твердофазный синтез метилина.
Автоматический твердофазный синтез метилина проводили согласно схеме 2 на синтезаторе Tribute-UV (Protein Technologies Inc., США), начиная с 0.83 г (0.5 ммоль) Fmoc-Phe-полимера (Sigma-Aldrich, США) с содержанием стартовой аминокислоты 0.60 ммоль/г. ТФС метилина в ручном варианте проводили, исходя из 2.5 г (1.5 ммоль) того же аминоацилполимера.
На приведенной выше схеме циклы ТФС пронумерованы в соответствии с последовательностью присоединения аминокислот. Протокол ТФС для циклов 1–7 был стандартным и включал следующие стадии: деблокирование α-аминогрупп раствором 5% 4-MePip и 2% DBU (1,8-диазабицикло[5.4.0]-ундец-7-ен) в DMF 5 мин (в программу была включена функция спектрофотометрического контроля полноты отщепления Fmoc-защиты); промывки DMF; присоединение 4-х кратного избытка Fmoc-AA в присутствии TBTU/NMM в DMF в течение 1.5 ч; промывки DMF. В циклах 8 и 10 для присоединения Fmoc-Arg-OH и Boc-NαMe-Arg-OH cоответственно проводили двойные конденсации. Вариант 1 – к раствору 2.0 ммоль Fmoc-Arg-OH и 2.0 ммоль HBr-Pyr в 15 мл смеси DMF/NMP 1 : 1, добавляли 2.0 ммоль DIC и 2.0 ммоль HOBt и эту смесь прибавляли к пептидилполимеру, перемешивали 1.5 ч, после чего пептидилполимер промывали DMF и конденсацию повторяли с теми же количествами реагентов. Вариант 2 – к раствору 2.0 ммоль Fmoc-Arg-OH и 4.0 ммоль HOBt в 15 мл смеси DMF/NMP 1 : 1, прибавляли 2.0 ммоль DIC и смесь прибавляли к пептидилполимеру, перемешивали 1.5 ч, пептидилполимер промывали DMF и конденсацию повторяли. Вариант 3 – смесь 2.0 ммоль Fmoc-Arg-OH + 2.0 ммоль BOP растворяли в 10 мл DMF в течение 4 мин, прибавляли к пептидилполимеру и перемешивали 1.5 ч, после промывок конденсацию повторяли. Кроме того, в двух заключительных циклах синтеза – 9 и 10 перед конденсацией в протокол была включена дополнительная промывка α-аминодеблокированного пептидилполимера 5% раствором HOBt в DMF в течение 5 мин. Деблокирование и отщепление додекапептида от полимера осуществляли действием смеси TFA–вода–TIBS (95 : 2.5 : 2.5), после осаждения пептида и промывок диэтиловым эфиром полученный продукт очищали с помощью ИОХ до 98%-й чистоты (по данным ВЭЖХ в вышеуказанных условиях). Структуру полученного пептида подтверждали данными 1Н-ЯМР-спектроскопии, приведенными в табл. 2. Масс-спектр: найдено m/z 1418.866; Mрасчет. = 1418.7.
Получение плазмы крови. Плазму получали из крови здорового донора путем осаждения клеточных элементов и ультрацентрифугирования в градиенте плотности NaBr в течении 48 ч при 4°С для удаления фракции липидов. Плазму без липидов диализовали в фосфатно-солевом буфере (2.68 мМ KCl, 1.47 мМ KH2PO4, 136.89 мМ NaCl, 8.1 мМ Na2HPO4, pH 7.1–7.4), лиофилизовали и растворяли в D2O.
1Н-ЯМР-анализ деградации пептидов. Химические сдвиги сигналов в 1Н-ЯМР-спектрах были измерены относительно внутреннего стандарта – натриевой соли 2,2-диметил-2-силапентан-5-сульфаната. Отнесение сигналов к определенным группам протонов аминокислотных остатков проводилось с помощью метода дифференциального двойного резонанса. Для выделения сигналов, принадлежащих протонам пептида, применялась разностная спектроскопия: из каждого спектра, полученного после добавления пептида в плазму крови, вычитался спектр свободной плазмы. Концентрация пептида в образце составляла 2–2.5 мг/мл.
Список литературы
Pitkin S., Maguire J., Bonner T., Davenport A. // Pharmacol. Rev. 2010. V. 62. P. 331–342.
Tatemoto K., Takayama K., Zou M.X., Kumaki I., Zhang W., Kumano K., Fujimiya M. // Regul. Pept. 2001. V. 99. P. 87–92.
Писаренко О.И., Шульженко В.С., Пелогейкина Ю.А., Студнева И.М., Кхатри А.Н., Беспалова Ж.Д., Азьмуко А.А., Сидорова М.В., Палькеева М.Е. // Кардиология. 2010. Т. 50. № 10. С. 44–49.
Писаренко О.И., Серебрякова Л.И., Пелогейкина Ю.А., Студнева И.М., Кхатри Д.Н., Цкитишвили О.В., Беспалова Ж.Д., Азьмуко А.А., Сидорова М.В., Палькеева М.Е. // Бюлл. эксп. биологии и медицины. 2011. Т. 152. № 7. С. 86–89.
Juhl C., Els–Heindl S., Schçnauer R., Redlich G., Haaf E., Wunder F., Riedl B., Burkhardt N., Beck-Sickinger A.G., Bierer D. // ChemMedChem. 2016. V. 11. P. 2378–2384.
Murza A., Belleville K., Longpré J.-M., Sarret P., Marsault É. // Biopolymers (Peptide Science). 2014. V. 102. № 4. P. 297–303.
Писаренко О.И., Шульженко В.С., Пелогейкина Ю.А., Палькеева М.Е., Сидорова М.В., Азьмуко А.А., Молокоедов А.С., Беспалова Ж.Д., Терещенко С.Н., Масенко В.П. Патент РФ № 2457216 от 21.12.2010, опубл. 27.07.2012. Бюл. № 21.
Сидорова М.В., Азьмуко А.А, Палькеева М.Е., Молокоедов А.С., Бушуев В.Н., Дворянцев С.Н., Шульженко В.С., Пелогейкина Ю.А., Писаренко О.И., Беспалова Ж.Д. // Биоорган. химия. 2012. Т. 38. № 1. С. 40–51. [Sidorova M.V., Az’muko A.A., Pal’keeva M.E., Molokoedov A.S., Bushuev V.N., Dvoryantsev S.N., Shulzhenko V.S., Pelogeykina Yu.A., Pisarenko O.I., Bespalova Zh. D. // Russ. J. Bioorgan. Chem. 2012. V. 38. № 1. P. 30–40.]
Лакомкин В.Л., Абрамов, А.А., Лукошкова Е.В., Лакомкин С.В., Грамович В.В., Выборов О.Н., Ундровинас Н.А, Ермишкин В.В., Цыплёнкова В.Г., Ширинский В.П., Капелько В.И. // Kaрдиология. 2015. Т. 55. № 6. С. 54—62.
Пелогейкина Ю.А., Серебрякова Л.И., Цкитишвили О.В., Студнева И.М., Писаренко О.И. // Кардиологический вестник. 2014. № 3. С. 54–63.
Synthetic Peptides: a User’s Guide / Ed. by Grant G.A. 2nd ed. New York, Oxford Univ. Press 2002.
Шредер Э., Любке К. Пептиды. Т. 1.: Пер. с англ. М.: Мир, 1967 (Schröder E., Lübke K. The Peptides. V. 1. New York; London: Academic Press, 1965).
Coy D., Branyas N. // Int. J. Pept. & Prot. Res. 1979. V. 14. P. 339–343.
Рубина А.Ю., Беспалова Ж.Д., Бушуев В.Н. // Биоорган. химия. 2000. Т. 26. С. 263–272. [Rubina A.Yu., Bespalova Zh.D., Bushuev V.N. // Russ. J. Bioorgan. Chem. 2000. V. 26. № 4. P. 235–244.]
Сидорова М. В., Молокоедов А.С., Овчинников М.В., Беспалова Ж.Д., Бушуев В.Н. // Биоорган. химия. 1997. Т. 23. С. 46–55. [Sidorova M.V., Molokoedov A.S., Ovchinnikov M.V., Bespalova Zh.D., Bushuev V.N. // Russ. J. Bioorgan. Chem. 1997. V. 23. P. 41–50.]
Xue C.-B, LeGrado W.F. // Tetrahedron Lett. 1995. V. 36. P. 55–58.
Castro B., Dormoy J.R., Evin G., Selve C. // Tetrahedron Lett. 1975. V. 14. P. 1219–1222.
Knorr R., Trzeciak A., Bannwarth W., Gillessen D. // Tetrahedron Lett. 1989. V. 30. P. 1927–1930.
Золотарев Ю.А., Дадаян А.К., Долотов О.В., Козик В.С., Кост Н.В., Соколов О.Ю., Дорохова Е.М., Мешавкин В.К., Иноземцева Л.С., Габаева М.В. и др. // Биоорган. химия, 2006. Т. 32. С. 183–191. [Zolo-tarev Yu.A., Dadayan A.K., Dolotov O.V., Kozik V.S., Kost N.V., Sokolov O.Yu., Dorokhova E.M., Meshavkin V.K., Inozemtseva L.S., Gabaeva M. et al. // Russ. J. Bioorgan. Chem. 2006. V. 32. P. 166–173.]
Золотарев Ю.А., Дадаян А.К., Кост Н.В., Воеводина М.Э., Соколов О.Ю., Козик В.С., Шрам С.И., Азев В.Н., Бочаров Э.В., Богачук А.П., Липкин В.М., Мясоедов Н.Ф. // Биоорган. химия. 2015. Т. 41. № 6. С. 644–656.
Esposito S., Mele R., Ingenito R., Bianchi E., Bonelli F., Monteagudo E., Orsatti L. // Anal. Bioanal. Chem. 2017. V. 409. P. 2685–2696.
Mesmin C., Dubois M., Becher F., Fenaille F., Ezan E. // Rapid Commun. Mass Spectrom. 2010. V. 24. P. 2875–2884.
Jardetzky O., Roberts G.C.K. NMR in Molecular Biology. New York; London: Acad. Press, 1981. 681 p.
King G.F., Kuchel P.W. // Biochem. J. 1984. V. 220. P. 553–560.
Исакова О.Л., Сепетов Н.Ф., Бушуев В.Н., Беспалова Ж.Д., Виноградов В.А., Рууге Э.К., Титов М.И. // Биоорган. химия 1986. Т. 12. С. 106–111.
Сахаров И.Ю., Исакова О.Л., Сепетов Н.Ф., Беспалова Ж.Д., Рууге Э.К. // Биохимия. 1987. Т. 52(2). С. 311–316.
Wüthrich K. NMR in Biological Research: Peptides and Proteins. New York: American Elsivier, 1976. 379 p.
Секридова А.В., Сидорова М.В., Азьмуко А.А., Молокоедов А.С., Бушуев В.Н., Марченко А.В., Щербакова О.В., Ширинский В.П., Беспалова Ж.Д. // Биоорган. химия. 2010. Т. 36. № 4. С. 498–504. [Sekridova A.V., Sidorova M.V., Az’muko A.A., Molokoedov A.S., Bushuev V.N., Marchenko A.V., Shcherbakova O.V., Shirinskii V.P., Bespalova Z.D. // Russ. J. BioorganicChemistry. 2010. V. 36, № 4. P. 461–467.]
Khapchaev A.Y., Kazakova O.A., Samsonov M.V., Sidorova M.V., Bushuev V.N., Vilitkevich E.L., Az’muko A.A., Molokoedov A.S., Bespalova Zh.D., Shirinsky V.P. // J. Pept. Sci. 2016. V. 22. P. 673–681.]
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия