

Биоорганическая химия, 2019, T. 45, № 4, стр. 374-379
Синтез и противоопухолевая активность конъюгатов на основе пептидного фрагмента PHE-D-TRP-LYS-THR соматостатина
Д. В. Авдеев 1, 2, *, М. В. Сидорова 1, М. В. Овчинников 1, Н. И. Моисеева 3, В. Н. Осипов 3, 4, А. Н. Балаев 4, Д. С. Хачатрян 5
1 НМИЦ кардиологии Минздрава России
121552 Москва, 3-я Черепковская, 15А, Россия
2 Высший химический колледж РАН
125047 Москва, Миусская площадь, 9, Россия
3 НМИЦ онкологии им. Н.Н. Блохина Минздрава России
115478 Москва, Каширское шоссе, 23, Россия
4 АО Фарм-синтез
121357 Москва, Верейская 29, стр. 134, Россия
5 НИЦ “Курчатовский институт” – ИРЕА
107076 Москва, Богородский вал, 3, Россия
* E-mail: mityaavdeev93@mail.ru
Поступила в редакцию 19.11.2018
После доработки 14.01.2019
Принята к публикации 12.02.2019
Аннотация
Синтезированы новые аналоги соматостатина, содержащие фрагменты адамантана, кумарина, тетрагидрокарбазола и пальмитиновой кислоты, общей формулой R-Phe-D-Trp-Lys(Boc)-Thr-OMe. Конъюгаты сочетают в структуре пептидный фрагмент, обладающий сродством к соматостатиновым рецепторам (sstr), и непептидный фрагмент, потенциально обладающий противоопухолевой активностью. Предположительно, синтезированные соединения являются агонистами sstr. Для создания амидной связи между пептидным и непептидным фрагментом применяли карбодиимидный метод и метод активированных эфиров. Структура полученных конъюгатов подтверждена методами масс-спектрометрии (ESI+) и 1H-ЯМР-спектроскопии. Противоопухолевая активность полученных конъюгатов была исследована с помощью MTT-теста на линиях клеток аденокарциномы легкого человека (А549), простаты человека (PC3), толстого кишечника человека (НСТ-116), молочной железы человека (MCF7) и острого Т-клеточного лейкоза человека (Jurkat). Выявлены соединения, селективно ингибирующие рост клеток А549.
ВВЕДЕНИЕ
В настоящее время используется несколько лекарств на основе фрагментов гормона соматостатина, ингибирующего множество физиологических функций в гипоталамусе, передней доле гипофиза, желудочно-кишечном тракте и поджелудочной железе [1–6]. Это такие препараты как Октреотид (торговое название “Sandostatin”), Ланреотид (“Somatuline”), Вапреотид (“Sanvar”) и Пасиреотид (“Signifor”). Действие всех этих препаратов обусловлено их связыванием с рецепторами соматостатина, гиперэкспрессия которых наблюдается в различных опухолевых клетках [2–4]. Создано много аналогов соматостатина и ведутся исследования по поиску новых соединений [5, 6]. Современные исследования посвящены разработке аналогов с улучшенными свойствами: повышенной метаболической устойчивостью, селективностью действия и увеличенным противоопухолевым эффектом, например за счет включения в молекулу известных цитостатических препаратов: доксорубицина, метотрексата, камптотецина, цис-платина [7]. Одним из современных подходов является конструирование химерных молекул, сочетающих пептидный и непептидный фрагменты. Пептидный фрагмент обеспечивает высокую селективность действия конъюгата за счет связывания с рецепторами, а непептидный – усиливает противоопухолевую активность. Кроме того, наличие пептидного фрагмента в молекуле снижает токсичность препарата [8]. Настоящая работа является продолжением исследований по синтезу и изучению цитостатической активности аналогов соматостатина, содержащих фрагменты адамантана и нафталина, некоторые из которых показали высокий цитостатический эффект на линиях опухолевых клеток MCF-7, PC3, HCT-116 [9, 10]. В этих исследованиях в качестве пептидного компонента выбрана тетрапептидная последовательность -Phe-D-Trp-Lys-Thr-, являющаяся фармакофорной в соматостатине [11].
Данная работа посвящена синтезу и изучению противоопухолевой активности новых аналогов соматостатина на основе последовательности Phe-D-Trp-Lys-Thr. В отличие от предыдущих исследований, в которых конъюгация осуществлялась через карбоксильную группу треонина, в нашей работе непептидный фрагмент конъюгировали с пептидом за счет образования амидной связи с его концевой α-аминогруппой.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Синтетический этап работы был посвящен получению химерных молекул путем конденсации защищенного тетрапептида H-Phe-D-Trp-Lys(Boc)-Thr-OМe и непептидного компонента по α-аминогруппе остатка фенилаланина (схема 1 ). Предполагается, что введение защитных групп в последовательность -Phe-D-Trp-Lys-Thr- и замена остатка L-Trp- на D-Trp должны увеличить метаболическую устойчивость тетрапептида. В качестве непептидного фрагмента были выбраны органические остатки, потенциально обладающие противоопухолевой активностью, перспективные при разработке противоопухолевых препаратов: производное карбазола (3-(1,2,3,4-тетрагидро-9Н-карбазол-9-ил) пропановая кислота) [12], производное адамантана и аналоги кумарина.

Общая схема синтеза конъюгатов.
Ранее показано, что конъюгаты адамантана с защищенным по аминогруппе дипептидом Boc-Tyr-D-Trp-OH обладают высокой противоопухолевой активностью на клетках линий HCT-116 [13], причем удаление Boc-защиты приводило к снижению активности полученных соединений [13]. Для производных кумарина известен ряд соединений, ингибирующих рост линий опухолевых клеток HCT-116, A-549, MCF-7 [14–16], кроме того, имеются данные о сравнимой с известными онкологическими препаратами 5-фторурацилом [16] и энтиностатом противоопухолевой активности кумаринамидов [17]. Также нами была предпринята попытка увеличения противоопухолевой активности H-Phe-D-Trp-Lys(Boc)-Thr-OМe за счет его конъюгирования с пальмитиновой кислотой [18].
Пептид H-Phe-D-Trp-Lys(Boc)-Thr-OМe в настоящей работе синтезировали по ранее описанной методике [19]. При получении соединений (I)–(IV) для создания амидной связи применяли карбодиимидный метод. Для синтеза соединения (V) использовали N-гидроксисукцинимидный эфир пальмитиновой кислоты, который получали с помощью трансэтерефицирующего реагента – N-трифторацетоксисукцинимида, поскольку карбодиимидный метод зачастую оказывался непригодным для получения высоколипофильных производных пептидов с жирными кислотами в органических растворителях [20]. Все соединения были получены с высокими выходами. Их структура подтверждена методами масс-спектрометрии (ESI+) и 1H-ЯМР-спектроскопии (см. экспериментальную часть).
Противоопухолевую активность полученных соединений (I)–(V) определяли с помощью МТТ-теста на клеточных линиях опухолей человека, экспрессирующих рецепторы соматостатина: аденокарциноме легкого (А549), простаты (PC3), колоректального рака (НСТ-116) и молочной железы (MCF7), а также острого Т-клеточного лейкоза человека (Jurkat) (табл. 1). В качестве контрольных псевдонормальных клеток использовались трансформированные фибробласты эмбриона человека (ФЭЧ).
Таблица 1.
Противоопухолевая активность (IC50, мкМ) для конъюгатов общей формулы RCO-Phe-D-Trp-Lys(Boc)-Thr-OМe
| Соеди-нение | R | Раковые клетки | ФЭЧ | IC50(ФЭЧ)/ IC50(A-549) | ||||
|---|---|---|---|---|---|---|---|---|
| HCT-116 | PC3 | MCF-7 | A-549 | Jurkat | ||||
| (I) | 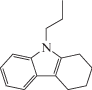 |
12 | 11 | 11 | 10 | 47 | 13 | 1.18 |
| (II) | 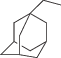 |
10 | 8.4 | 65 | 8.2 | 8.4 | 17 | 2.07 |
| (III) |  |
16 | 17 | 25 | 5 | 16 | 46 | 9.2 |
| (IV) |  |
75 | 72 | >100 | 11 | 71 | >100 | 9.09 |
| (V) | CH3(CH2)14 | 50 | >100 | >100 | >100 | 87 | 90 | 0.9 |
Стоит отметить, что исходный пептид H-Phe-D-Trp-Lys(Boc)-Thr-OМe не демонcтрировал противоопухолевой активности на вышеперечисленных линиях клеток, а показал лишь незначительную цитотоксичность на клетках рака молочной железы MCF-7 (IC50 = 75 мкМ).
Согласно результатам исследований противоопухолевой активности соединений (I)–(V), конъюгат (V), содержащий пальмитиновую кислоту, оказывает довольно слабое цитостатическое действие по сравнению с другими аналогами. Соединение (I) показало высокий противоопухолевый эффект практически на всех клеточных линиях, кроме Jurkat, однако его селективность (IC50(ФЭЧ)/IC50(опухолевая клетка)) близка к 1, что свидетельствует о потенциальной токсичности этого конъюгата. Наилучшие результаты показали соединения (II)–(IV), особенно при ингибировании роста клеток аденокарциномы легкого человека (А549). Кроме того, соединения (III), (IV) проявляют почти на порядок большую эффективность действия на опухолевые клетки (A549), чем на контрольные (IC50(ФЭЧ)/IC50(A549) > 9). По значениям IC50 соединения (III) и (IV) сравнимы с известным противоопухолевым препаратом 5-фторурацилом (11.13 мкМ) на линии клеток А549 [16]. Известно, что 5-фторурацил обладает высокой токсичностью, обусловленной низкой селективностью действия, чтоне отмечено для соединений (III), (IV). Соединение (III) показало также высокое цитотоксическое действие против первичной глиобластомы (IC50 13.5 мкМ), одной из самых агрессивных опухолей.
В результате проведенных исследований получены новые аналоги соматостатина, содержащие фрагменты адаманта, кумарина, тетрагидрокарбазола и пальмитиновой кислоты. Выявлены соединения, обладающие in vitro-цитотоксической активностью и выраженной селективностью действия по отношению к аденокарциноме легкого человека (А549).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовались производные L/D-аминокислот DMF, DCC, NMM, HOBt, дихлорметан, метанол (Fluka, Швейцария). Спектры 1Н-ЯМР11 регистрировали на спектрометре “AVANCE III NanoBay” в DMSO-d6 при 300 МГц и 300 Kс использованием в качестве внутренних стандартов DMSO-d6 (δ = 2.500); концентрация соединений составляла 2–3 мг/мл. Приведены значения химических сдвигов (δ, м.д.) и константы спин-спинового взаимодействия (J, Гц). Масс-спектры регистрировали на приборе “AmazonBruker” методом электрораспылительной ионизации (ESI) в режиме регистрации положительных и отрицательных ионов (напряжение на капилляре – 3500 В). Диапазон сканирования масс – m/z – 70–2200. Применяли шприцевой ввод образца, растворенного в метаноле либо в ацетонитриле. Газ-распылитель – азот, температура интерфейса – 100°C. Аналитическую ВЭЖХ проводили на хроматографе “Shimadzu” (Япония), использовали колонки Reprosil – PurBasic C18 (5 мкм), 4.6 × 240 мм, подвижная фаза: буфер А – 0.1% TFA в воде, буфер Б – 0.1% TFA в ацетонитриле, элюирование градиентом концентрации буфера Б в буфере А от 5 до 100% за 30 мин; скорость потока 1 мл/мин, детекция при 220 нм.
Общая методика получения соединений (I)–(IV)
В 10 мл DMF последовательно растворяли 0.69 г (1 ммоль) H-Phe-D-Trp-Lys(Boc)-Thr-OМe, 1.03 ммоль соответствующей органической кислоты (см. схему) и 0.14 г (1.033 ммоль) HOBt. К охлажденной до 0°C реакционной смеси добавляли по каплям раствор 0.21 г (1 ммоль) DCC в 1.67 мл DMF, перемешивали 2 ч при 0°C, затем еще 12 ч при комнатной температуре. Выпавший осадок дициклогексил мочевины отфильтровывали, промывали 2 мл DMF. К объединенному фильтрату добавляли 100 мл 5% раствора лимонной кислоты. Выпавший осадок отфильтровывали, промывали водой (2 × 20 мл), 5% раствором бикарбоната калия (10 мл), водой (2 × 20 мл), переосаждали из смеси С2Н5ОН–Н2О, 0.5 : 1 (1 × 10 мл), осадок промывали смесью С2Н5ОН–Н2О, 1 : 1 (1 × × 10 мл), высушивали на воздухе до постоянного веса.
Nα-[3-(1,2,3,4-Тетрагидро-9Н-карбазол-9-ил) пропаноил]-Phe-D-Trp-Lys(Boc)-Thr-OМe (I) синтезировали из 0.25 г (1.03 ммоль) 3-(1,2,3,4-тетрагидро-9Н-карбазол-9-ил) пропановой кислоты. Получили 0.9 г (0.98 ммоль, 97.8%) белого порошка. Масс-спектр ESI, m/z (Iотн, %): 920.6 (100) [M]+, 820.5(5) [M-Boc]+, 410.8 (2) [(M-Boc)/2]+. Чистота (ВЭЖХ): 92.47%. Спектр 1H-ЯМР: 1.06 (д, 3H, J 6.3, γCH3, Thr), 1.10–1.32 (м, 4H, γCH2, δCH2, Lys), 1.35 (с, 9H, -Boc), 1.4–1.67 (м, 2H, βCH2, Lys), 1.68–1.87 (м, 4H, (CH2)2, Сrb), 2.24–2.46 (м, 2H, –NCH2CH2, Prp), 2.51–3.28 (м, 10H, βCH2, D-Trp; (CH2)2, Сrb; εCH2, Lys; βCH2, Phe), 3.60 (с, 3H, –OCH3, Thr(OMe)), 4.01–4.13 (м, 3H, –NCH2CH2(O), Prp; αCH, Phe), 4.25 (дд, 1H, J 8.2, 3.4, βCH, Thr), 4.37 (кв, 1H, J 7.5, αCH, Lys), 4.57 (дт, 1H, J 13.1, 6.6, αCH, DTrp), 4.95 (д, 1H, J 5.7, –OH, Thr), 6.66–6.81 (м, 2H, HAr, Phe; εNH, Lys), 6.85–7.12 (м, 8H, HAr, Phe; HAr, DTrp), 7.16 (д, 1H, J2.3, HAr, Crb),7.20–7.38 (м, 3H, HArCrb, 7.69 (д, 1H, J 7.7, HAr, DTrp,), 7.97 (т, 2H, J 8.1, αNH, Lys; αNH, DTrp), 8.21 (д, 1H, J 8.1, αNH, Phe), 8.38 (д, 1H, J 8.2, αNH, Thr), 10.77 (с, 1H, NH, D-Trp).
Nα-[2-((3S,5S,7S)-Адамантан-1-ил) ацетил]-Phe-D-Trp-Lys(Boc)-Thr-OМe (II) синтезировали из 0.20 г (1.03 ммоль) 2-((3S,5S,7S)-адамантан-1-ил) уксусной кислоты. Получили 0.70 г (0.92 ммоль, 89.5%) белого порошка. Масс-спектр, m/z (Iотн, %): 893.7 (100) [M + Na]+, 909.62(45) [M + K]+. Чистота (ВЭЖХ): 89.84%. Спектр 1H-ЯМР: 1.07 (д, 3H, J 6.4, γCH3, Thr), 1.13–1.27 (м, 4H, γCH2, δCH2, Lys), 1.35 (c, 9H, -Boc), 1.40–1.86 (м, 17H, βCH2, Lys; CH15, Adm), 1.86–2.08 (м, 2H, CH2, AcAdm), 2.65–2.74 (м, 1H, β'-CH2, Phe), 2.75–3.00 (м, 4H, βCH2, D-Trp; εCH2, Lys), 3.12 (дд, 1H, J 14.6, 5.0, β''-CH2, Phe), 3.61 (c, 3H, –OCH3, Thr(OMe)), 4.10 (д, 1H, J 7.0, αCH, Thr), 4.25–4.36 (м, 2H, αCH, DTrp; αCH, Lys), 4.46–4.62 (м, 2H, αCH, Phe; βCH, Thr), 5.17 (с, 1H, –OH, Thr), 6.72 (т, 1H, J 5.7, εNH, Lys), 7.21–6.91 (м, 8H, HAr, Phe; HAr, D-Trp), 7.29 (д, 1H, J 7.9, HAr, D-Trp), 7.63 (д, 1H, J 7.8, HAr, D-Trp), 7.79 (д, 1H, J 8.3, αNH, Lys), 8.09 (д, 1H, J 8.3, αNH, D-Trp), 8.27 (д, 1H, J 8.3, αNH, Phe ), 8.39 (д, 1H, J 8.4, αNH, Thr), 10.80 (с, 1H, ‒NH, D-Trp).
Nα-2-((4-Метил-2-оксо-2Н-хромен-7-ил)окси) бутаноил-Phe-D-Trp-Lys(Boc)-Thr-OМe (III) синтезировали из 0.271 г (1.03 ммоль) 2-((4-метил-2-оксо-2Н-хромен-7-ил) окси) бутановой кислоты. Получили 0.85 г (0.92 ммоль, 91.8%) белого порошка. Масс-спектр ESI, m/z (Iотн, %): 939.5 (100) [M]+, 839.5(3) [M-Boc]+. Чистота (ВЭЖХ): 92.83% Спектр 1H-ЯМР (DMSO-d6): 0.77 (кв, 3H, J 7.4, –CH3СH2(Chr)), 1.03 (д, 3H, J 6.4, γCH3, Thr), 1.07–1.31 (м, 4H, γCH2, δCH2, Lys), 1.35 (с, 9H, –Boc), 1.38–1.77 (м, 4H, βCH2, Lys; –CH3СH2(Chr)), 2.37 (д, 3H, J 8.6, –CH3(Chr)), 2.71 (д, 1H, J 11.6, β'-CH2, Phe), 2.77–2.98 (м, 4H, βCH2, D-Trp; εCH2, Lys), 3.07 (д, 1H, J 14.7, β''-CH2, Phe), 3.60 (с, 3H, –OCH3, Thr(OMe)), 3.98–4.11 (м, 1H, αCH, Thr), 4.18–4.27 (м, 1H, βCH, Thr), 4.31–4.41 (м, 1H, αCH, Lys), 4.45–4.76 (м, 2H, αCH, DTrp; αCH, Phe), 4.98 (с, 1H, –OH, Thr), 6.21 (д, 1H, J 8.9, 3H, (Chr)), 6.57–7.19 (м, 11H, HAr, Phe; HAr, D-Trp; εNH, Lys; HAr, Chr), 7.29 (д, 1H, J 8.2, HAr, D-Trp), 7.55 (дд, 1H, HAr(Chr)), 7.68 (д, 1H, J 7.7, HAr, D-Trp), 8.00 (д, 1H, J 8.4, αNH, Lys), 8.19–8.40 (м, 2H, αNH, Phe; αNH, Thr), 10.78 (с, 1H, NH, D-Trp).
Nα-(9-Этил-4-метил-2-оксо-2Н-фуро[2,3-h] хромен-8-карбонил)-Phe-D-Trp-Lys(Boc)-Thr-OМe (IV) синтезировали из 0.05 г (0.18 ммоль) 9-этил-4-метил-2-оксо-2Н-фуро[2,3-h] хромен-8-карбоновой кислоты. Получили 0.11 г (0.11 ммоль, 62.5%) белого порошка. Масс-спектр ESI, m/z (Iотн, %): 949.9 (100) [M]+, 242.6(3) [M/4 + 5H]+, 397.5 (2.2) [M/3 + DMSO + 2H]+. Чистота (ВЭЖХ): 90.12%. Спектр ЯМР 1H (DMSO-d6): 0.98–1.23 (м, 8H, γCH3, Thr; –CH3CH2(FuroChr), γCH2, Lys), 1.26–1.8 (м, 12H, δCH2, Lys; –Boc; βCH2, Lys), 2.7–3.02 (м, 8H, βCH2, Phe; βCH2, D-Trp; εCH2, Lys; –CH3CH2(FuroChr), 3.09–3.23 (м, 1H, αCH, Phe), 3.58 (с, 3H, –OCH3, Thr(OMe)), 4.01–4.15 (м, 1H, αCH, Thr), 4.23 (дд, 1H, J 8.0, 3.5, βCH, Thr), 4.31–4.44 (м, 1H, αCH, Lys), 4.58–4.86 (м, 1H, αCH, D-Trp), 4.94 (д, 1H, J 5.7, -OH, Thr), 6.41 (с, 1H, 3H(FuroChr)), 6.70 (с, 1H, εNH, Lys), 6.87–7.23 (м, 8H, HAr, Phe; HAr, D-Trp), 7.3 (д, 1H, J 7.9, HAr, D-Trp), 7.59 (д, 1H, J 7.7, HAr(FuroChr)), 7.71 (д, 1H, J 7.7, HAr, D-Trp), 7.82 (д, 1H, J 8.8, HAr(FuroChr)), 7.98 (д, 1H, J 8.1, αNH, Lys), 8.13 (д, 1H, J 8.1, αNH, DTrp), 8.26 (д, 1H, J 8.0, αNH, Phe), 8.56 (д, 1H, J 8.1, αNH, Thr), 10.80 (с, 1H, –NH, D-Trp).
Nα-Пальмитоил-Phe-D-Trp-Lys(Boc)-Thr-OМe (V). В круглодонной колбе на 150 мл растворяли 2.0 г (17.4 ммоль) N-гидроксисукцинимида в 5 мл (26 ммоль) трифторуксусного ангидрида. Через 15 мин полученный раствор упаривали досуха, промывали гексаном, растворитель декантировали. Остаток высушивали в вакууме. Получили 3.6 г (17 ммоль, 98%) бесцветных кристаллов N-трифторацетоксисукцинимида (TFS), к которым прибавляли раствор 4 г (15 ммоль) пальмитиновой кислоты в 15 мл хлористого метилена и 2 мл пиридина. Реакционную массу перемешивали 40 мин при комнатной температуре. Растворитель удаляли в вакууме (40°С 15 мм. рт. ст.), к остатку добавляли 100 мл воды. Выпавший осадок отфильтровали, промывали водой (2 × 10 мл), 5% раствором бикарбоната натрия (10 мл), смесью С2Н5ОН/Н2О 1/1 (1 × 10 мл), высушивали на воздухе до постоянного веса. Получили 5 г (14.14 ммоль, 94.2%) белого порошка N-гидроксисукцинимидного эфира пальмитиновой кислоты (Palm-ONSu).
К раствору 0.50 г (0.72 ммоль) H-Phe-D-Trp-Lys(Boc)-Thr-OМe в 5.00 мл DMF добавляли 120 мкл NMM до pH 8, затем 0.26 г (0.69 ммоль) Palm-ONSu и перемешивали при комнатной температуре 18 часов. В реакционную смесь добавляли 70 мл 2% раствора лимонной кислоты. Выпавший осадок отфильтровали, промывали водой (2 × 20 мл), 5% раствором бикарбоната натрия (10 мл), переосаждали из смеси С2Н5ОН/Н2О 0.5/1 (1 × 10 мл), осадок промывали смесью С2Н5ОН/Н2О 1/1 (1 × 10 мл), высушивали на воздухе до постоянного веса. Получили 0.40 г (0.43 ммоль, 59.5%) белого порошка. Rf = 0.6 (хлороформ–метанол–уксусная кислота, 9 : 1 : 0.5). Масс-спектр ESI, m/z (Iотн, %): 955.87(100) [M + Na]+, 971.79 (14) [M + K]+. Спектр ЯМР 1H (DMSO-d6): 0.85 (т, 3H, J 6.5, –CH3(Palm)), 0.95–1.54 (м, 42H, γCH3, Thr; γCH2, δCH2, Lys; –Boc; –CH2(Palm)), 1.54–1.71 (м, 2H, βCH2, Lys), 1.88–2.05 (м, 2H, –CH2C(O)(Palm)), 2.68 (д, 1H, J 10.5, β'-CH2, Phe), 2.76–2.99 (м, 4H, βCH2, DTrp; εCH2, Lys), 3.12 (дд, 1H, J 14.6, 5.0, β''-CH2, Phe), 3.61 (с, 3H, OCH3, Thr(OMe)), 4.02–4.17 (м, 1H, αCH, Thr), 4.26 (дд, 1H, J 8.3, 3.5, βCH, Thr), 4.3–4.4 (м, 1H, αCH, Lys), 4.44–4.66 (м, 2H, αCH, DTrp; αCH, Phe), 5.0 (д, 1H, J 5.4, –OH, Thr), 6.70 (т, 1H, J 5.7, εNH, Lys), 6.9–7.19 (м, 8H, HAr, Phe; HAr, DTrp), 7.29 (д, 1H, J 7.9, HAr, DTrp), 7.66 (д, 1 H, J 7.7, HAr, DTrp), 7.83 (д, 1H, J 8.2, αNH, Lys), 7.99 (д, 1H, J 8.2, αNH, D-Trp), 8.18 (д, 1H, J 8.0, αNH, Phe), 8.33 (д, 1H, J 8.2, αNH, Thr), 10.78 (с, 1H, NH, D-Trp).
Противоопухолевая активность новых аналогов соматостатина исследовалась с помощью MTT-теста на линиях клеток аденокарциномы легкого человека (А549), аденокарциномы простаты человека (PC3), колоректального рака человека (НСТ-116), аденокарциномы молочной железы человека (MCF7), первичной культуре глиобластомы, любезно предоставленной Е.Ю.Рыбалкиной , и острого Т-клеточного лейкоза человека (Jurkat). В качестве контрольных клеток использовались трансформированные фибробласты эмбриона человека (ФЭЧ). Клетки А549, MCF-7, клетки первичной глиобластомы и ФЭЧ культивировались на среде DMEM (ПанЭко, Россия), а клетки НСТ-116, PC3 и Jurkat на среде RPMI1640 (ПанЭко, Россия) с добавлением 10% эмбриональной бычьей сыворотки (HyClone, США) и гентамицина 40 мкг/мл.
Адгезивные культуры опухолевых клеток рассевались на 96-луночное плато в количестве 5 × 103 клеток на лунку, клетки Jurkat – в количестве 15 × 103. На следующий день к клеткам добавляли исследуемые вещества в конечных концентрациях от 100 до 5 мкМ. После инкубации в течение 72 часов в лунки вносили МТТ-реагент в концентрации 5 мг/мл по 20 мкл в лунку. После инкубации 3–4 часа полученные кристаллы формазана растворяли 60 мкл DMSО и определяли оптическую плотность раствора на спектрофотометре MultiSkanFC (ThermoScientific, США) при длине волны 540 нм.
Список литературы
Guillemin R., Gerich J.E. // Annu. Rev. Med. 1976. V. 27. P. 379–388.
Patel Y.C. // FrontNeuroendocrinol. 1999. V. 20. P. 157–198.
Lamberts S.W., van der Lely A.J., Hofland L.J. // European Journal of Endocrinology. 2002. V. 146. P. 701–705.
Barnett P. // Endocrine. 2003. V. 20. P. 255–264.
Janecka A., Zubrzycka M., Janecki T. // J. Peptide Res. 2001. V. 58. P. 91–107.
Weckbecker G., Lewis I., Albert R., Herbert A. Schmid, Hoyer D., Bruns C. // Nature Reviews. 2003. V. 2. P. 999–1017.
Novohradsky V., Zamoria A.M., Gandioso A., Brabec V., Ruiz J., Marchan V. // Chem. Commun. 2017. V. 53. P. 5523–5526.
Srinivasarao M., Chris V. Galliford, Philip S. Low // Nature Reviews. 2015. V. 14. P. 203–219.
Балаев А.Н., Осипов В.Н., Охманович К.А., Ручко Е.А., Барышникова М.А., Хачатрян Д.С. // Известия Академии наук. Серия химическая. 2016. T. 11. C. 2766–2769.
Балаев А.Н., Осипов В.Н., Охманович К.А., Ручко Е.А., Барышникова М.А., Хачатрян Д.С. // Известия Академии наук. Серия химическая. 2016. T. 12. C. 2948–2951.
Modlin I.M., Pavel M., Kidd M., Gustafsson B.I. // Aliment Pharmacol. Ther. 2010. V. 31. P. 169–188.
Sambasivarao K., Mohammad Saifuddin, Vikas R. Aswar // Org. Biomol. Chem. 2016. V. 14. P. 9868–9873.
Kuriyama I., Miyazaki A., Tsuda Y. // Anticancer Res. 2010. V. 30. P. 4841–4849.
Nathaniel Edward Bennett Saidu, Sergio Valente, Emilie Bana, Gilbert Kirsch,Denyse Bagrel, Mathias Montenarh // Bioorganic & Medicinal Chemistry. 2012. V. 20. P. 1584–1593.
Fatma A. El-Samahy, Hayam A. Abd El Salam, Naglaa F. El-Sayed, Elsayed M. Shalaby, Mahmoud F. Dondeti // Z. Naturforsch. 2017. V. 72. P. 705–716.
Rina Soni, Shweta Umar, Nirav N. Shah, Suresh Balkrishnan, Shubhangi S. Somana // J. Heterocyclic Chem. 2017. V. 49. P. 3232–2510.
Tooba Abdizadeh, Mohammad Reza Kalani, Khalil Abnous, Zahra Tayarani-Najaran, Bibi Zahra Khashyarmanesh, Rahman Abdizadeh, Razieh Ghodsi, Farzin Hadizadeh // European Journal of Medicinal Chemistry. 2017. V. 132. P. 42–62.
Veronika Mäde, Sylvia Els-Heindl, Annette G. Beck-Sickinger // Beilstein J. Org. Chem. 2014. V. 10. P. 1197–1212.
Балаев А.Н., Осипов В.Н., Охманович К.А., Федоров В.Е., Решетников Е.В. // Российский биотерапевтический журнал. 2011. T. 4. C. 43–46.
Андреев С.М., Сидорова М.В., Ракова О.А., Цветков Д.Е., Фонина Л.А. // Биоорганическая химия. 1987. T. 13. C. 696–700.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия