

Биоорганическая химия, 2019, T. 45, № 5, стр. 545-554
Новый флуоресцентный аналог ингибитора обратного захвата дофамина GBR12909
А. В. Лаврова 1, *, Н. М. Грецкая 1, М. Г. Акимов 1, В. В. Безуглов 1
1 Институт биоорганической химии им. М.М. Шемякина и Ю.А. Овчинникова РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
* E-mail: alinalavrova1@gmail.com
Поступила в редакцию 24.10.2018
После доработки 19.12.2018
Принята к публикации 19.02.2019
Аннотация
Обратный захват дофамина из синаптического пространства осуществляет его белок-транспортер, интегрированный в пресинаптическую мембрану. Для некоторых нейродегенеративных заболеваний, в том числе для болезни Паркинсона, характерна дегенерация дофаминергических нейронов, приводящая к снижению концентрации дофамина в синапсах. Для терапии таких нейродегенеративных патологий потенциальной мишенью является транспортер дофамина. Недостаток информации о распределении и регуляции самого дофаминового транспортера делает актуальным создание молекулярных инструментов на основе его ингибиторов для проведения научных исследований. Ранее были описаны серии ингибиторов обратного захвата дофамина, наиболее распространенные из которых получены на основе производных тропана и пиперазина. Среди них эталонным является производное N-замещенного пиперазина – GBR12909, селективное по отношению к дофаминовому транспортеру, но не к транспортерам серотонина и норадреналина. Это свойство делает GBR12909 перспективным для синтеза специфически функционализированных производных на основе его структуры. Нами впервые синтезирован флуоресцентный аналог ингибитора дофаминового транспортера GBR12909 с BODIPY-FL-флуорофором. Для этого разработана модульная схема синтеза базового ядра молекулы с линкерным фрагментом, несущим защищенную аминогруппу на ее дистальном конце, позволяющую присоединять целевые функционально значимые фрагменты. На первом этапе синтеза целевого соединения получают отдельно 1-(2-(бис(4-фторфенил)метокси)этил)пиперазин и 4-(3-иодопропил)фениламид 6-(трет-бутилоксикарбонил)аминогексановой кислоты и затем “собирают” эти фрагменты в одну молекулу. После снятия защиты с аминогруппы ее ацилируют флуоресцентной BODIPY-FL-C3-кислотой. Полученный флуоресцентный аналог использовали в эксперименте по исследованию его интернализации в клетки феохромоцитомы PC12, экспрессирующие дофаминовый транспортер. На культуре этих клеток продемонстрировано специфическое накопление флуоресцентного аналога, опосредованное транспортером обратного захвата дофамина.
ВВЕДЕНИЕ
Обратный захват дофамина осуществляет трансмембранный белок-транспортер (DAT), который регулирует концентрацию нейромедиатора в синаптическом пространстве. Транспортер является представителем семейства симпортеров, осуществляющих обмен нейротрансмиттера на ионы Na+, это так называемое SLC6-семейство, или Na+/Cl–-сопряженный транспортер [1–3]. Некоторые нейродегенеративные заболевания, в том числе болезнь Паркинсона, сопровождающиеся гибелью дофаминергических нейронов, приводят к снижению концентрации дофамина в синапсах и нарушению дофаминергической регуляции. Дисфункции в работе самого транспортера также приводят к развитию ряда заболеваний, в том числе болезни Паркинсона, депрессии, наркомании. В терапии подобных заболеваний белок-транспортер дофамина является перспективной мишенью. Однако недостаток информации о структуре, регуляции и распределении DAT затрудняет создание эффективных терапевтических средств.
Как правило, при создании новых функционализированных лигандов DAT, используют тропановые ингибиторы (кокаин и его производные) [4, 5]. Однако более селективным и высокоаффинным ингибитором обратного захвата дофамина, причем без побочного психотропного эффекта, является GBR12909 (ваноксерин) [6, 7], что делает его структуру (рис. 1) привлекательной в качестве основы для создания новых молекулярных инструментов при исследовании функционирования транспортера дофамина. Ранее С.Дж. Розенталь и соавт. [8] использовали шаблон GBR12909 для синтеза высокоаффинного производного GBR12909, имеющего тиольный фрагмент для конъюгации с квантовыми точками “ядро-оболочка”.
Рис. 1.
Структуры ваноксерина (GBR12909) и его флуоресцентного аналога GBR-BODIPY. Обозначены синтоны А–D.
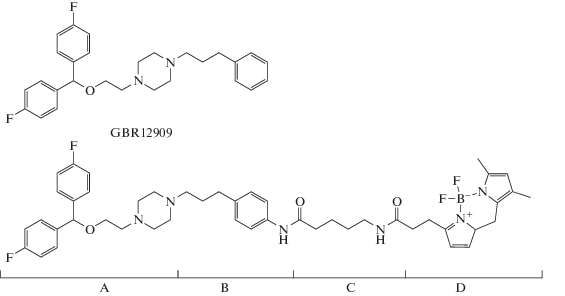
Цель данной работы – получение нового флуоресцентного производного на основе антагониста DAT – GBR12909 (рис. 1), которое станет удобным молекулярным инструментом для исследования локализации и динамики транспортера в пресинаптической мембране.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Основной проблемой синтеза аналогов ваноксерина является введение дополнительной функциональной группы в фенилпропановый домен B GBR12909 (рис. 1). В работе [8] был синтезирован аналог GBR12909, пригодный для связывания с квантовыми точками, путем введения в структуру ингибитора остатка пара-нитрофенилпропионовой кислоты вместо остатка фенилпропионовой кислоты, с последующим восстановлением нитрогруппы до аминогруппы. Трудности выделения амина на последней стадии приводили к низкому выходу целевого амино-аналога GBR12909. При этом низкая реакционная активность введенной пара-аминогруппы в планируемых реакциях присоединения требует довольно жестких условий, что в дальнейшем ограничивает введение функционализированных остатков, например производных BODIPY-флуорофора [9].
Предложенная нами схема синтеза целевого соединения предполагала первоначальный синтез домена А (1-(2-(бис(4-фторфенил)метокси)этил)пиперазин) (II) и домена BС (4-(3-иодопропил)фениламид 6-(трет-бутилоксикарбонил)аминогексановой кислоты) (VII) с последующей “сборкой” этих фрагментов в одну молекулу (рис. 1, схема 1). В зависимости от научных задач линкер C может видоизменяться. Такая последовательность синтетических стадий позволяет получить фрагмент BC, удобный для дальнейших модификаций, с приемлемым выходом.
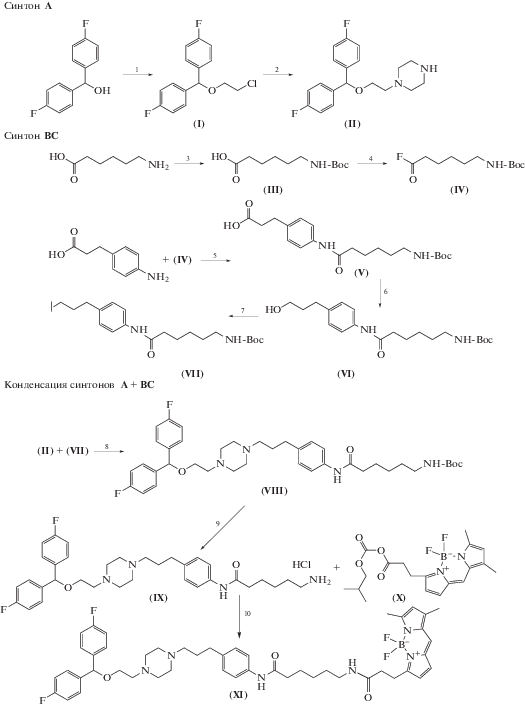
Схема 1. Синтез флуоресцентного аналога GBR-BODIPY: 1) 2-хлорэтанол, п-толуолсульфокислота, толуол, 110°С, 2 ч; 2) пиперазин, Cs2CO3, толуол; 110 °С, 4 ч; 3) Boc2O, ТEА, ацетонитрил/вода 1 : 1; 23°С, 30 мин; 4) цианурфторид, пиридин, ацетонитрил, 0°С, 2 ч; 5) ТEА, ацетонитрил/DMF 3 : 1; 23°С, 30 мин; 6) изобутилхлорформиат, ТEА, ацетонитрил; 23°С 30 мин; NaBH4, THF/вода; 0°С, 15 мин; 7) ТEА, метансульфонилхлорид, CH2Cl2, 0°С, 30 мин; KI, ацетон, 56°С, 1.5 ч; 8) толуол, 110°С, 2 ч; 9) 4 М HCl/диоксан, диоксан, 0°С, 30 мин; 10) ТEА, ацетонитрил, 23°С, 1 ч.
Для получения синтона А бис(4-фторфенил)метанол превращали в эфир (I) реакцией с хлорэтанолом в присутствии пара-толуолсульфокислоты с выходом до 96% [10]. Конденсация (2‑хлорэтокси)-бис(4-фторфенил)метан (I) с 4‑ кратным избытком пиперазина по методу [11] давала соединение (II) – (синтон А) с выходом до 60%, причем реакция проходила за 3–4 ч против 15 ч по методике работы [11]. Нами было установлено, что пролонгирование реакции не приводит к увеличению выхода продукта.
В качестве линкера С была выбрана 6-аминогексановая кислота, которую после предварительной защиты аминогруппы и превращения во фторангидрид (IV) конденсировали с 3-(4-аминофенил)пропионовой кислотой, что позволило получить производное (V) с выходом 60%. Наиболее подходящим для активации карбоксильной группы 6-Boc-аминогексановой кислоты (III) для стадии 4 оказался фторангидридный метод, тогда как в реакции с тионилхлоридом происходило частичное деблокирование аминогруппы и, как следствие, увеличение количества трудноотделяемых побочных продуктов в реакционной среде. Применение более мягких методов активации, например метода смешанных ангидридов, ограничивалось низкой реакционной способностью аминогруппы 3-(4-аминофенил)пропионовой кислоты.
Смешанный ангидрид карбоновой кислоты (V), полученный по методу [12], восстанавливали далее боргидридом натрия в спирт (VI) (стадия 6) по методу [13] с 85% выходом. Превращение спирта (VI) в галоген-производное (VII) осуществляли через промежуточный мезилат (выход 92%) по реакции с метансульфонилхлоридом [14], который после реакции с иодидом калия давал иодид (VII) или синтон ВС с выходом 54%.
Превращение спирта (VI) в иодид (VII) другими методами, например через замену активированной N,N'-карбонилдиимидазолом спиртовой группы соединения (VI) на иод кипячением в толуоле с метилиодидом в присутствии триэтиламина по методу [15] или замещением гидроксильной группы (VI) на иод в присутствии трифенилфосфина по методу [16], приводило в первом случае к деблокированию аминогруппы, а во втором – к загрязнению трудноотделимой примесью трифенилфосфиноксида и, как следствие, низким выходам (8–10%) целевого соединения (VII).
В результате конденсации соединений (II) и (VII) получали производное GBR12909 (VIII) с линкером в виде N-Boc-защищенной 6-аминогексановой кислоты, которое после деблокирования давало соединение (IX) со свободной аминогруппой. Удаление Boc-защиты раствором 4 М HCl в диоксане при комнатной температуре [17] приводило к быстрому образованию белого осадка гидрохлорида соединения (IX). Стандартный способ деблокирования Boc-защищенной аминогруппы трифторуксусной кислотой в хлористом метилене [18] в нашем случае приводил к образованию неоднородного желтого маслообразного продукта с низкой реакционной способностью. Деблокирование аналога GBR12909 (VIII) соляной кислотой в этаноле при комнатной температуре проходило не полностью, а при кипячении – приводило к полному гидролизу производного в течение 15 мин.
Далее BODIPY-FL-C3-кислоту превращали в смешанный ангидрид (X) с изобутилхлорформиатом в присутствии триэтиламина и конъюгировали с гидрохлоридом (IX) в присутствии триэтиламина с образованием флуоресцентного производного (XI) с выходом 32%. Спектр поглощения содержал максимумы, характерные для поглощения BODIPY-флуорофора (λ = = 510 нм, ε510 = 94 829 М–1 см–1) и бензольных колец остова GBR12909 (λ = 234 нм, ε234 = 27 340 М–1 см–1). Структура целевого соединения подтверждена с помощью масс-спектрометрии.
Полученный флуоресцентный аналог (XI) использовали в эксперименте по исследованию его захвата клетками феохромоцитомы PC12, которые характеризуются экспрессией DAT [19], в присутствии и в отсутствие ингибитора транспорта дофамина GBR12909 (рис. 2). Было показано накопление флуоресцентного аналога клетками феохромоцитомы РС12 (10 мин), однако при этом 30-минутная предынкубация клеток с 5 мкМ раствором ингибитора GBR12909 приводит к практически полному ингибированию накопления флуоресцентного GBR12909. Это свидетельствует о конкуренции меченого и немеченого вещества за место связывания на транспортере DAT.
Рис. 2.
Интернализация комплекса DAT-GBR-BODIPY в клетки феохромоцитомы РС12 при разных концентрациях. Время инкубации 10 мин. Увеличение 200×, масштабная линейка 100 нм.

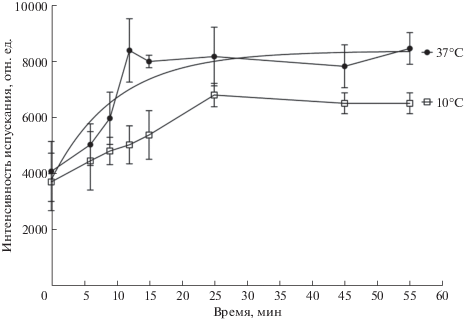
Изучена кинетика накопления флуоресцентного аналога (XI) в клетках при 37 и 10°С. При высокой температуре кривая представляла собой участок быстрого накопления вещества до 12 мин (τ1/2 = 10 ± 3 мин) с последующим выходом на плато, тогда как при понижении температуры характер кривой изменялся в сторону линейной зависимости, что характерно для ферментативного процесса (рис. 3). Из этого можно сделать вывод о том, что флуоресцентный аналог (XI) накапливается в клетках при участии белкового переносчика.
Рис. 3.
Накопление 1 мкМ конъюгата (XI) в клетках PC12 при 37 и 10°С; детектирование интенсивности испускания, среднее ± стандартное отклонение.
Для подтверждения участия транспортера в накоплении флуоресцентного аналога (XI) была проведена серия опытов по изучению концентрационной зависимости данного процесса и влияния на него ингибиторов (рис. 4). В частности, были использованы ингибиторы дофаминового транспортера GBR12909 и ингибитор накопления норадреналина гидроксибупропион (5 мкМ с предынкубацией в течение 30 мин), который при этом является слабым ингибитором накопления дофамина [20]. В этих экспериментах инкубацию с веществом проводили в течение 10 мин, основываясь на данных по кинетике накопления. В диапазоне концентраций 10–1000 нМ было зафиксировано дозозависимое увеличение флуоресценции клеток. GBR12909 полностью блокировал накопление флуоресцентного аналога (XI) в клетках; гидроксибупропион также был активен, но ингибировал захват только на 39%. Полученные данные полностью согласуются с гипотезой об интернализации соединения (XI) дофаминовым транспортером.
Рис. 4.
Накопление конъюгата (XI) в клетках PC12 в зависимости от его концентрации в отсутствие ингибиторов (а) и накопление 100 нМ конъюгата в присутствии 5 мкМ ингибиторов транспортера обратного захвата дофамина (б). Время инкубации с (XI) 10 мин, при 37°С, предынкубация с ингибиторами 30 мин. * – Статистически достоверное отличие от контроля (0 нМ), ** – статистически достоверное отличие от действия 100 нМ конъюгата (XI) без ингибиторов, дисперсионный анализ с пост-тестом Тьюки. Оценка по результатам измерения интенсивности испускания клеток при микроскопии, среднее ± среднеквадратичная ошибка.

Таким образом, нами синтезирован новый флуоресцентный аналог GBR12909 и показано специфическое накопление (интернализация) этого соединения клетками культуры феохромоцитомы РС12 через транспортер обратного захвата дофамина.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали следующие реактивы и растворители: изобутилхлорформиат (Fluka, Швеция), ацетон, ацетонитрил, диметилформамид, DMSO, иодид калия, метанол метансульфонилхлорид, пара-толуолсульфокислота, пентаоксид фосфора, тетрагидрофуран, трифторуксусная кислота, триэтиламин, толуол, хлорид натрия, хлористый метилен, хлороформ, этилацетат (Химмед, Россия), ди-трет-бутилдикарбонат, пиперазин (Acros Organic, Германия), 3-(4-аминофенил)пропионовая кислота, бычий сывороточный альбумин, GBR 12909, гидроксибупропион, D-глюкоза, пиридин, 9-флуоренилметоксикарбонилхлорид, цианурфторид, 4 М HCl в диоксане (Sigma-Aldrich, США), BODIPY-FL-C3-кислота (Invitrogen, США), карбонат цезия (Новосибирск, Россия), сульфат натрия (Merck, Германия), амфортерицин B, L-глутамин, пенициллин, раствор Версена, раствор Хэнкса, HEPES, RPMI 1640, стрептамицин, трипсин (Панэко, Россия), коровья эмбриональная сыворотка (Biosera, Франция). Очистку растворителей проводили по стандартным методикам [21]. ТСХ выполняли на пластинках Kieselgel 60 F254 (Merck, Германия) в системах хлороформ–метанол 19 : 1 (система А) и хлороформ–метанол 10 : 1 (система Б). Колоночную хроматографию осуществляли на силикагеле Silica Gel 60 (Merck, Германия). Чистоту синтезированных веществ определяли на жидкостном микроколоночном хроматографе “Милихром А-02” (“ЭкоНова”, Россия), колонка для ВЭЖХ ProntoSil 120-5-C18 AQ 5 мкм (2.0 × 75 мм) (Bischoff Leonber, Германия). Для полученных целевых и промежуточных соединений чистота составляла не менее 80%. УФ-спектры регистрировали на УФ-спектрометре Agilent Technologies Cary 60 UV-Vis (США); спектры 1Н-ЯМР (δ, м.д.) регистрировали на приборе Bruker Fourier 300 HD (Германия); масс-спектры получали на масс-спектрометре Bruker q-TOF Mass-Spectrometer Maxis Impact (Германия).
(2-Хлорэтокси)-бис(4-фторфенил)метан (I). К раствору 500 мг (2.27 ммоль) бис(4-фторфенил)метанола в 5 мл сухого толуола прибавляли 393 мкл (4.82 ммоль) хлорэтанола и 36 мг (0.20 ммоль) пара-толуолсульфокислоты и кипятили с обратным холодильником в течение 2 ч. Температуру реакционной смеси доводили до комнатной, удаляли растворитель на роторном испарителе, остаток разбавляли этилацетатом (150 мл) и последовательно промывали водой (2 × × 75 мл) и насыщенным раствором хлорида натрия (1 × 75 мл). Органическую фазу сушили безводным Na2SO4. Осушитель отфильтровывали, фильтрат упаривали. Выход 632.6 мг (97%, желтое масло). 1Н-ЯМР: 3.66–3.75 (4Н, –СН2–СН2–), 5.32–5.41 (1Н, –СН<), 7.01–7.08 (2Н, аром.), 7.28–7.34 (2Н, аром.).
1-(2-(Бис(4-фторфенил)метокси)этил)пиперазин (II). К раствору 366 мг (4.25 ммоль) безводного пиперазина в 20 мл сухого толуола прибавляли 300 мг (1.06 ммоль) соединения (I), 731 мг (2.25 ммоль) карбоната цезия, молекулярные сита А4 и кипятили с обратным холодильником в течение 4 ч. После охлаждения до комнатной температуры реакционную смесь разбавляли бензолом (100 мл), последовательно промывали водой (2 × 50 мл) и рассолом (50 мл). Органическую фазу сушили безводным Na2SO4. Осушитель отфильтровывали, фильтрат упаривали. Остаток растворяли в 2 мл хлороформа и хроматографировали на колонке с силикагелем в системе хлороформ-метанол в градиентном режиме. Выход 211 мг (60%, желтое масло). 1H-ЯМР: 2.034–2.036 (1H, NH), 2.458–2.481 (4H, пиперзин), 2.618–2.648 (2H, –CH2–), 2.869–2.892 (4H, пиперазин), 3.531–3.561 (2H, CH2), 5.333 (1H, –СН<), 6.965–7.068 (4H, аром.), 7.156–7.265 (4H, аром.).
6-(трет-Бутоксикарбонил)аминогексановая кислота (III). К раствору 1000 мг (7.6 ммоль) 6‑аминогексановой кислоты в 10 мл смеси ацетонитрил–вода (1 : 1, v/v) прибавили 660 мкл (11.44 ммоль) ди-трет-бутилкарбоната и 0.417 мл (11.44 ммоль) триэтиламина. Перемешивали при 23°C в течение 30 мин. Реакционную смесь разбавляли ацетонитрилом (90 мл) и промывали гексаном (3 × 50 мл). Слой гексана отбрасывали, а к ацетонитрилу прибавляли 30 мл воды. Упаривали ацетонитрил из фазы ацетонитрил–вода, остаток разбавляли этилацетатом (150 мл) и последовательно промывали 0.5 М раствором бисульфата натрия (50 мл), водой (2 × 75 мл) и рассолом (75 мл). Органическую фазу сушили безводным Na2SO4. Осушитель отфильтровывали, фильтрат упаривали. Выход 1357 мг (77%, белые кристаллы). 1Н-ЯМР: 1.386–1.409 (2H, CH2), 1.459 (9H, CH3), 1.495–1.518 (2H, CH2), 1.673–1.698 (2H, CH2), 2.370 (2H, CH2), 3.128 (2H, CH2).
Фторангидрид 6-(трет-бутоксикарбонил)аминогексановой кислоты (IV). К раствору 225 мг (0.97 мкл) кислоты (III) в 3 мл ацетонитрила прибавляли 134 мкл (1.65 ммоль) пиридина и 138 мкл (1.65 ммоль) цианурфторида и перемешивали при 0°С в течение 2 ч. Отфильтровывали осадок, из фильтрата упаривали ацетонитрил. Остаток разбавляли этилацетатом (75 мл) и последовательно промывали водой (2 × 30 мл) и рассолом (30 мл). Органическую фазу сушили безводным Na2SO4. Осушитель отфильтровывали, фильтрат упаривали. Выход 143 мг (63%, желтое масло).
3-(4-(6-(трет-Бутоксикарбониламино)гексанамидо)фенил)пропионовая кислота (V). К раствору 143 мг (0.61 ммоль) кислоты (IV) в 2 мл ацетонитрила прибавляли 99 мг (0.60 ммоль) 3-(пара-аминофенил)пропионовой кислоты, растворенной в 1 мл диметилформамида, и перемешивали при 23°С в течение 30 мин. Растворители удаляли в вакууме масляного насоса, остаток разбавляли этилацетатом (75 мл) и последовательно промывали водой (2 × 30 мл) и рассолом (30 мл). Органическую фазу сушили безводным Na2SO4. Осушитель отфильтровывали, фильтрат упаривали. Выход 136 мг (60% в виде коричневых кристаллов).
4-(3-Гидроксипропил)фениламид 6-(трет-бутилоксикарбонил)аминогексановой кислоты (VI). К раствору 137 мг (0.36 ммоль) кислоты (V) в 3 мл ацетонитрила прибавляли 75 мкл (0.54 ммоль) триэтиламина и 70 мкл (0.54 ммоль) изобутилхлорформиата и перемешивали при 23°С 30 мин. Ацетонитрил упаривали, остаток растворяли в этилацетате (50 мл) и последовательно промывали водой (2 × 25 мл) и рассолом (25 мл). Органическую фазу сушили безводным Na2SO4. Осушитель отфильтровывали, фильтрат упаривали. Выход 160 мг (98%, желтое масло).
К раствору 160 мг (0.35 ммоль) полученного смешанного ангидрида в 2 мл тетрагидрофуране добавляли раствор 27 мг (0.72 ммоль) боргидрида натрия в воде (10 мг в 100 мкл) и инкубировали при 0°С. Через 40 мин добавили 2 мл воды, оставляли реакционную смесь еще на 20 мин. Упаривали растворитель до половины объема, разбавляли этилацетатом (50 мл) и последовательно промывали водой (2 × 25 мл) и насыщенным раствором хлорида натрия (25 мл). Органическую фазу сушили безводным Na2SO4. Осушитель отфильтровывали, фильтрат упаривали. Выход 110 мг (85%, желтое масло). 1Н-ЯМР: 1.398 (9H, CH3), 1.493 (2H, CH2, aHxA), 1.515 (2H, CH2, aHxA), 2.351 (2H, CH2, aHxA), 2.590–2.616 (2H, CH2, phPrA11), 2.945–2.881 (2H, CH2, phPrA), 3.067 (1H, NH), 7.164–7.193 (2H, аром.), 7.594 (2H, аром.).
(4-(3-Иодопропил)фениламид 6-(трет-бутилоксикарбонил)аминогексановой кислоты) (VII). К раствору 105 мг (0.288 ммоль) (VI) в 2 мл свежеперегнанного над P2O5 хлористого метилена прибавляли 120 мкл триэтиламина (0.864 ммоль) и 33 мкл (0.432 ммоль) метансульфонилхлорида и перемешивали при охлаждении (0°С) 30 мин. Разбавляли реакционную смесь хлороформом (50 мл) и последовательно промывали водой (2 × × 25 мл) и рассолом (25 мл). Органическую фазу сушили безводным Na2SO4. Осушитель отфильтровывали, фильтрат упаривали.
К раствору 117 мг 3-(4-(6((трет-бутоксикарбонил)амино)гексанамидо)фенил)пропил метансульфоната в 2 мл ацетона прибавляли 191 мг (1.152 ммоль) иодида калия и кипятили с обратным холодильником в течение 90 мин. Удаляли растворитель на роторном испарителе, разбавляли реакционную смесь этилацетатом (50 мл) и последовательно промывали водой (2 × 25 мл) и рассолом (25 мл). Органическую фазу сушили безводным Na2SO4. Осушитель отфильтровывали, фильтрат упаривали. Остаток растворяли в 2 мл хлороформа и хроматографировали на колонке с силикагелем с элюцией хлороформом в изократическом режиме. Выход 63 мг (46%, белые кристаллы). МС, m/z: 475.15 [M + H]+, 497.1327 [M + + Na]+. 1Н-ЯМР: 1.454 (9H, CH3; 2H, СH2, aHxA), 1.520–1.544 (2H, СH2, aHxA), 1.743–1793 (2H, СH2, aHxA), 2.09–2.138 (2H, СH2, phPrA), 2.363–2.388 (2H, СH2, phPrA), 2.684–2.732 (2H, СH2, phPrA), 3.145–3.169 (4H, СH2, aHxA , СH2, phPrA), 7.144–7.163 (4H, аром.), 7.221 (1H, NH).
6-(трет-Бутилоксикарбонил)амино-N-(4-(3-(4-(2-(бис(4-фторофенил)метокси)этил)пиперазин-1-ил)пропил)фенил)гексанамид (VIII). К раствору 28 мг (0.08 ммоль) соединения (II) в 2 мл толуола прибавляли 9 мг (0.06 ммоль) карбоната калия и 10 мг (0.02 ммоль) соединения (VII) и кипятили в присутствии молекулярных сит 4 Å с обратным холодильником в течение 2 ч. Растворитель упаривали, остаток растворяли в 1 мл хлороформа и хроматографировали на колонке с силикагелем в системе хлороформ–метанол в градиентном режиме. Фракции, содержащие продукт, объединяли и упаривали, остаток сушили в вакууме. Выход 14 мг (98%, желтое масло). МС, m/z: 679.4069 [M + H]+, 701.3819 [M + Na]+.
Гидрохлорид 6-амино-N-(4-(3-(4-(2-(бис(4-фторофенил)метокси)этил)пиперазин-1-ил)пропил)фенил)гексанамида (IX). К 14 мг (0.02 ммоль) Boc-конъюгата (VIII) в 0.5 мл диоксана прибавляли 1 мл 4 М HCl в диоксане при охлаждении (0°С) и перемешивали 30 мин. Растворитель упаривали, остаток сушили в вакууме. Выход 13 мг (99%, желтое масло).
Смешанный ангидрид BODIPY-C3-кислоты и изобутилхлорформиата (X). К раствору 1 мг (0.003 ммоль) BODIPY-C3-кислоты в 1 мл ацетонитрила добавляли 47 мкл (0.03 ммоль) триэтиламина и 45 мкл (0.03 ммоль) изобутилхлорформиата и оставляли при 4°C на 10 мин. Разбавляли реакционную смесь этилацетатом (20 мл) и последовательно промывали водой (2 × 10 мл), рассолом (10 мл). Органическую фазу сушили безводным Na2SO4. Осушитель отфильтровывали, фильтрат упаривали. Выход 1 мг (75%, красные кристаллы).
Флуоресцентный аналог GBR12909 (XI). К раствору 4.2 мг (6 мкмоль) конъюгата (IX) в 0.5 мл ацетонитрила добавили 1.7 мкл (6 мкмоль) триэтиламина и 1 мг (2.5 мкмоль) смешанного ангидрида (X) и перемешивали при комнатной температуре в течение 1 ч. Растворитель упаривали, остаток растворяли в 1 мл хлороформа и хроматографировали на колонке с силикагелем в системе хлороформ–метанол в градиентном режиме. Фракции, содержащие продукт, объединяли и упаривали, остаток сушили в вакууме. Выход 0.93 мг (32%). МС, m/z: 853.476 [M + H]+, 833.478 [M – F]+.
Определение захвата флуоресцентного аналога GBR клетками феохромоцитомы PC12 Клетки культивировали в среде RPMI 1640 с добавлением 2 мМ L-глутамина, 7% эмбриональной коровьей сыворотки (FBS), 100 ед./мл пенициллина, 100 мкг/мл стрептамицина и 2.5 мкг/мл амфотерицина B при 37°С в атмосфере 5% CO2 при 95% влажности; клетки пересевали после обработки трипсином после промывки раствором Версена. Клетки высевали в 96-луночный планшет с плотностью 10 тыс. на лунку (при детектировании с помощью микроскопии) или 60 тыс. на лунку (при детектировании с помощью планшетного ридера) накануне эксперимента. Перед экспериментом среду культивирования убирали и добавляли вещества в виде раствора в среде инкубации (раствор Хэнкса с добавлением 20 мМ HEPES (pH 7.4), 1 мг/мл обезжиренного бычьего сывороточного альбумина и 10 мМ D-глюкозы). Исходные растворы веществ готовили в DMSO, финальная концентрация DMSO не превышала 0.5%. При обработке ингибиторами сначала клетки предынкубировали с ингибиторами 30 мин, затем добавляли раствор веществ и инкубировали 10 мин. Инкубацию проводили при 37°С с перемешиванием при атмосферной влажности и концентрации CO2. При изучении кинетики захвата растворы веществ для варианта инкубации при 10°С предварительно охлаждали до этой температуры для уменьшения температурных флуктуаций при добавлении. В конце инкубации клетки трижды промывали 200 мкл среды инкубации и либо фотографировали с помощью микроскопа Nikon Ti-S (куб фильтров Semrock GFP-3035D, увеличение 200×), залив 50 мкл среды инкубации, либо растворяли в 100 мкл DMSO и измеряли флуоресценцию с помощью аппарата Hidex Sense Beta Plus (λвозб. = 485 нм, λисп. = 535 нм). Каждый вариант обработки клеток был представлен в трех повторах. При количественном анализе данных микроскопии использовали программный комплекс Nikon NIS-Elements BR 4.0; для каждых условий анализировали не мене 50 клеток. Каждый эксперимент повторяли три раза. Статистическую обработку данных проводили с помощью программы GraphPad Prizm 6.0, достоверными считали отличия при p ≤ 0.05.
ФОНДОВАЯ ПОДДЕРЖКА
Работа поддержана грантом Президиума РАН “Фундаментальные исследования для биомедицинских технологий”, тема: “Создание фундаментальной основы (методологии) инновационной технологии адресной доставки лекарственных средств к дегенерирующим дофаминергическим нейронам при болезни Паркинсона”.
Список литературы
Gether U., Andersen P.H., Larsson O.M., Schousboe A. // Trends Pharmacol. Sci. 2006. V. 27. P. 375–383.
Torres G.E., Amara S.G. // Curr. Opin. Neurobiol. 2007. V. 17. P. 304–312.
Zomot E., Bendahan A., Quick M., Zhao Y., Javitch J.A., Kanner B.I. // Nature. 2007. V. 449. P. 726–730.
Eriksen J., Rasmussen G. F., Rasmussen T.N., Vaegter C.G., 1 Cha J.H., Zou M.F., Newman A.H., Gether U. // J. Neurosci. 2009. V. 29. P. 6794–6808.
Kovtun O., Tomlinson I.D., Sakrikar D.S, Chang J.C., Blakely R.D., Rosenthal S.J. // ACS Chem. Neurosci. 2011. V. 2. P. 370–378.
Andersen P.H. // Eur. J. Pharm. 1989. V. 166. P.493–504.
Izenwasser S., Werling L.L., Cox B.M. // Brain Res. 1990. V. 520. P. 303–309.
Tomlinson I.D., Mason J., Burton J.N., Blakely R., Rosenthal S.J. // Tetrahedron. 2003. V. 59. P. 8035–8047.
Tomlinson I.D., Mason J.N., Blakely R.D., Rosenthal S.J. // Bioorg. Med. Chem. Lett. 2006. V. 16. P. 4664–4667.
Buzas A., Champagnac A., Dehnel A., Lavielle G., Pommier M. // J. Med. Chem. 1980. V. 23. P. 149–153.
Lewis D.B., Matecka D., Zhang Y., Hsin L.W., Dersch C.M., Stafford D., Glowa J.R., Rothman R.B., Rice K.C. // J. Med. Chem. 1999. V. 42. P. 5029–5042.
Куклев. Д. В., Безуглов В. В. // Биоорганическая химия. 1994. T. 20. C. 679. (Kuklev, D.V., Bezuglov. V.V. // Russ. J. Bioorg. Chem. 1994. V. 20. P. 679.)
Rodriguez M., Linares M., Doulut S., Heitz A., Martinez J. // Tetrahedron Lett. 1991. V. 32. P. 923–926.
Camps P., Lozano D., Font-Bardia M. // Eur. J. Org. Chem. 2015. V. 2015. P. 5013–5020.
Kamijo T., Harada H., Iizuka K. // Chem. Pharm. Bull. 1983. V. 31. P. 4189–4192.
Rokhum L., Vanlalveni C., Lalfakzuala R. // IJETCAS. 2013. V. 4. P. 370–376.
Han G., Tamaki M., Hruby V.J. // J. Peptide Res. 2001. V. 58. P. 338–341.
Wuts P.G.M. // Greene’s Protective Groups in Organic Synthesis / Eds. Wuts P.G.M., Greene T.W. John Wiley & Sons, 2007. P. 1120.
Kadota T., Yamaai T., Saito Y., Akita Y., Kawashima S., Moroi K., Inagaki N., Kadota K. // J. Histochem. Cytochem. 1996. V. 44(9). P. 989–996.
Damaj M.I., Carroll F.I., Eaton J.B., Navarro H.A., Blough B.E., Mirza S., Lukas R.J., Martin B.R. // Mol. Pharmacol. 2004. V. 66. P. 675–682.
Гордон А., Форд Р. // Спутник химика. М.: Мир, 1976. 540 с.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия