

Биоорганическая химия, 2019, T. 45, № 6, стр. 633-639
Предварительная структурная характеристика полисахаридов, экстрагируемых из каллусной ткани стебля борщевика Сосновского Heracleum sosnowskyi Manden водным раствором оксалата аммония
Е. Н. Гордина 1, С. П. Кузнецов 2, В. В. Головченко 3, А. А. Злобин 1, *
1 Вятский государственный университет
610000 Кировская обл., Киров, ул. Московская, 36, Россия
2 ФГБУН Институт химии Коми научного центра Уральского отделения РАН
167982 Республика Коми, Сыктывкар, ул. Первомайская, 48, Россия
3 Институт физиологии Коми научного центра Уральского отделения Российской академии наук, ФИЦ Коми НЦ УрО РАН
167982 Республика Коми, Сыктывкар, ул. Первомайская, 50, Россия
* E-mail: vgtu.aua@yandex.ru
Поступила в редакцию 11.06.2019
После доработки 14.06.2019
Принята к публикации 23.06.2019
Аннотация
Из каллусной ткани стебля борщевика Сосновского Heracleum sosnowskyi Manden выделены и охарактеризованы полисахариды, экстрагируемые 0.7%-ным водным раствором оксалата аммония. Показано, что они представлены пектиновыми полисахаридами, которые отличаются от пектиновых полисахаридов нативного растения меньшим содержанием остатков D-галактуроновой кислоты и более высоким содержанием остатков арабинозы и галактозы. Результаты, полученные сочетанием методов ионообменной хроматографии, ферментативного гидролиза, периодатного окисления, метилирования и спектроскопии ЯМР, указывают на присутствие в пектиновых полисахаридах каллуса участков низкоэтерифицированного метил-1,4-α-D-галактуронана и рамногалактуронана-I, а также арабинана и галактана или арибиногалактана, в составе углеводных цепей которых идентифицированы терминальные, 1,2- и 1,5-связанные остатки арабинофуранозы, терминальные, 1,3-, 1,6-связанные и 3,6-замещенные остатки галактопиранозы и 1,4-связанные остатки гексопиранозы (галактопиранозы или глюкопиранозы). Присутствующие в спектрах ЯМР транс-гликозильные корреляционные пики метильных групп 2,4-ди-О-замещенных остатков рамнозы, косвенно свидетельствуют о присоединении боковых арабинановых и галактановых углеводных цепей к остаткам рамнопиранозы главной углеводной цепи по С4-положению.
ВВЕДЕНИЕ
Ранее [1, 2] было показано, что пектиновые полисахариды, экстрагируемые из надземной части борщевика Сосновского Heracleum sosnowskyi Manden водным раствором оксалата аммония, включающие участки частично этерифицированного метил-1,4-α-галактуронана и разветвленного рамногалактуронана-I (RG-I) с боковыми цепями из 1,4- и 1,6-связанных остатков β-D-галактопиранозы, оказывают стимулирующее действие на рост проростков и корней Triticum aestivum L.
Известно, что биологическая активность пектиновых полисахаридов определяется макромолекулярными характеристиками, а также составом и строением углеводных цепей этих биополимеров, которые зависят от вида, стадии развития, условий произрастания растения, а также части растения, из которой они экстрагируются [3]. С этой точки зрения в отличие от нативных растений культуры дифференцированных растительных клеток и тканей благодаря отсутствию организменного контроля и независимости от почвенно-климатических условий могут быть использованы для синтеза пектиновых полисахаридов с относительно постоянными характеристиками. Более того, изменяя условия культивирования, в клеточных стенках растений in vivo можно добиться модификации структуры углеводных цепей синтезируемых полисахаридов и синтеза полисахаридов с требуемой структурой, обладающих определенным спектром биологической активности [4–6].
Цель данной работы – выделение и предварительная структурно-химическая характеристика пектиновых полисахаридов каллусной ткани стебля борщевика Сосновского.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Экстракцию полисахаридов проводили согласно ранее описанной методики [4]. Для этого лиофильно высушенную каллусную ткань стебля борщевика подвергали последовательной исчерпывающей экстракции сначала дистиллированной водой (фракция водорастворимых полисахаридов HScI), затем, после гидролиза протопектинового комплекса клеточных стенок разбавленным раствором хлористоводородной кислоты (рН 3.8–4.0), – 0.7%-ным водным раствором оксалата аммония (фракция HScII).
С помощью хроматомасс-спектрометрии (ГЖХ-МС) триметилсилиловых эфиров моносахаридов, после полного гидролиза полисахаридов 2 М раствором трифторуксусной кислоты (TFA), в составе фракции HScI из гликуроновых кислот (GliA) идентифицированы остатки глюкуроновой и галактуроновой кислот, а в составе HScII – остатки только галактуроновой кислоты. Результаты, полученные при определении содержания остатков нейтральных моносахаридов в полисахаридах полученных фракций с помощью ГЖХ-МС в виде перацетатов полиолов соответствующих моносахаридов, свидетельствуют о том, что главными моносахаридными остатками в HScI являются остатки галактозы (Gal), арабинозы (Ara), глюкозы (Glc) и ксилозы (Xyl), в HScII – остатки арабинозы и галактозы (табл. 1). Все полученные фракции полисахаридов включают менее 1% остатков маннозы (Man).
Таблица 1.
Выход и состав фракций полисахаридов каллуса борщевика*
| Фракция | Выход, % | Содержание, % | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MeO1 | GliA2 | Ara2 | Gal2 | Rha2 | Xyl2 | Glс2 | ∑ (мс)1 | ||
| HScI | 3.43 | 1.0 | 26.6 | 20.8 | 37.0 | 0.6 | 6.3 | 8.7 | 68.2 |
| HScII | 2.33 | 2.3 | 65.7 | 20.9 | 7.8 | 2.6 | 1.5 | 1.5 | 84.0 |
| HSсI-u | 72.84 | 0.7 | 28.0 | 18.0 | 38.7 | 2.4 | 5.2 | 7.7 | 66.2 |
| HSсII-u | 41.64 | 2.4 | 67.3 | 14.9 | 11.4 | 3.2 | 1.6 | 1.6 | 84.4 |
| HSсII-u-f | 66.74 (48.5) | 3.0 | 53.6 | 24.8 | 13.7 | 4.6 | 1.3 | 2.0 | 82.7 |
| HSсII-u-1d | 30.64 (22.2) | 2.7 | 70.1 | 17.1 | 7.7 | 2.0 | 1.4 | 1.7 | 84.5 |
| HSсII-u-2d | 26.04 (18.9) | 2.1 | 76.0 | 12.7 | 4.2 | 3.1 | 1.3 | 2.7 | 83.4 |
| HSсII-u-s | 15.14 (10.9) | Н.о. | 34.3 | 44.8 | 14.7 | 5.3 | 0.1 | 0.8 | 85.1 |
Как было показано ранее [1, 2, 7, 8], из надземной части борщевика Сосновского водой экстрагируются главным образом протеогликаны с арабиногалактанами в углеводной части молекулы, а водным раствором оксалата аммония в условиях, аналогичных использованным в данной работе, – пектиновые полисахариды с незначительным содержанием арабиногалактана.
Моносахаридный состав фракции водорастворимых полисахаридов каллуса свидетельствует о наличии в HScI арабинанов и/или арабиногалактанов, типичных для культур каллусных тканей растений. Следует отметить, что из каллусной ткани экстракцией 0.7%-ным водным оксалатом аммония получены полисахариды, со значительно более низким выходом, чем из нативного растения. Кроме того, они отличаются более высоким содержанием остатков нейтральных моносахаридов (арабинозы и галактозы) и меньшим содержанием остатков галактуроновой кислоты. Такое “переключение” на биосинтез нейтральных арабиногалактанов показано ранее для полисахаридов каллусной ткани смолевки татарской Silene tatarica L. и апиогалактуронана каллуса ряски малой Lemna minor L. [9].
Для удаления низкомолекулярных фрагментов полисахариды фракций HScI и HScII подвергали ультрафильтрации на мембранах с размером пор, обеспечивающих транспорт молекул со средневесовой молекулярной массой (Mw) 100 кДа. В результате получены фракции HSсI-u и HSсII-u, состав которых практически идентичен составу соответствующих исходных полисахаридов (табл. 1).
При фракционировании HSсII-u на DEAE-целлюлозе (Cl–-форма) получены две близкие по моносахаридному составу фракции HSсII-u-1d и HSсII-u-2d, элюирующиеся 0.2 и 0.3 М растворами NaCl соответственно (табл. 1).
Все полученные фракции содержат до 5% белковых компонентов, которые не отделяются в ходе ультрафильтрации и ионообменной хроматографии. Вероятно, белки образуют с полисахаридами прочные комплексы или ковалентно связаны с ними.
При ферментативном гидролизе HSсII-u 1,4-α-эндо-полигалактуроназой Rhizopus sp. была получена устойчивая к действию фермента фракция HSсII-u-f, содержащая 4.3% белков. Образование свободной галактуроновой кислоты, идентифицированной в ферментолизате методом ГЖХ-МС в виде Me3Si-производных, свидетельствует о наличии в составе HSсII-u участков низкоэтерифицированного метил-1,4-α-D-галактопиранозилуронана. Моносахаридный состав полученной фракции HSсII-u, а также повышенное содержание в ней остатков рамнозы (Rha) характерны для пектиновых полисахаридов, содержащих участки RG-I [3].
Среди продуктов метилирования HSсII-u по методу Хакамори [10] идентифицированы производные 2,3-ди-О-метил-, 3,5-ди-О-метил- и 2,3,5-три-О-метил-арабинозы, 2,4-ди-О-метил-, 2,4,6-три-О-метил- и 2,3,4-три-О-метил-, 2,3,4,6-тетра-О-метил-галактозы, 2,3,6-три-О-метилгексозы (галактозы или глюкозы), 2,3,4,6-тетра-О-метил-глюкозы, а также 2,3,4-три-О-метил-ксилозы. Полученные результаты указывают на присутствие в составе HSсII-u терминальных, 1,2- и 1,5-связанных остатков арабинофуранозы, терминальных, 1,3-, 1,6-связанных и 3,6-замещенных остатков галактопиранозы, 1,4-связанных остатков гексопиранозы (галактопиранозы или глюкопиранозы), терминальных остатков глюкозы, а также терминальных остатков ксилозы.
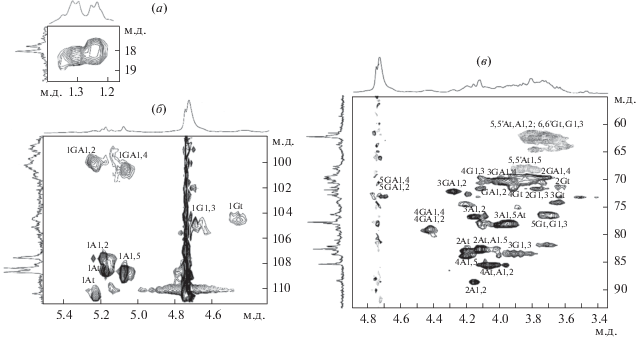
Для частичного разрушения гликозидных связей в полисахариде HSсII-u и получения образца с меньшей молекулярной массой, хорошо растворимого в воде, использовали периодатное окисление по Смиту [11]. В результате была получена фракция HSсII-u-s с Mw – 26.7 кДа и степенью полидисперсности (Mw/Mn) – 2.6, отличающаяся от исходной фракции меньшим содержанием остатков D-галактуроновой кислоты и бóльшим содержанием остатков нейтральных моносахаридов (табл. 1).
Для выявления структурных элементов и типов гликозидных связей в полисахариде HSсII-u-s использовали спектроскопию ЯМР. Значения сигналов углерода и протонов были установлены с использованием двумерных гомоядерных экспериментов 1H/1H COSY, ROESY, все соответствующие им корреляционные пики были идентифицированы в 1H/13C HSQC (рис. 1в, табл. 2).
Таблица 2.
Химические сдвиги сигналов в 1H и 13С спектрах ЯМР фрагмента HSсII-u-s
| Остатки | Химические сдвиги, м.д. (δC ацетон 31.45 м.д.; δH ацетон 2.225 м.д.) | |||||
|---|---|---|---|---|---|---|
| C1 H1 |
C2 H2 |
C3 H3 |
C4 H4 |
C5 H5; H5' |
C6 H6 |
|
| →4)-α‑D-GalpA‑(1→2 (GA1,2) | 99.77 5.25 |
70.32 3.91 |
71.84 4.27 |
78.95 4.42 |
72.85 4.70 |
176.73 |
| →4)‑α‑D-GalpA‑(1→4 (GA1,4) | 100.78 5.08 |
69.81 3.75 |
70.32 3.99 |
78.95 4.42 |
72.85 4.80 |
176.20 |
| β-Galp-(1→ (Gt) | 104.34 4.47 |
71.83 3.62 |
73.87 3.65 |
70.32 3.92 |
76.41 3.70 |
62.70 3.77; 3.77 |
| β-Galp-(1→ (G1,3) | 104.85 4.67 |
71.83 3.76 |
83.52 3.88 |
70.32 4.11 |
76.41 3.70 |
62.70 3.77; 3.77 |
| →2)-α-Araf-(1→ (A1,2) | 107.79 5.19 |
88.60 4.14 |
76.41 4.14 |
85.55 4.06 |
62.70 3.84; 3.73 |
|
| α‑Araf-(1→ (Аt) | 108.40 5.17 |
82.50 4.12 |
77.93 3.94 |
85.55 4.06 |
62.70 3.84; 3.73 |
|
| →5)-α‑Araf-(1→ (A1,5) | 108.91 5.08 |
82.50 4.12 |
78.44 4.00 |
83.52 4.20 |
67.78 3.87; 3.80 |
|
| α‑Araf-(1→ (Аt) | 110.43 5.24 |
83.01 4.19 |
77.93 3.94 |
85.55 4.06 |
62.70 3.84; 3.73 |
|
Аномерная область 13C-спектра (рис. 1б) содержит восемь сигналов различной интегральной интенсивности, два из которых сильно уширены. В 1H/13C HSQC-спектре HSсII-u-s присутствуют соответствующие этим сигналам транс‑гликозильные корреляционные пики. Сигналы при δC/H 99.77/5.25 и 100.79/5.07 м.д. были идентифицированы как соответствующие C1/Н1 остаткам галактуроновой кислоты в фрагментах →4)-GalpA-(1→4)-GalpA-(1→ (остатки обозначены GA1,4) и →4)-GalpA-(1→2)-Rhap-(1→ (остатки обозначены GA1,2); сигнал при δC/H 104.34/4.47 м.д. идентифицирован как соответствующий терминальным остаткам галактозы (остатки обозначены Gt), при δC/H 104.85/4.67 – как соответствующий 1,3-связанным остаткам галактозы (остатки обозначены G1,3), при δC/H 107.9/5.19; 108.91/5.07; 108.40/5.17; 110.43/5.22, м.д. – как соответствующие остаткам арабинозы: 1,2- (остатки обозначены A1,2), 1,5-связанным (остатки обозначены A1,5) и двум различным терминальным (остатки обозначены Аt) (рис. 1б, табл. 2).
Положение сигналов Н2 для каждого остатка, за исключением остатков галактозы, было установлено из корреляции с аномерным протоном в COSY-спектре. Для остатков арабинозы и галактуроновой кислоты GalA1,4 положение сигналов всех протонов было определено последовательно из COSY-спектра. Низкая интенсивность сигнала аномерного протона остатка галактозы и перекрывание с более интенсивными сигналами не позволила установить точное значение протонов. Для остатков GalA1,2 в COSY-спектре была установлена корреляция только между Н1 и Н2. Положения сигналов протонов и атомов углерода для этих остатков были определены по аналогии с ранее опубликованными данными [12, 13].
В ROESY-спектре были идентифицированы только транс‑гликозильные корреляционные пики между отдельными протонами в некоторых остатках арабинозы.
В 1H/13C HSQC-спектре полисахарида HSсII-u-s присутствуют транс‑гликозильные корреляционные пики при δ 18.01/1.25 м.д. и δ 18.52/1.30 м.д., характерные для атомов углерода и протонов метильных групп 1,2‑связанных и 2,4-ди-О-замещенных остатков α‑рамнозы соответственно (рис. 1a). Другие C/H корреляции для остатков рамнозы в спектрах не были обнаружены, что вероятно связано с их низким содержанием или с высокой степенью разветвленности полученного фрагмента. Наличие в спектрах сигналов атомов углерода и протонов 1,2-связанных и 2,4-ди‑О‑замещенных остатков α‑рамнозы и остатков GA1,2 указывает на присутствие в составе пектина участков RG-I. К сожалению, из-за наложения сигналов аномерных протонов остатков галактуроновой кислоты и арабинозы в спектрах ЯМР не удалось оценить соотношение остатков GA1,4 и GA1,2. Учитывая моносахаридный состав и мольные соотношения остатков GalpA : Rhap и Σ(Araf + Galp) : Rhap, которые cоставляют примерно 7:1 и 11:1 соответственно, можно заключить, что в HSсII-u-s каждый седьмой остаток галактуроновой кислоты связан с остатком рамнозы и на него приходится по одиннадцать остатков нейтральных моносахаридов. Полученные данные свидетельствуют о том, что нейтральные боковые цепи в RG-I HSсII-u-s образованы 1,2- и 1,5-связанными остатками арабинозы и 1,3-связанными остатками галактозы.
Сравнение полученных и ранее опубликованных данных указывает на существенные структурные различия пектиновых полисахаридов, синтезируемых нативным растением H. sosnowskyi и каллусной культурой. Идентифицированные в полисахариде HSсII-u-s остатки арабинозы и галактозы могут являться как элементами нейтральных полисахаридов, так и боковых цепей RG-I. В данном исследовании, равно как и во всех предыдущих, наличия ковалентной связи между остатками главной углеводной цепи и боковыми цепями показано не было, и этот вопрос до настоящего времени обстается дискуссионным.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Растительное сырье. Каллусную ткань стебля H. sosnowskyi Manden культивировали в темноте при 26°С на питательной среде Мурасиге-Скуга [4] с добавлением витаминов и фитогормонов (мг/л): В1 – 1.0; В2 – 0.5; В3 – 2.0; В5 (пантотената кальция) – 1.0; В6 – 1.0; В7 – 1.0; В9 – 0.5; В12 – 0.0015; 2,4-дихлорфеноксиуксусной кислоты – 2.0 и 6-бензиламинопурина – 0.1. Длительность культивирования – 21 сут.
Общие методы. Растворы упаривали под вакуумом при 40–50°С на роторном испарителе IKA HB 10 basic (Германия). Центрифугирование растворов проводили при 3000–9000 об./мин в течение 10–30 мин на центрифуге Sigma 2-16 PK (Германия). Для термостатирования проб использовали водяную баню LB-200 (Россия) и суховоздушный термостат Thelco 6559 (США). Растворы диализовали с помощью диализных пленок (Cellu⋅Sep H1, Бельгия) с отсекаемыми молекулярными массами 1–8 кДа. Для высушивания растворов образцов использовали лиофильную сушилку ALPHA 2-4LD plus (Германия).
Количественное определение гликуроновых кислот проводили реакцией с 3,5-диметилфенолом (≥99%, Sigma-Aldrich, США) [14] по калибровочному графику для D-галактопиранозилуроновой кислоты (≥97%, Sigma-Aldrich, США), белка – по методу [15] с калибровочным графиком для бычьего сывороточного альбумина (≥96%, Sigma-Aldrich, США), метоксильных групп – по методу [16]. Для спектрофотометрических измерений использовали спектрофотометр Shimadzu UV-mini 1240 (Япония).
Идентификацию и количественное определение моносахаридов проводили ГЖХ-МС частично ацетилированных производных, триметилсилиловых эфиров моносахаридов и перацетатов полиолов [17] на газовом хроматографе G2589A (Agilent Tech., США) с капиллярной колонкой HP-5MS (0.25 мм × 30 м) (Hewlett-Packard, США). Газ-носитель – гелий (1 мл/мин; деление потока 20 : 1). Температура испарителя – 250°С. Температурные режимы термостата колонки: триметилсилиловые эфиры и перацетаты полиолов – 175→250°С (градиент – 3°С/мин); ацетаты частично метилированных моносахаридов – 130→250°С (градиент – 5°С/мин). Масс-спектрометр 5973 INERT (Agilent Tech., США): температура интерфейса – 230°С; развертка – от m/z 44 до m/z 550; энергия ионизирующих электронов – 70 эВ; частота сканирования – 1 скан/с.
Количественное содержание моносахаридных остатков рассчитывали с использованием молярных коэффициентов и мио-инозита (≥99%, Sigma-Aldrich, США) в качестве внутреннего стандарта.
ЯМР-спектроскопия. Полисахарид HSсII-u-s (25 мг) растворяли в 600 мкл D2O и лиофилизовали, процедуру повторяли три раза, затем образец растворяли в 500 мкл D2O, к полученному раствору добавляли 0.5 мкл ацетона (≥99.9, Sigma Aldrich, США) в качестве внутреннего стандарта (δн 2.225 м.д., δс 31.45 м.д.). Все гомо- и гетероядерные спектры ЯМР снимали на спектрометре Bruker DRX Avance II 300 (Германия) при температуре зонда, обеспечивающей минимальное перекрытие сигнала D2O с сигналами полимера (313 К). Отнесение сигналов в 1Н и 13С спектрах ЯМР осуществляли на основании данных двумерных гомоядерных (1Н/1Н COSY, ROESY) и гетероядерных (1Н/13С HSQC) экспериментов. Для проведения 1Н/1Н-экспериментов использовали стандартные методики фирмы “Bruker”. Эксперименты ROESY выполняли со временем смешивания 200 мс. Положения замещения и последовательность моносахаридных остатков устанавливали с помощью 2D-экспериментов по определению ядерных эффектов Оверхаузера (ROESY).
Определение молекулярно-массовых характеристик. Образец полисахарида HSсII-u-s (3.0 мг) растворяли в 1 мл 0.15 М NaCl, содержащего 0.02% NaN3, фильтровали и использовали для измерения. Молекулярно-массовые характеристики полисахаридов определяли с помощью хроматографической системы, состоящей из насоса LC-20AD, термостата CTO-10AS и рефрактометрического детектора RID-10A (Shimadzu, Япония) с использованием предколонки Shodex GS-2G 7B (7.6 мм × 5.0 см) и колонки Shodex OH-pak SB-804 HQ (7.6 мм × 30 см) (Shimadzu, Япония). Режим разделения: температура – 40°C; скорость подачи элюента (0.15 M раствор NaCl, содержащий 0.02% NaN3) – 0.3 мл/мин. Средневесовую молекулярную (Mw) и среднечисловую (Mn) молекулярные массы рассчитывали с помощью программы LCsolutionGPC (LCsolution, версия 1.24 SP1). Для калибровки колонки использовали пуллуланы (Fluka, Германия) с Mw (кДа): 1; 3; 6; 12; 22; 50; 110; 200; 400; 800.
Метилирование. Метилирование пектиновых полисахаридов каллуса проводили по методу Хакамори [10] с последующим гидролизом 2 М раствором TFA (99%, Acros organics, CША) при 100°С и переводом частично метилированных моносахаридов в ацетаты или триметилсилильные производные и анализом методом ГЖХ-МС.
Окисление по Смиту [11]. Окисление полисахарида HSсII-u (272.6 мг) 0.03 М раствором периодата натрия (200 мл) проводили при 4°С в темноте до прекращения изменения оптического поглощения раствора при 305 нм. К раствору приливали 10 мл 10% раствора этиленгликоля, выдерживали при комнатной температуре в течение 1 ч, добавляли 1 г боргидрида натрия и оставляли на ночь для восстановления. Избыток боргидрида натрия разрушали добавлением концентрированной уксусной кислоты. Раствор диализовали и лиофильно высушивали. К полученному полиспирту (196.4 мг) приливали 100 мл 2% уксусной кислоты и проводили гидролиз при 100°С в течение 2 ч. Гидролизат упаривали до объема 3 мл и наносили на колонку (46 × 2.2 см) с сефадексом G-25 (Рharmacia Fine Chemicals, Швеция). В качестве элюента использовали дистиллированную воду (объемный расход – 20 мл/ч). Выход углеводов из колонки контролировали качественной реакцией по методу [18]. Элюат, содержащий продукты окисления, лиофилизовали. В результате получили фракцию HSсII-u-s (41.2 мг).
Выделение полисахаридов. Для выделения полисахаридов лиофильно высушенную каллусную ткань (36.79 г) обрабатывали кипящей смесью хлороформа и метилового спирта (2 : 1 по объему). Последовательную истощающую экстракцию полисахаридов [4] проводили дистиллированной водой при 68°С и 0.7% водным раствором оксалата аммония при 68°С после обработки остатка материала разбавленным раствором HCl (50°С, 1 ч) при рН 3.8–4.0. Полноту экстракции полисахаридов из ткани контролировали качественной реакцией по фенол-сернокислотному методу [18]. Экстракты упаривали, диализовали, осаждали полисахариды добавлением 4-х кратного объема 96% этилового спирта, растворяли в дистиллированной воде и лиофилизовали. В результате получили фракцию водорастворимых полисахаридов НSсI (1248.0 мг) и фракции пектиновых полисахаридов НSсII (848.0 мг).
Ультрафильтрация. Ультрафильтрацию растворов полисахаридов HScI (885.6 мг) и HScII (830.0) проводили с помощью мембраны Vivacell 250, ПЭС (Владисарт, Россия) с отсекаемой Mw 100 кДа. Концентрированные растворы лиофильно высушивали. В результате получили фракции фракции HSI-u (644.7 мг) и HSII-u (345.3 мг) соответственно.
Ионообменная хроматография. Образец полисахарида HSсII-u (29.7 мг) растворяли в 5 мл 0.01 М раствора NaCl и наносили на колонку (37 × × 1.5 см) с DEAE-целлюлозой (Fluka, Швеция) в Cl–-форме. Ступенчатое элюирование проводили 0.01–0.5 М растворами NaCl (объемный расход элюента – 30 мл/ч). Выход фракций полисахаридов из колонки контролировали качественной реакцией по методу Смита [18]. В результате главные фракции полисахаридов, элюируемые 0.2 и 0.3 М растворами NaCl, соответствующие отдельным пикам на выходных кривых для каждого элюента, собирали отдельно, концентрировали, диализовали и лиофилизовали. В результате получили HSсII-u-1d (9.1 мг) и HSсII-u-2d (7.9 мг).
Полный кислотный гидролиз. Для определения моносахаридного состава к навеске полисахаридов (1–3 мг) приливали 1 мл 2 М раствора TFA, содержащей в качестве внутреннего стандарта мио-инозит (0.1 мг/мл), и выдерживали в запаянных ампулах при 100°С в течение 4–5 ч. Кислоту удаляли многократным упариванием досуха с метиловым спиртом. Моносахариды определяли в виде соответствующих производных методом ГЖХ-МС.
Ферментативный гидролиз. Навеску полисахарида HSсII-u (3.8 мг) растворяли в 29 мл дистиллированной воды и добавляли 1 мл раствора, содержащего 25 мг α-1,4-эндо-полигалактуроназы Rhizopus sp. (620 U/G; Sigma, США). Ферментативный гидролиз проводили при комнатной температуре до прекращения увеличения концентрации восстанавливающих сахаров в растворе, которую определяли по методу Шомодьи-Нельсона [19]. Фермент инактивировали нагреванием на водяной бане при 100°С в течение 10 мин. Раствор центрифугировали и осаждали продукты ферментативного гидролиза 4-х кратным объемом 96% этилового спирта. Полученный осадок промывали 96% этиловым спиртом, растворяли в дист. воде и лиофилизовали. В результате получили фракцию HSсII-u-f (25.9 мг). Идентификацию моносахаридов в ферментолизате проводили в виде Me3Si-производных методом ГЖХ-МС.
Список литературы
Shakhmatov E.A., Mikhailova E.A., Makarova E.N. // Chem. Var. Raw Materials. 2015. № 4. P. 15–22.
Shakhmatov E.G., Atukmaev K.V., Makarova E.N. // Carbohydr. Polym. 2016. V. 136. P. 1358–1369.
Ovodov Yu.S. // Bioorganic Chemistry. 2009. V. 35. № 3. P. 293–310.
Gunter E.A., Popeiko O.V., Ovodov Yu.S. // Biochemistry. 2007. V. 72. № 9. P. 1238–1247.
Geshi N., Jorgensen B., Scheller H.V., Ulvskov P. // I. Planta. 2000. V. 210. P. 622–629.
Rubery P.H., Northcote D.H. // Biochim. Biophys. Acta. 1970. V. 222. P. 95–108.
Shakhmatov E.G., Toukach P.V., Kuznetsov S.P., Makarova E.N. // Carbohydr. Polym. 2014. V. 102. P. 521–528.
Makarova E.N., Shakhmatov E.G., Belyy V.A. // Carbohydr. Polym. 2016. V. 153. P. 66–77.
Gunter E.A. // Chem. Var. Raw Materials. 2001. № 2. P. 63–67.
Hakomori S. // J. Biochem. 1964. V. 55. № 2. P. 205–208.
Goldstein I.J., Hamilton J.K., Montgomery R., Smith F. // J. Am. Chem. Soc. 1957. V. 79. P. 6469.
Polle A.Ya., Ovodova R.G., Shashkov A.S., Ovodov Yu.S. // Carbohydr. Polym. 2002. V. 49. P. 337–344.
Khramova D.S., Golovchenko V.V., Shashkov A.S., Otgonbayar D., Chimidsogzol A., Ovodov Yu.S. // Food Chemistry. 2011. V. 126. P. 870–877.
Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. // J. Biol. Chem. 1951. V. 193. P. 265–275.
Usov A.I., Bilan M.I., Klochkova N.G. // Bot. Marina. 1995. V. 38. P. 43–51.
Wood P.J., Siddiqui I.R. // Analyt. Biochem. 1971. V. 39. P. 418–428.
York W.S., Darvil A.G., McNeil M., Stevenson T.T. // Meth. Enzymol. 1986. V. 118. P. 3–40.
Dubois M., Gilles K.A., Hamilton J.K., Rebers P.A., Smith F. // Analyt. Chem. 1956. V. 28. P. 350–356.
Sinitsyn A.P., Chernoglazov V.M., Gusakov A.V. // M.: VINITI, 1990. V. 25. P. 30–37.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия