

Биоорганическая химия, 2019, T. 45, № 6, стр. 588-614
С-метилированные аналоги спермина и спермидина: синтез и биологическая активность
М. А. Хомутов 1, И. В. Михура 2, С. Н. Кочетков 1, А. Р. Хомутов 1, *
1 Институт молекулярной биологии им. В.А. Энгельгардта РАН
119991 Москва, ул. Вавилова, 32, Россия
2 ФГБУН Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
117997 г. Москва, ул. Миклухо-Маклая 16/10, Россия
* E-mail: alexkhom@list.ru
Поступила в редакцию 10.04.2019
После доработки 25.04.2019
Принята к публикации 13.05.2019
Аннотация
Биогенные полиамины спермин и спермидин присутствуют в клетках эукариот в микро- и миллимолярных концентрациях, что определяет множественность их функций и необходимость для поддержания нормального роста клеток. Аналоги и производные спермина и спермидина широко используются в биохимии полиаминов, с их помощью получен ряд принципиально значимых для области результатов. С-метилированные аналоги полиаминов представляют собой особую группу соединений, так как среди этих веществ найдены функционально активные in vitro и in vivo миметики спермидина и спермина, биохимические свойства которых можно регулировать, перемещая метильную группу по скелету полиамина и/или, изменяя конфигурацию хирального центра. Рассматриваются особенности взаимодействия С-метилированных аналогов полиаминов с ферментами их метаболизма, активность в культуре клеток и обсуждаются методы синтеза этих соединений.
СОДЕРЖАНИЕ
1. Введение
2. Биологическая активность С-метилированных аналогов спермина и спермидина
3. Синтезы С-метилированных аналогов спермина и спермидина
3.1. Методы избирательной защиты первичных и вторичных аминогрупп в химии полиаминов
3.2. Синтезы С-метилированных аналогов полиаминов в растворе
3.2.1. Методы алкилирования
3.2.2. Присоединение по Михаэлю и восстановление нитрилов
3.2.3. Реакция Мицунобу
3.2.4. Восстановление амидов, азидов, оснований Шиффа
4. Заключение
1. ВВЕДЕНИЕ
Биогенные полиамины спермин (1,12-диамино-4,9-диазадодекан, Spm), спермидин (1,8-диамино-4-азаоктан, Spd) и их предшественник путресцин (1,4-диаминобутан, Put) присутствуют в клетках эукариот в микро- и миллимолярных концентрациях, участвуя в регуляции дифференцировки и нормального роста клеток [1, 2]. Высокое внутриклеточное содержание легко взаимопревращающихся и частично взаимозаменяемых Spm и Spd a priori определяет множественность и разнообразие их клеточных функций, многие из которых на молекулярном уровне остаются все еще малоизученными, что и обеспечивает поступательное развитие этой области биохимии [3]. Клеточные функции Spm и Spd, протонированных при физиологическом значении рН, и их природных аналогов описываются, как правило, в терминах специфического взаимодействия с отрицательно заряженными макромолекулами, нуклеопротеидными комплексами, участками мембран и рецепторов. Spm и Spd служат природными регуляторами топоизомераз, рестриктаз, а также ферментов биосинтеза ДНК и РНК [4]. Полиамины участвуют в регуляции транспорта ионов Са2+ в митохондрии [5], являются модуляторами N-метил-D-аспартатного рецептора [6] и эффекторами транспорта ионов K+ [7].
В растениях, бактериях и грибах спектр полиаминов более разнообразен и включает в себя нор-Spd (1,7-диамино-4-азагептан) и гомо-Spd (1,9-диамино-4-азанонан), 1,3-диаминопропан, агматин ((4-аминобутил)гуанидин), кадаверин (1,5-диаминопентан) и даже разветвленные полиамины, тогда как Spm во многих случаях отсутствует [8]. В E. coli около 90% всего Spd и около 50% всего Put связаны с РНК и являются важными элементами ее архитектуры, причем концентрации этих полиаминов в E. coli составляют около 7 и 30 мM, соответственно. Полиамины способствуют правильной сборке 30S-субъединиц рибосом E. coli и, тем самым, увеличивают синтез белка в 1.5–2.0 раза. Более того, Spd стимулирует синтез 20 различных белков на уровне трансляции (гены, кодирующие эти белки, объединяются в так называемый “polyamine modulon”), которые активно регулируют рост и жизнеспособность клеток [9]. Уменьшение концентрации Spm и Spd приводит к торможению роста или даже гибели клеток. Все это позволяет рассматривать эти полимины в качестве низкомолекулярных регуляторов клеточного метаболизма.
В дополнение к другим клеточным функциям, Spd служит еще и единственным донором аминобутильного остатка в биосинтезе аминокислоты гипузин {N-ε-(4-амино-2-гидроксибутил)-L-лизин, Hyp}, образующейся в результате двухстадийной посттрансляционной модификации ε-аминогруппы Lys-50 в высококонсервативном участке эукариотического фактора инициации трансляции (eIF-5A). Гипузинилированный eIF-5A (eIF5AHyp) необходим для трансляции пролин-богатых пептидов [10], его биосинтез идет постоянно и нарушается лишь при практически полном истощении внутриклеточного пула Spd [11]. Именно поэтому Spd, а не Spm, играет ключевую роль в поддержке роста клеток с хроническим дефицитом полиаминов.
В отличие от Spd, подобные уникальные функции на уровне культур клеток для Spm не найдены. Делеция гена сперминсинтазы (SMS) или ингибирование фермента существенно не влияют на рост клеток, а лишь изменяют их восприимчивость к лекарствам [12–14], ответ на окислительный стресс или реакцию на УФ-облучение [15]. Однако мутации в гене SMS человека, расположенного на хромосоме Xp22.11, приводят к значительному снижению уровня Spm, увеличению содержания Spd и вызывают редкое заболевание – синдром Шнайдера-Робинсона, для которого характерны умственная отсталость, остеопороз, гипотония, нарушения речи и судороги [16]. Недавно удалось показать, что инкубация клеток, полученных от пациента, страдающего синдромом Шнайдера-Робинсона, с Spm нормализует соотношение Spd/Spm в этих клетках [17]. Таким образом можно говорить об уникальной роли Spm у млекопитающих.
Биосинтез Spm и Spd тесно связан с превращениями орнитина, метионина и S-аденозилметионина и включает в себя несколько ферментов (рис. 1), ключевыми из которых являются пиридоксаль-5'-фосфат-зависимая орнитиндекарбоксилаза (ODC) и пируват-зависимая декарбоксилаза S-аденозилметионина (AdoMetDC). Спермидин/спермин-N1-ацетилтрансфераза (SSAT) и сперминоксидаза (SMOX) определяют скорость катаболизма полиаминов (рис. 1). Биосинтез и деградация этих четырех короткоживущих ферментов легко индуцируются, в том числе и в ответ на изменение внутриклеточной концентрации Spm и Spd. Клетки оснащены системой активного транспорта полиаминов, которая вносит существенный вклад как во внутриклеточное содержание полиаминов, так и в регуляцию их метаболизма. Важное место в регуляции гомеостаза полиаминов в клетке занимает короткоживущий белок антизим (AZ). Его биосинтез индуцируется в ответ на повышение содержания полиаминов, и AZ регулирует транспорт полиаминов в клетки и активность в них ODC [18].
Рис. 1.
Метаболизм полиаминов. AdoMet – S-аденозилметионин; AdoMetDC – S-аденозилметиониндекарбоксилаза; AZ – антизим; PAOX – ацетилполиаминооксидаза; N1-Ас-Spd – N1-ацетилспермидин; N1-Ас-Spm – N1-ацетилспермин; DHS – дезоксигипузинсинтаза; DOHH – дезоксигипузингидроксилаза; deAdoMet – декарбоксилированный S‑аденозилметионин; AZI – ингибитор антизима; Orn – орнитин; ODC – орнитиндекарбоксилаза; SpdSy – спермидинсинтаза; SpmSy – сперминсинтаза; SSAT – спермидин/спермин-N1-ацетилтрансфераза; SMOХ – сперминоксидаза. (Схема метаболизма как в работе [19].)
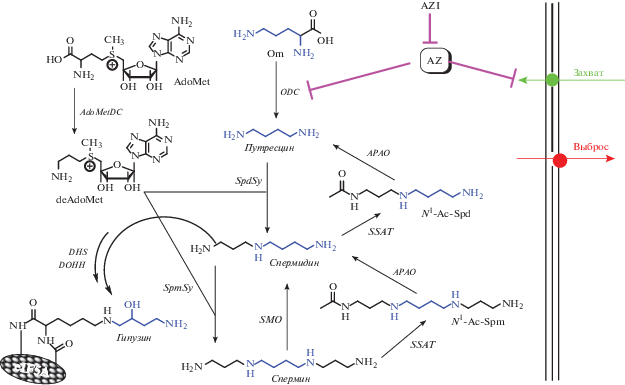
Опухолевые клетки имеют повышенный, по сравнению с нормальными, уровень Spm и Spd, и на протяжении последних десятилетий поиск эффективных и избирательных подходов к снижению внутриклеточного содержания полиаминов служит одним из важнейших факторов, стимулирующих развитие не только фундаментальных, но и прикладных аспектов биохимии этого класса соединений [20]. Известен широкий спектр ингибиторов биосинтеза полиаминов [21] и индукторов ферментов катаболизма Spm и Spd [22, 23], эффективно подавляющих рост опухолевых клеток. Более того, α-дифторметилорнитин (DFMO, Eflornithine®), суицидальный ингибитор ODC, в сочетании с другими противоопухолевыми препаратами применяется, например, для предотвращения рецидива в послеоперационный период при опухолях прямой кишки [24, 25]. Сперминоксидаза (SMOХ) служит ключевым ферментом катаболизма полиаминов и расщепляет Spm с образованием Spd, молекулы акролеина и молекулы перекиси водорода (рис. 1). SMOХ легко индуцируется при воспалении (этиология эпителиального рака в 20–30% случаев связана с воспалением [26]), и у больных, страдающих язвенным колитом, отмечено снижение уровня Spm за счет повышения активности SMOХ [27]. Инфицирование Helicobacter pylori сопровождается многократной индукцией SMOХ в слизистой оболочке желудка, что приводит к массовому образованию активных форм кислорода, которые, в свою очередь, служат одним из триггеров злокачественных трансформаций, приводящих к раку желудка [28, 29]. У позвоночных, помимо белка eIF5AHyp-1, обнаружена и его близкородственная изоформа eIF5AHyp-2 [30]. В норме белок eIF5AHyp-2 не детектируется, но его эксперессия наблюдается в ряде раковых клеток [31, 32]. Анализ уровней мРНК и белка eIF5AHyp-2 при резекции опухоли предстательной железы показал, что уровень этого белка связан со стадией опухоли и, если его содержание высоко, то можно ожидать агрессивный рост опухоли [33].
Нарушения метаболизма полиаминов ассоциированы с развитием также многих других заболеваний [34], включая пониженный иммунный ответ [35], некоторые формы панкреатита [36] и даже диабет 2-го типа [37]. Недавно установлена корреляция между содержанием эндогенных полиаминов в тканях сердца человека и клиническими параметрами у пациентов с сердечной недостаточностью [38], также показано, что Spd продлевает жизнь мышей и обладает кардиопротекторными свойствами, уменьшающими гипертрофию сердца и сохраняющими диастолическую функцию желудочков сердца у пожилых мышей [39].
2. БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ С-МЕТИЛИРОВАННЫХ АНАЛОГОВ СПЕРМИНА И СПЕРМИДИНА
Аналоги и производные Spm и Spd традиционно служат одними из основных инструментов исследования клеточных функций полиаминов. Среди множества описанных в литературе подобных веществ наиболее известны, пожалуй, терминально N,N'-бисалкилированные производные Spm, которые обладают широким спектром противоопухолевой активности [21–23]. Соединения этого класса обладают также противопаразитарной [40], а некоторые его представители кроме того и антибактериальной активностью, в том числе, и в отношении устойчивых и мультиустойчивых штаммов бактерий [41]. Более того, на основе скелета Spm были созданы эпигенетические модуляторы полиаминной природы, неконкурентно ингибирующие лизин-специфическую деметилазу1, например, 1,15-бис-{N5-[3,3-(дифенил)пропил]-N1-бисгуанидил}-4,12-диазапентадекан [42]. Соответственно, N-замещенные производные Spm и Spd с подобным спектром активности не способны выполнять клеточные функции полиаминов и представляют собой их биологически активные антагонисты.
Вторую, менее многочисленную и менее изученную группу алкильных производных полиаминов, составляют С-замещенные аналоги Spd и Spm. Первые представители этого семейства, гем-диметилированные производные Spd (рис. 2), появились еще в середине 80-х годов [43]. Однако оказалось, что два метильных заместителя при одном углеродном атоме практически полностью уничтожают “спермидиновую природу” аналога и гем-диметильные производные Spd не являются субстратами ферментов SpmSy и SSAT, что обеспечивает их катаболическую устойчивость [44]. Однако, в отличие от Nα,Nω-бисалкильных производных Spm, эти аналоги Spd способны поддерживать рост клеток c острым дефицитом Put и Spd, возникающим при кратковременной (3 сут) инкубации с α-дифторметилорнитином (DFMO) (ингибитор ODC), но не способны поддерживать рост клеток с хроническим дефицитом полиаминов, возникающим в результате длительной (12 сут и более) инкубации клеток с DFMO, что связано с отсутствием у гем-диметилированных производных субстратных свойств Spd в DHS-реакции [44].

Оказалось, что 1-МеSpd (1,8-диамино-5-азанонан), как и 1,1-Me2Spd (1,8-диамино-8-метил-5-азанонан), не ацетилировался SSAT [45], но, в отличие от 1,1-Me2Spd, поддерживал рост клеток с хроническим дефицитом полиаминов (инкубация клеток с 5'-(((Z)-4-амино-2-бутенил)метиламино)-5'-дезоксиаденозином (AbeAdo), ингибитором AdoMetDC, в течение 12 сут) подобно природному Spd [46]. Способность 1-MeSpd выполнять основные функции Spd in vitro обусловлена его субстратными свойствами в DHS-реакции, обеспечивающей посттрансляционную модификацию фактора инициации eIF-5A, что принципиально отличает его от гем-диметильных производных Spd, не являющихся субстратами этого ключевого фермента биосинтеза eIF5AHyp [47]. Кроме того, 1-MeSpd, 1-MeSpm (1,12-диамино-4,9-диазатридекан) и 1,12-Me2Spm (2,13-диамино-5,10-диазатетрадекан) взаимодействуют с ДНК аналогично Spd и Spm [48, 49]. Такая совокупность свойств позволяет рассматривать 1-MeSpd и 1,12-Me2Spm (оба аналога не являются субстратами SSAT) как катаболически устойчивые миметики Spd и Spm и делает их полезным инструментом для исследования метаболизма полиаминов.
Дальнейшее изучение биохимического потенциала 1-MeSpd и 1,12-Me2Spm (рис. 3) лимитировалось их малой доступностью [45], и свое развитие эти исследования получили лишь после того, как были разработаны удобные методы синтеза, позволяющие получать аналоги в препаративных (граммовых) количествах [50, 51]. Уже первые эксперименты показали, что 1-МеSpd способен эффективно предотвращать развитие острого некротизирующего панкреатита, вызванного дефицитом Spd [52], у крыс с регулируемой экспрессией гена спермидин/спермин-N1-ацетилтрансферазы (SSAT-трансгенные крысы [53]). Таким образом, удалось впервые установить, что последствия активации катаболизма полиаминов у SSAT-трансгенных крыс обусловлены именно дефицитом Spd в поджелудочной железе, а в распоряжении исследователей появилась возможность не только индуцировать панкреатит (индукторы SSAT, SSAT-трансгенные животные), но предотвращать его. Это свидетельствует о достаточной адекватности наших представлений о системе в целом [52].

Для изучения клеточных функций полиаминов и регуляции их метаболизма были использованы не только их 1-метилированные производные, но и система С-метилированных аналогов Spd и Spm (рис. 3) [54, 55]. Оказалось, что эффективность и продуктивность взаимодействия этих аналогов с ферментами биосинтеза и катаболизма полиаминов определяется положением метильной группы.
Так, SSAT не ацетилирует 1-MeSpd, а 2-MeSpd (1,8-диамино-2-метил-4-азаоктан) оказался неплохим субстратом этого фермента с близким для Spd значением Km, но заметно сниженным, по сравнению со Spd, значением Vmax. Перемещение метильной группы еще на одно положение, приводящее к 3-MeSpd (1,8-диамино-3-метил-4-азаоктан), вновь лишает аналог субстратных свойств в SSAT-реакции, а 8-MeSpd (1,8-диамино-4-азанонан) ацетилируется столь же эффективно, как и природный Spd. Такие, на первый взгляд, парадоксальные зависимости субстратных свойств от положения метильных групп достаточно хорошо согласуются с данными рентгеноструктурных исследований комплекса фермента с конъюгатом Spm с коэнзимом А (HS-СоА), моделирующим бисубстратный комплекс [56], а также бинарным комплексом HS-CoA–Spm [57] и отражают особенности узнавания полиаминов SSAT.
Изучение взаимодействия N1-ацетильных производных 1-MeSpd, 2-MeSpd, 3-MeSpd и 8-MeSpd с ацетилполиаминооксидазой (PAOX) показало, что лишь N1-Ас-3-MeSpd (N1-(ацетил)-1,8-диамино-3-метил-4-азаоктан) не расщепляется этой FAD-зависимой оксидазой до Put и N-ацетил-4-аминобутанона-2. По-видимому, метильная группа в третьем положении Spd стерически препятствует отрыву протона и промежуточному образованию в активном центре PAOX основания Шиффа [58]. Таким образом, катаболическая устойчивость 1-MeSpd определяется лишь невозможностью его ацетилирования, тогда как в случае 3-MeSpd существует “двойной блок” – как на стадии ацетилирования, так и на стадии собственно окислительного расщепления.
Исследование транспорта С-метилированных аналогов Spd (рис. 3) в клетки DU145 показало, что все они конкурируют с [14C]-Spd, однако лишь 2-MeSpd был столь же эффективен, как и “холодный“ Spd, тогда как остальные производные оказались несколько менее активными [57]. Судьба попавших в клетки С-метилированных Spd’s принципиально отличалась. Оказалось, что 1-MeSpd, 2-MeSpd и 8-MeSpd превращались в 1-MeSpm (1,12-диамино-4,9-диазатридекан), 2-MeSpm (1,12-диамино-2-метил-4,9-диадодекан) и 5‑MeSpm (1,12-диамино-5-метил-4,9-диадодекан), что не происходило в клетках дефицитных по сперминсинтазе (SpmSy), тогда как 3-MeSpd не проявлял субстратных свойств в отношении этого фермента [56, 57]. Причины столь кардинальных отличий в субстратных свойствах аналогов в отношении SpmSy не совсем понятны, но полученная зависимость прямо указывает на существование высокой комплементарности между соответствующим участком активного центра SpmSy и центральным фрагментом молекулы Spd, обеспечивающим продуктивное связывание субстрата и его аналогов в активном центре фермента.
Сбалансированная работа ферментов синтеза и деградации полиаминов служит одним из основных факторов, поддерживающих гомеостаз полиаминов в клетке. Активность, биосинтез и деградация ферментов метаболизма полиаминов зависят от многих факторов, в том числе и от внутриклеточного содержания Spm и Spd. Оказалось, что 2-MeSpd, в противоположность всем остальным метилированным производным, практически не снижал активность ODC ни после 24, ни после 72 ч инкубации клеток DU145 с ним [57]. Подобная зависимость активности аналога от положения метильной группы связана, скорее всего, с низкой эффективностью индукции производным 2-MeSpd биосинтеза AZ. Практически все аналоги Spm и Spd вызывают +1-сдвиг рамки считывания мРНК AZ и индуцируют биосинтез этого короткоживущего белка, обеспечивающего быструю деградацию ODC и ингибирующего транспорт полиаминов в клетки [59]. Возможность регулировать экспрессию AZ простым перемещением метильной группы вдоль углеродного скелета Spd является совершенно неожиданной, уникальной и не имеющей аналогий в литературе.
Способность восстанавливать рост клеток с истощенным пулом полиаминов также зависела от положения метильной группы в аналогах. При остром дефиците полиаминов (инкубация клеток DU145 c DFMO в течение 3 сут) все монометилированные Spd, подобно гем-диметильным производным Spd, восстанавливали рост клеток, а в случае хронического дефицита полиаминов (инкубация клеток DU145 c DFMO в течение 14 сут и более) такой способностью обладали лишь 1-MeSpd и 2-MeSpd [57]. Эти различия, скорее всего, связаны со способностью аналогов служить донором аминобутильной группы в DHS-реакции. Действительно, рацемические 1-MeSpd, 2-MeSpd и 3-MeSpd были субстратами DHS [60], но рацемический 3-MeSpd не восстанавливал рост клеток. Это кажущееся противоречие нашло свое объяснение при исследовании рост-восстанавливающей способности энантиомеров 3-MeSpd (см. ниже). Уместно отметить, что лишь 2-MeSpd, в отличие от других С-метилированных аналогов Spd, восстанавливал рост Leishmania donovani [61], что, по-видимому, связано с отсутствием у остальных С‑метилированных аналогов Spd субстратных свойств в трипанотионсинтетазной реакции (N1,N8-бис(глутатионил)спермидин – жизненно важный метаболит Plasmodium falciparum, Leishmania donovani, Trypanosoma brucei gambiense и др., вызывающих малярию, лейшманиоз и сонную болезнь соответственно [62]).
В последние годы различными группами исследователей было показано, что образование “биопленок” (biofilms) бактериями регулируется, в том числе, и Spd, при этом нор-Spd способен также выполнять функции самого Spd [63 и ссылки в ней], что предполагает важность трехуглеродного фрагмента Spd/нор-Spd для этих процессов. Использование коммерчески доступных аналогов Spd и его C-метилированных производных подтвердило ранее полученные данные о том, что для бактерий консервативным участком Spd является трехуглеродный фрагмент [64], тогда как для эукариот консервативным оказывается четырехуглеродный фрагмент, служащий донором аминобутильного остатка в пострансляционной модификации белка eIF5A.
Таким образом, удалось показать, что биохимические свойства аналогов можно регулировать, изменяя положение метильного заместителя, а 3‑MeSpd оказался первым функционально активным метаболически устойчивым миметиком Spd, пригодным для изучения клеточных функций легко взаимопревращающихся и частично взаимозаменяемых Spm и Spd.
Введение метильной(ых) групп в ахиральные Spm, Spd и их N1-ацетильные производные приводит к появлению в этих молекулах хиральных центров, однако до начала наших исследований в литературе отсутствовали какие-либо данные о стереоспецифичности ферментов метаболизма полиаминов. Вместе с тем, исходя из общих соображений, а также основываясь на различиях в активности хиральных аналогов иммунодепрессанта дезоксиспергуалина [65] и гидроксилированных производных диэтил-нор-спермина (3,7,11,15-тетраазагептадекан), являющихся индукторами SSAT [66], можно было предположить существование стереоспецифичности у ферментов метаболизма полиаминов.
Исследование взаимодействия энантиомеров N1-Ac-1-MeSpd с FAD-зависимой ацетилполиаминооксидазой (РАОX) показало, что этот фермент предпочтительно расщепляет R-изомер N1‑Ac-1-MeSpd и, тем самым, было впервые показано, что РАОX, природными субстратами которой служат ахиральные N1-Ac-Spd и N1-Ac-Spm, обладает скрытой стереоспецифичностью [67]. Известно, что РАОX способна в присутствии ароматических альдегидов эффективно окислять и неацетилированный Spd [68]. Оказалось, что бензальдегид катализирует расщепление преимущественно (R)-1-MeSpd, а в присутствии пиридоксаля субстратом служит уже S-изомер. Напротив, 4-пиридинальдегид не осуществляет стереоконтроль реакции и одинаково эффективен в случае как R-, так и S-изомеров 1-MeSpd [69]. Таким образом, при помощи ахиральных ароматических альдегидов был найден изящный способ регуляции стереоспецифичности РАОX, позволяющий избирательно окислять аналоги субстрата только с R-, или только с S-конфигурацией.
FAD-зависимая сперминоксидаза (SMOХ) окисляет Spm в Spd без предварительного N1-ацетилирования [70]. Поскольку родственная FAD-зависимая РАОX обладает стереоспецифичностью, то было исследовано взаимодействие R,R- и S,S-диастереомеров 1,12-Me2Spm с рекомбинантной SMOХ. Оказалось, что субстратными свойствами обладал лишь (S,S)-1,12-Me2Spm, а эффективность его превращения в Spd была даже несколько выше, чем у Spm. Таким образом, впервые было показано, что SMOX также обладает стереоспецифичностью, но предпочтительно расщепляет (S,S)-1,12-Me2Spm [71], тогда как субстратом РАОX служит (R)-N1-Ac-1-MeSpd.
Известно, что полиамины могут медленно расщепляться и другими FAD-зависимыми аминооксидазами, например полиаминоксидазой дрожжей Fms1. Недавно было показано, что, варьируя структуру ароматического альдегида, вносимого в субстратную смесь, удается регулировать региоселективность расщепления Spd (табл. 1), происходящего предпочтительно по Е1-сайту [72]. Внесение в субстратную смесь 4-гидроксибензальдегида увеличивает региоселективность почти в 10 раз, в то время как 3-гидроксибензальдегид имеет прямо противоположный эффект и расщепление происходит предпочтительно по Е2-сайту (табл. 1). Использование (R)-, и (S)-1-MeSpd в качестве субстратов Fms1 показало, что внесение в субстратную смесь пиридин-альдегидов открывает возможность регулировать не только региоселективность, но и стереоспецифичность расщепления этих субстратов под действием Fms1. Для этого оказалось достаточным лишь изменять положение альдегидной группы в пиридиновом ядре [72].
Таблица 1.
Кинетические параметры (kcat/Km) и региоселективность (E1/E2) расщепления Spd под действием FAD-зависимой полиаминоксидазы дрожжей (Fms1) в зависимости от строения добавленного в субстратную смесь альдегида
 |
Расщепление по Е1-сайту | Расщепление по Е2-сайту | |
|---|---|---|---|
| Spd или Spd и альдегид | соотношение Е1/Е2 | kcat/Km (M–1 s–1) | kcat/Km (M–1 s–1) |
| Spd | 12 | 505 | 42 |
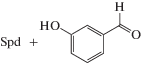 |
0.21 | 9657 | 46 078 |
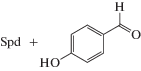 |
115 | 83 333 | 723 |
Как отмечалось выше, rac-1-MeSpd поддерживал рост клеток с хроническим дефицитом полиаминов, а rac-3-MeSpd не поддерживал, что плохо коррелирует с субстратными свойствами рацематов обоих аналогов в DHS-реакции. Для устранения этого кажущегося противоречия были исследованы субстратные свойства энантиомеров 1-MeSpd и 3-MeSpd в отношении DHS и оказалось, что фермент предпочтительно использует (S)-1-MeSpd [60] и (R)-3-MeSpd [73] в качестве доноров аминобутильного остатка. Исследование рост восстанавливающей активности энантиомеров 1-MeSpd и 3-MeSpd показало, что (S)-1-MeSpd [60] и (R)-3-MeSpd [73] восстанавливают рост клеток DU145 с хроническим дефицитом полиаминов, а (R)-1-MeSpd [60] и (S)-3-MeSpd [73] не обладали подобными свойствами. Отсутствие у rac-3-MeSpd рост-восстанавливающей активности было связано с тем, что неактивный (S)-3-MeSpd ингибировал транспорт (R)-3-MeSpd, который служил субстратом DHS [73, 74]. Таким образом, было показано, что DHS и система транспорта полиаминов обладают стереоспецифичностью, что существенно для направленного создания биологически активных производных Spm и Spd.
3. СИНТЕЗЫ С-МЕТИЛИРОВАННЫХ АНАЛОГОВ СПЕРМИНА И СПЕРМИДИНА
Биогенные полиамины Spm и Spd (рис. 1) и их аналоги имеют, как правило, достаточно простую структуру, тем не менее, их синтез весьма сложен и трудоемок. Это объясняется монотонностью структуры целевых соединений, необходимостью проведения селективных трансформаций по первичной или вторичной аминогруппам в одной молекуле, используемых для построения скелета С-метилированного аналога. Кроме того, высокая полярность конечных и многих промежуточных соединений создает дополнительные проблемы при их выделении и очистке. В связи с перспективностью использования аналогов спермина и спермидина в качестве инструмента исследования метаболизма и клеточных функций полиаминов, а также учитывая возможную практическую составляющую подобных работ, были разработаны эффективные подходы к синтезу этих соединений, а разнообразные методы органической химии, используемые для создания C–N-связи, были адаптированы к конкретным структурам сперминовой или спермидиновой природы.
3.1. Методы избирательной защиты первичных и вторичных аминогрупп
Одной из основных проблем, возникающих при синтезе полиаминов и их аналогов, оказывается необходимость проведения превращений только по первичной или вторичной аминогруппам, реакционная способность которых весьма близка. Известно несколько реагентов, которые избирательно реагируют с первичной аминогруппой в присутствии вторичной. Самым простым из них является этиловый эфир трифторуксусной кислоты (схема 1 ), селективно ацилирующий первичные аминогруппы при 0°С [75]. Обработка Spd этилтрифторацетатом приводит к терминально дизащищенному производному (I), которое далее может быть модифицировано по вторичной аминогруппе, что используется, например, для получения аналогов полиаминов разветвлeнного строения. Напротив, защита вторичной аминогруппы (Boc, Cbz, и т.п.) и последующее селективное удаление трифторацетильных групп приводит к N4-Boc- или N4-Cbz-Spd’s, которые являются удобными интермедиатами в синтезе линейных аналогов полиаминов. Преимуществами этилтрифторацетата служат его доступность и высокая селективность по отношению к первичной аминогруппе.
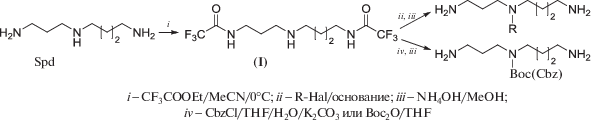
Схема 1 .
Использование 2-гидроксибензальдегидов для защиты первичной аминогруппы лежит в основе другой схемы селективной модификации вторичной аминогруппы в присутствии первичной [76]. Обработка первичного амина эквимольным количеством салицилового альдегида при 20°C приводит к основанию Шиффа, стабилизированному водородной связью (схема 2 ). Вторичная аминогруппа в этих условиях в реакцию не вступает. Высокая устойчивость подобных арилиденовых производных позволяет модифицировать вторичную аминогруппу. Главным преимуществом этого метода оказывается возможность удаления салицилиденовой защитной группы в очень мягких условиях (напр., CH3ONH2-основание при 20°С), что делает ее полезной при модификации молекул с основно- и кислотолабильными группами. Кроме того, все превращения проводятся “one-pot” с высоким выходом. Этот подход с успехом был использован в синтезе N1-Ac-8-MeSpd (схема 2 ). Cbz-триамин (II) вводился в реакцию с 10% мольным избытком салицилового альдегида в THF, образовавшееся салицилиденовое производное (III) вводили в реакцию с Вос2О и затем обрабатывали избытком H2NOCH3-основания, что позволило получить Boc-Cbz-триамин (IV) “one-pot” c выходом 73%, из которого в три стадии был получен целевой N1-Ac-8-MeSpd [58].
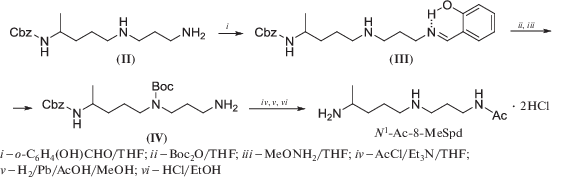
Схема 2 .
Салицилиденовая защитная группа с успехом использовалась для очистки промежуточных продуктов в синтезе bis-Et-SpmTrien, “зарядодефицитного” аналога диэтилспермина. При алкилировании избытка N-этил-1,3-диаминопропана метансульфонатом (V) наряду с целевым соединением линейного строения (VI) образовывалось некоторое количество продукта алкилирования по вторичной аминогруппе (схема 3 ). Для удаления этой примеси неочищенный пентамин (VI) обрабатывали ~20% мольным избытком салицилового альдегида, что приводило к образованию устойчивого основания Шиффа c первичной аминогруппой примеси, которое затем легко отделялось от основного продукта (VI). После удаления Cbz-защитных групп целевой bis-Et-SpmTrien был получен с суммарным выходом 7.5%, считая на N‑этиламиноэтанол [77, 78].
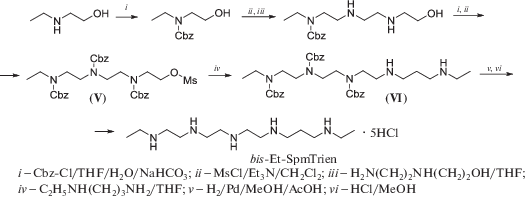
Схема 3 .
Достаточно распространeнным и удобным в работе реагентом для защиты первичных аминогрупп в синтезе полиаминов служит 2-{[(трет-бутоксикарбонил)окси]имино}-2-фенилацетонитрил (Boc-ON) [79]. Этот реагент (схема 4 ) позволяет селективно защищать первичные аминогруппы для последующей модификации вторичных. Следует отметить, что Boc-ON медленно реагирует и со вторичными аминогруппами – модификация N-(2-цианоэтил)-1,4-диаминобутана проходит за 72 ч при 20°С на 70% [80].

Схема 4 .
Интересным примером десимметризации структуры полиаминов оказывается замыкание под действием формальдегида гексагидро-1,3-пиримидинового цикла, который, после проведения необходимых трансформаций, может быть раскрыт по ретрореакции Кляйзена обработкой малоновой кислотой в присутствие пиридина (схема 5 ). Этот метод позволяет дискриминировать аминопропильную и аминобутильную группы Spd и открывает удобный путь получения его N1- и N8-замещенных производных [81].
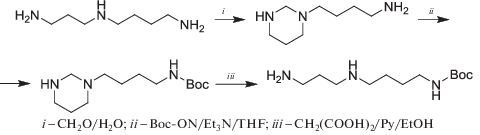
Схема 5 .
Обычно введение N-защитной группы не предусматривает дальнейших трансформаций по этой же аминогруппе. Однако сульфонамидные производные стоят несколько особняком. Если в молекуле присутствуют первичные и вторичные аминогруппы, то исчерпывающее сульфонамидинилирование защищает вторичные аминогруппы и активирует первичные, которые становятся весьма реакционноспособны, и их алкилирование галоидными алкилами представляет собой общий и удобный способ превращения первичных аминов во вторичные. В работе [82] для избирательной защиты вторичной и активации первичной аминогрупп был предложен 2-нитрофенилсульфонилхлорид (NsCl). Такая защитная группа имеет ряд преимуществ по сравнению с другими сульфониламидами, так как она легко удаляются меркаптанами [51, 82–84], а достаточно высокая селективность NsCl по отношению к первичной аминогруппе (схема 6 ) позволяет, варьируя соотношение олигоамин/NsCl и проводя реакцию в водном THF, избирательно нозилировать первичные аминогруппы, или получать полностью нозилированные производные [83]. Нозильная защита оказывается комплементарной широко используемым Вос- и Cbz-группам, что делает ее весьма удобной для получения аналогов полиаминов.

Схема 6 .
Таким образом, в литературе описан достаточно широкий спектр реагентов, избирательно защищающих первичную и/или вторичную аминогруппу, что позволяет проводить в дальнейшем различные трансформации, необходимые для синтеза целевых полиаминов.
3.2. Синтез полиаминов и их аналогов
Для получения С-метилированых аналогов полиаминов и их производных, в основном, используют: 1) методы алкилирования; 2) присоединение по Михаэлю и восстановление нитрилов; 3) реакцию Мицунобу; 4) восстановление азидов, амидов, оснований Шиффа, нитросоединений.
3.2.1. Методы алкилирования
Наиболее распространенным методом создания C–N-связи и построения скелета аналогов Spm и Spd служит N-алкилирование, которое позволяет легко наращивать полиметилениминовую цепь. При этом в роли нуклеофилов могут выступать как свободные амины, так и N-анионы сульфамидов, фосфамидов, карбоксамидов и т.д. Алкилирование первичного амина может приводить к смеси вторичного, третичного аминов и четвертичной аммониевой соли. Для получения продукта моноалкилирования в реакцию вводят большой избыток амина или генерируют анион N-защищенного производного. Ниже приведено несколько примеров, иллюстрирующих различные варианты алкилирования в синтезе С-метилированных аналогов полиаминов.
При использовании в качестве алкилирующих средств галоидных алкилов для получения вторичных аминов обычно в реакцию вводят избыток незащищенного амина или диамина, а в случае алкилхлоридов и бромидов – добавляют 20–25 мольных % NaJ/KJ, и реакции часто проводят при нагревании. Этот подход позволил с успехом получить R- и S-изомеры 1-MeSpm (схема 7 ), исходя из коммерчески доступных энантиомеров Вос-аланинола [84]. На одной из промежуточных стадий синтеза избыток 1,3-диаминопропана алкилировали йод-производными (VII)/(VIII) в THF, что привело Boc,Ns-тетрааминам (IX) и (X) с выходами 92 и 90%, соответственно. При этом происходило незначительное (около 2%) образование 2-нитрофенил-1,3-диаминопропана в результате активированного нуклеофильного замещения сульфамидной группы под действием 1,3-диаминопропана. Это наблюдение вполне согласуется с тем, что Ns-группа легко удаляется под действием алкил- и арилсульфид-анионов. После удаления защитных групп целевые (R)- и (S)-1-MeSpm были получены с суммарными выходами 32 и 33%, считая на соответствующий Вос-аланинол [84].
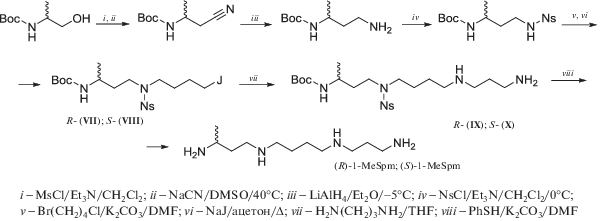
Схема 7 .
Для получения защищенного 8-MeSpd, который использовался в синтезе модифицированных производных 15-дезоксиспергуалина, обладающих иммуносупрессорной активностью, избыток 1,3-диаминопропана алкилировали 5-хлорпентанолом-2 в кипящем н-BuOH в присутствии 25 мольн. % KJ (схема 8 ), что привело к 1-амино-8-гидрокси-4-азанонану с выходом лишь 37% [65].

Схема 8 .
Напротив, алкилирование (R)-N2-(трет-бутилоксикарбонил)-2-амино-6-фенил-5-азагексана (XI) 4-хлорбутиронитрилом по вторичной аминогруппе в кипящем н-BuOH в присутствии 20 мольн. % KJ в течение 20 ч (схема 9 ) привело к защищенному (R)-N8-(трет-бутилоксикарбонил)-N5-(бензил)-8-нонилнитрилу (выход 98%), из которого был получен защищенный триамин (XII) [65].
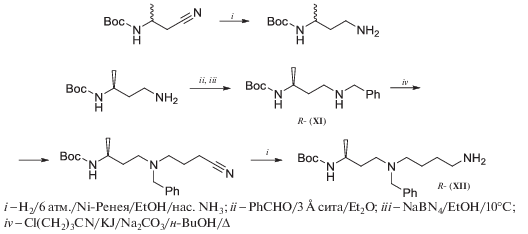
Схема 9 .
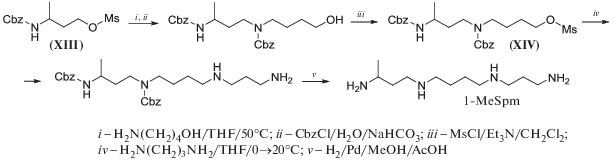
Аминоспирты, как правило, более доступны по сравнению с аминоалкилгалогенидами, а алкил(арил)сульфонаты соответствующих N-защищенных аминоспиртов представляют собой эффективные алкилирующие средства [65, 85]. Поэтому ранее труднодоступный (см. далее схему 28 ) рацемический 1-MeSpd был получен, исходя из этилового эфира 3-аминомасляной кислоты (схема 10 ). Ключевая стадия этого синтеза – алкилирование избытка 1,4-диаминопропана метансульфонатом N-Cbz-3-аминобутанола (XIII). В результате целевой тригидрохлорид 1-MeSpd синтезирован в 5 стадий с суммарным выходом 43%, считая на этиловый эфир 3-аминомасляной кислоты [50].

Схема 10 .
Для построения скелета 1-MeSpm полиаминную цепь последовательно наращивали, алкилируя сначала 4-аминобутанол-1 метансульфонатом (XIII), а потом 1,3-диаминопропан метансульфонатом (XIV) (схема 11 ). Все это позволило получить 1-MeSpm с суммарным выходом 41%, считая на метансульфонат (XIII) [51] (схема 11).
Энантиомеры 2-MeSpd синтезированы, исходя из коммерчески доступных R- и S-изомеров 3-бромпропанола-1, из которых ыли получены N-фталоильные производные. Их сначала превращали в соответствующие 3-амино-2-метилпропанолы-1, а затем в метансульфонаты (XVIII) и (XIX) (схема 12 ). Алкилирование этими метансульфонатами избытка 1,4-диаминобутана и последующее удаления Cbz-групп позволило синтезировать целевые (R)- и (S)-2-MeSpd с суммарными выходами 47 и 34%, считая на соответствующие N-фталоильные производные [86].

Схема 12 .
Следует отметить, что для алкилирования диаминов можно с успехом использовать и метансульфонаты N-защищенных вторичных спиртов. Так, алкилирование избытка 1,4-диаминобутана метансульфонатом N-(трет-бутилоксикарбонил)-4-аминобутанола-2 проходит достаточно гладко, 3-MeSpd выделен с 31% выходом, считая на исходный 4-аминобутанол-2 [54].
Алкилирование 4-аминобутанола-1 мезилатом 3-азидо-3-метилбутанола-1 в кипящем н‑BuOH в присутствии каталитических количеств DBU (схема 13 ) привело с 79%-м выходом к азидоаминоспирту (XVII), промежуточному соединению в синтезе ортогонально защищенного 1,1-Me2Spd [65].
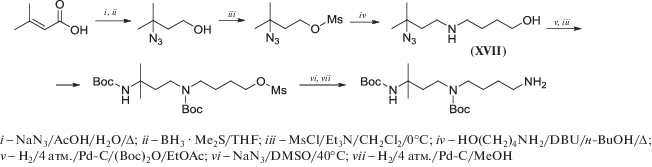
Схема 13 .
Таким образом, активация гидроксильной группы путем превращения ее в эфиры сульфокислот дает возможность использовать для построения полиаминометиленовой цепи легкодоступные N-защищенные аминоспирты вместо существенно более труднодоступных N-защищенных бромалкиламинов.
Для создания C–N-связи использовали и восстановительное алкилирование алифатических азидов дихлорборанами (схема 14 ). Исходя из 3‑хлор-1-аминопропанола в 4 стадии был получен 1,7-диамино-4-азагептан (2-Me-nor-Spd) [87]. Однако в дальнейшем этот метод не нашел широкого применения.

Схема 14 .
Алкилирование сульфонамидов (рис. 4) галоидными алкилами представляет собой общий и удобный способ превращения первичных аминов во вторичные. Ранние работы были выполнены с использованием тозильных (Tоs) производных [88–90], однако жесткие условия гидролиза Tоs-амидов привели к тому, что Tоs-производные были практически полностью вытеснены более лабильными мезитильными (Mts) [91–94]. В литературе имеются единичные сообщения об успешном использовании еще более кислотолабильных Pmc-производных [95, 96].
Рис. 4.
Сульфамидные группы, наиболее часто используемые в химии полиаминов: 4-толуолсульфонил- (Tos), мезитилен-2-сульфонил- (Mts), 2-нитрофенисульфонил- (Ns); 2,2,5,7,8-пентаметилхроман-6-сульфонил- (Pmc).
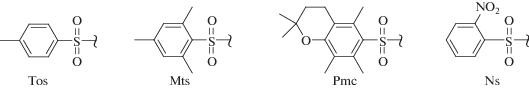
2-Нитрофенисульфонильные (Ns) производные амидов стоят несколько особняком от Tоs-, Mts- и Pmc-производных. Эта защитная группа имеет ряд преимуществ по сравнению с другими сульфониламидами, так как она легко удаляется в результате обработки меркаптанами (меркаптоэтанол, тиогликолевая кислота и, в особенности, тиофенол) в DMF в присутствии поташа [51, 55, 77, 82–84].
Для получения 1,12-Me2Spm описанная выше линейная стратегия (cхемы 10 и 11) оказалась малоэффективна, а симметрия молекулы 1,12-Me2Spm располагала к проведению конвергентного синтеза (cхема 15), ключевой стадией которого являлось алкилирование N1,N4-бис(2-нитрофенилсульфонил)-1,4-диаминобутана (bis-Ns-Put) в DMF при помощи N-Cbz-3-аминобутилбромида, что после удаления всех защитных групп дает 1,12-Me2Spm с суммарным выходом 57%, считая на N-Cbz-3-аминобутанол-1 [51]. Следует отметить, что алкилирование bis-Ns-Put мезилатом или тозилатом спирта (XV) приводит к искомому продукту лишь с низким выходом.

Схема 15 .
Удобный способ получения 2,11-Me2Spm и 3,10-Me2Spm заключается в использовании в качестве исходных соединений спиртов (XV) и (XVI), которые превращают в нозилаты (XVIII) и (XIX) и затем алкилируют их стехиометрическим количеством 1,4-дииодбутана (cхема 16). Перед удалением Ns-групп тиофенолом в реакционную смесь прибавляют бензилбромид (20 мольн. %) для превращения не вступивших в реакцию соединений (XVIII) и (XIX) в соответствующие N‑бензильные производные, что существенно упрощает выделение производных (XX) и (XXI). Последующее удаление Cbz-групп привело к целевым 2,11-Me2Spm и 3,10-Me2Spm с выходами 21 и 18% (8 стадий), считая на 3-амино-2-метилпропанол-1 и 4-аминобутанол-2 соответственно [97].

Схема 16 .
Исходными соединениями для синтеза R- и S-изомеров 3-MeSpd служили Boc-диамины (XXII) и (XXIII), из которых были получены нозилаты (XXIV)/(XXV) и (XXVI)/(XXVII). Алкилирование N-(4-йодбутил)фталимидом энантиомерно чистых Вос- и Cbz-защищенных нозилатов (XXIV)/(XXV) и (XXVI)/(XXVII) соответственно, представляло собой ключевую стадию синтеза R- и S-изомеров 1‑MeSpd и 3-MeSpd (схема 17 ). Существенным, как и в синтезах 2,11-Me2Spm и 3,10-Me2Spm (схема 16 ), было выведение из реакционной смеси не вступивших в реакцию нозилатов (XXIV)/(XXV) и (XXVI)/(XXVII). Последующее удаление защитных групп привело к R- и S-изомерам 1-MeSpd с суммарными выходами 45 и 47%, (5 стадий), считая на диамины (XXII) и (XXIII), соответственно [98] и в 7 стадий - к (R)- и (S)-изомерам 3-MeSpd с суммарными выходами 30% и 25, считая на производные (XXII) и (XXIII) соответственно [74] (схема 17).

Алкилирование Ns2-Put при помощи N-(фталоил)-3-амино-2-хлорметилпропена-1 (XXVIII) проходит гладко (схема 18 ), и с высоким выходом приводит к N1,N12-бис(фталоил)-1,12-диамино-2,11-бис(метилиден)-4,9-диазадодекану (XXIX). Вторичные аминогруппы бисфтального производного (XXIX) защищают Boc-группой и лишь после этого удаляют фталильные защиты, что позволяет легко очистить промежуточный бис-Вос-тетрамин и, после удаления Вос-защитных групп, получить целевой 2,11-бис(метилиден)Spm с суммарным выходом 27%. Следует отметить, что 2,11-бис(метилиден)Spm имеет аномально высокую хроматографическую подвижность по сравнению с 2,11-Me2Spm [99] (схема 18).
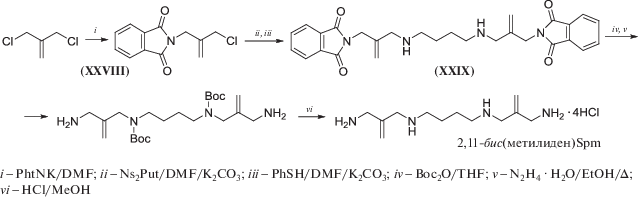
Таким образом, нозильная защита оказывается комплементарной широко используемым Вос-, Cbz- и Pht-группам и очень удобной для получения препаративных количеств различных C-метилированных производных Spm и Spd.
3.2.2. Присоединение по Михаэлю и восстановление нитрилов
Весьма эффективным и широко распространeнным подходом в синтезе аналогов Spm и Spd оказывается присоединение по Михаэлю. Донорами в реакции Михаэля служат первичные или вторичные амины, реже – N-анионы, а в качестве акцепторов используют акрилонитрил, метакрилонитрил, кротононитрил, производные акриловой кислоты или метилвинилкетон. На заключительной или одной из промежуточных стадий продукты цианэтилирования восстанавливают каталитическим гидрированием. Например, Вос-нитрилы гладко восстанавливаются водородом над Ni-Ренея в спирте, насыщенным аммиаком (схема 9 ), что приводит к соответствующим энантиомерно чистым N3-(трет-бутилоксикарбонил)-1,3-диаминобутанам с выходом 90–96% [65]. Восстановление нитрилов с помощью LiAlH4 также широко используется в синтезе метилированных аналогов полиаминов, хотя природа N-защитных групп в молекуле аминонитрилов накладывает определенные ограничения на условия реакций, традиционно используемые для восстановления нитрилов комплексными гидридами. Так, восстановление Вос-нитрилов (схема 7 ) с помощью LiAlH4 в эфире при –5...–2°С протекает без рацемизации и приводит к энантиомерно чистым N3-(трет-бутилоксикарбонил)-1,3-диаминобутанам c практически количественным выходом [74, 84].
При присоединении эквивалента первичного амина к акцепторам в реакции Михаэля, как правило, образуется смесь продуктов в результате повторного присоединения вторичного амина. Для построения скелета N1,N4-бис(трет-бутилоксикарбонил)-защищенных 2-MeSpd и 3-MeSpd авторы работы [65] использовали присоединение 4‑аминобутанола-1 к метакрилонитрилу, или кротононитрилу (схема 19 ) и получили соединения (XXX) и (XXXI), которые далее использовались без дополнительной очистки. Восстановление нитрилов (XXX) и (XXXI) проводили каталитическим гидрированием над PtO2 (схема 19 ) и получили искомые диаминоспирты. Выходы на стадии восстановления составляли около 80% [65].
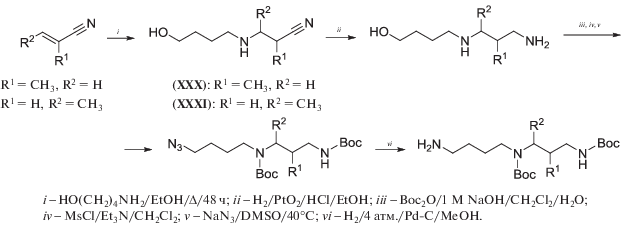
Схема 19 .
Присоединение избытка Put к метакрилонитрилу и кротононитрилу приводит к диаминонитрилам (XXXII) и (XXXIII) с 47%-м (условия реакции не оптимизированы) и 84%-м выходами соответственно (схема 20 ). Однако восстановление нитрилов водородом над Ni-Ренея или PtO2 сопровождалось образованием набора минорных побочных продуктов, и эти C-метилированные аналоги Spd были получены с низкими суммарными выходами [57]. Для повышения суммарных выходов нитрилы (XXXII) и (XXXIII) превращали в соответствующие ди-Вос-производные, которые гладко и с хорошими выходами восстанавливали с помощью LiAlH4 в эфире до ди-Вос-триаминов (XXXIV) и (XXV). Последующее удаление защитных групп привело к искомым 2-MeSpd и 3-MeSpd с суммарными выходами 27 и 45%, считая на метакрилонитрил и кротононитрил, соответственно [100]. Промежуточные ди-Вос-амины (XXXIV) и (XXXV) использовали в качестве исходных соединений в синтезах N1-ацетильных производных 2-MeSpd и 3-MeSpd (схема 20 ). Последовательное ацетилирование свободной аминогруппы и удаление Вос-защитных групп привели к N1-Ас-2-MeSpd и N1-Ас-3-MeSpd с очень высокими выходами [58].

Схема 20 .
Один из способов синтеза 2,11-Me2Spm и 3,10-Me2Spm предусматривал использование в качестве исходных соединений динитрилов (XXXVI) и (XXXVII). Их получение предпочтительно проводить присоединением избытка соединений (XXXII) и (XXXIII) к метакрилонитрилу и кротононитрилу, соответственно, а не вводить в реакцию с Put два эквивалента нитрила (схема 21 ). Восстановление ди-Вос-диаминодинитрилов (XXXVIII) и (XXXIX) до ди-Вос-тетрааминов при помощи L-iAlH4 оказалось не столь тривиальным, как представленное на схеме 20 восстановление ди-Вос-триаминов. Оказалось, что ни соединение (XXXVIII), ни соединение (XXXIX) не растворялись в эфире, а восстановление соединения (XXXVIII) в THF позволило получить соответствующий ди-Вос-тетраамин лишь с 15% выходом из-за конкурирующей ретро-реакции Михаэля, приводящей, в основном, к N1,N4-бис(трет-бутилоксикарбонил)-1,4-диаминобутану, а также к N4,N8-бис(трет-бутилоксикарбонил)-2-метил-1,8-диамино-4-азаоктану (16%). Последующее удаление Вос-защитных групп привело к целевому 2,11-Me2Spm (схема 21 ) с суммарным выходом лишь 6.5% (4 стадии). При восстановлении соединения (XXXIX) LiAlH4 в THF ди-Вос-3,11-Me2Spm не удалось выделить из реакционной смеси, а основным продуктом оказался N1,N4-бис(трет-бутилоксикарбонил)-1,4-диаминобутан [97].

Схема 21 .
Синтез разветвленных и непредельных аналогов 1-гуанидино-7-аминогептана (GC7), самого эффективного ингибитора дезоксигипузинсинтазы (DHS), потребовал восстановления нитрилов различного строения, включая и функционально замещенные [101]. Для этих целей использовались BH3/THF (схема 22 ), что с хорошими выходами привело к искомым диаминам, которые затем были превращены в целевые аминогуанидины (XL) и бисгуанидины (XLI).
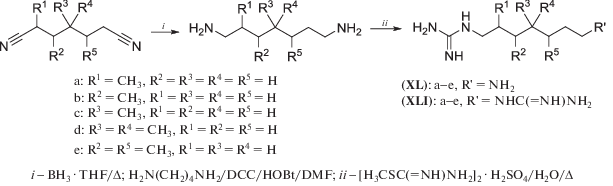
Схема 22 .
Синтез ортогонально защищенных 1-замещенных производных Spd является одной из важнейших задач при получении 1-замещенных Spm и Spd блочным методом. В работе [65] описано получение семейства таких 1-замещенных Spd, исходя из легко доступных винилкетонов (схема 23 ). Присоединение дибензиламина к винилкетонам, восстановление карбонильной группы и удаление одной бензильной группы давало N‑бензиламиноспирты (XLII), аминогруппу которых затем алкилировали хлорбутиронитрилом в присутствии 10% KJ, и полученные таким образом спирты превращали в азид через промежуточный метансульфонат (схема 23 ). Избирательное восстановление азидогруппы проводили гидрированием над Pd/С, а нитрил восстанавливали на последней стадии водородом над Ni-Ренея в спирте, насыщенным аммиаком. В результате, N5-(бензил)-N8-(трет-бутилоксикарбонил)-1-амино-5-азанонан был получен в 8 стадий с суммарным выходом 30%, считая на метилвинилкетон [65].
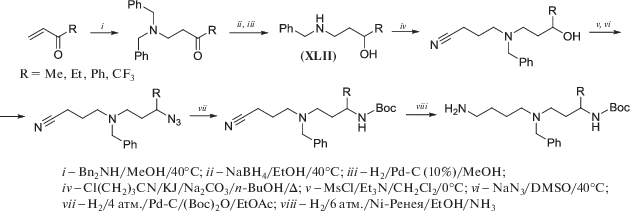
Схема 23 .
В работе [102] 1,8-диаминооктан присоединяли к кротононитрилу с последующим восстановлением образовавшегося динитрила водородом над PtO2 (схема 24 ), что позволило получить тетраамин (XLIII) с 29% выходом.

Схема 24 .
Исходным соединением в синтезе 4,5-Me2Spd служил вторичный амин (XLIV), который присоединяли к акрилонитрилу, азидонитрил (XLV) выделили с 82%-м выходом (схема 25 ). Одновременное восстановление азида и нитрила водородом над Ni-Ренея и привело к 4,5-Me2Spd в две стадии c суммарным выходом 60% [87].

Схема 25 .
Синтез 3,3,-Me2Spd проводили, исходя из 2,2-диметилакрилонитрила. Присоединение к нему 1,4-диаминобутана потребовало жестких условий (100°С, 8 дней), искомый нитрил (XLVI) синтезирован с выходом 52% (схема 26 ). Для восстановления нитрила (XLVI) использовали LiAlH4 (выход 22%) и NaBH4/CoCl2 (выход 27%), но оптимальным восстановителем оказался BH3 · THF [43].
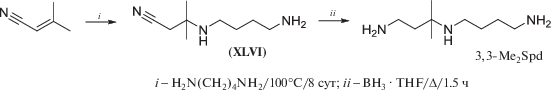
3.2.3. Реакция Мицунобу
Реакция Мицунобу – конденсация соединений, содержащих подвижный атом водорода, с первичными или вторичными спиртами в присутствии диалкилазадикарбоксилата и трифенилфосфина [103] – иногда используется в синтезе аналогов полиаминов. Так, в работе [104] для получения оптически активного тетраамина (XLVII) использовали конденсацию N-Tf-N-метил-3-аминобутанола с N1,N7-бис-Tf-1,7-диаминогептаном, которая приводила к тетра-Tf-производному с 80% выходом (схема 27 ). После удаление трифлильных групп (Tf-) действием натрия в смеси NH3/THF/t-BuOH при –70°С терамин (XLVII) очищали в виде тетра-Вос-производного, что после удаления Вос-защитных групп позволило получить целевое соединение (XLVII) с 31% выходом.
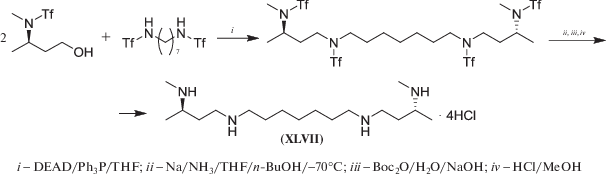
Схема 27 .
Восстановление амидов
Наиболее распространeнными реагентами для восстановления амидов в синтезах полиаминов являются комплексы диборана с THF или диметилсульфидом. Для получения 1-MeSpd использовали ацилирование N-Cbz-3-аминомасляной кислотой N-Cbz-путресцина в присутствии DCC и гидроксибензотриазола (HOBt) (схема 28 ). Защитные группы удаляли каталитическим гидрированием и полученный амид восстанавливали BH3 · THF, что приводило к 1-MeSpd, но лишь с выходом 15%, считая на N-Cbz-3-аминомасляную кислоту [45].
Рацемический 1,12-Me2Spm был получен конденсацией Put с двумя эквивалентами N-Cbz-3-аминомасляной кислоты (схема 28 ) с последующим удалением защитных групп и восстановлением амидных функций BH3 ⋅THF, но лишь с выходом 18%, считая на N-Cbz-3-аминомасляную кислоту [45]. Проблемы с растворимостью промежуточных соединений, неожиданно низкие выходы на стадии восстановления амидных групп, а также сложные и трудоемкие процедуры выделения полярных 1-MeSpd и 1,12-Me2Spm на последней стадии делали нецелесообразным масштабирование этих схем синтеза. Поэтому для получения препаративных количеств 1-MeSpd и 1,12-Me2Spm целесообразно использовать cхемы 10 и 15.

2,2-Me2Spd синтезирован, исходя из 2-циано-2-метилпропионовой кислоты, которой ацилировали избыток 1,4-диаминобутана, что позволило получить амид (XLVIII) с 59% выходом (cхема 29), после чего амидную и нитрильную функции в соединении (XLVIII) оодновременно восстанавили при помощи BH3 · THF, выход составил 49% [43].

Схема 29 .
Синтез 8,8-Me2Spd [43] проводили, исходя из 4‑метил-4-нитровалериановой кислоты (XLIX), которую получали присоединением 2-нитропропана к метилакрилату с последующим омылением сложного эфира. Конденсация кислоты (XLIX) с фумаратом 3-аминопропионитрила в присутствии DCC давала амид (L) с 97%-м выходом (схема 30 ). Одновременное восстановление нитрильной и амидной функций проводили при помощи BH3 · THF с выходом 92%. Восстановление стерически затрудненной нитрогруппы при помощи Pd/C–HCO2NH4 проходило лишь с 21%-м выходом, тогда как восстановление при помощи NiCl2 ⋅6H2O/NaBH4 [105] оказалось более успешным и позволяло синтезировать целевое соединение с выходом 70%.

Схема 30 .
Применение LiAlH4 для восстановления амидных связей накладывает серьезные ограничения на группы для защиты аминофункций. Однако известно, что тритильная защита устойчива даже при кипячении с LiAlH4 в THF (схема 31 ), что и было использовано для получения ряда N3,N8-алкилированных производных Spm [106].

Этот подход может рассматриваться в качестве не самой плохой альтернативы мезилатному и нозилатному путям получения (схемы 10, 15 и 16 , см. выше) рацематов 1-MeSpd, 1,12-Me2Spm и 2,11-Me2Spm, исходя из коммерчески доступных 3-аминомасляной и 3-аминоизомасляной кислот.
Восстановление азидов
Алифатические азиды легко и различными способами (Ph3P; меркаптаны, включая дитиотреитол; каталитическое гидрирование; BH3 и т.д.) восстанавливаются в амины. Как правило, реакции проходят в мягких условиях и с высоким выходом [107, 108]. Ниже приведены некоторые примеры, иллюстрирующие применение алифатических азидов для получения С-алкилированных аналогов полиаминов.
Восстановление азидов водородом над Pd/C (4 атм.) проходит с практически количественным выходом, что было с успехом использовано при получении ди-Вос-защищенных производных 8-MeSpd (схема 8 ), 1,1-Me2Spd (схема 13 ), 2-MeSpd и 3-MeSpd (схема 19 ), а также защищенного 5-MeSpd (схема 34 , см. ниже) [65].
В работе [101] описаны синтезы многочисленных непредельных и разветвленных аналогов 7-амино-1-гуанидиногептана (GC7), ингибирующего DHS в концентрации 10–7 М. Одна из стадий синтеза предусматривала восстановление азидов при помощи Ph3P/Н2О (схема 32 ), что с высокими выходами приводило к аминам [101]. Традиционно вместо воды для разложения азидофосфоранов используют водный аммиак [107, 108], а применение водного CH3NH2 (схема 16 ) оказывается даже эффективнее водного аммиака [97]. Одновременное восстановление тройной С–С-связи и азида в соединении (LI) (схема 32 ) проводили, гидрируя его над PtO2 [101].

Схема 32 .
Метаболически устойчивый 1,1-Me2Spd (схема 33 ) синтезировали, исходя из 3-метилкротоновой кислоты, к которой присоединяли HN3, полученной 3-азидо-3-метилмасляной кислотой (LII) ацилировали Put и затем амидную и азидогруппы одновременно восстанавливали кипячением с BH3 ⋅ THF. Трудности с восстановлением на последней стадии (выход 30–35%) и проблемы с очисткой конечного продукта привели к тому, что целевой 1,1-Me2Spd был получен лишь с 16% суммарным выходом [43].

Схема 33 .
Восстановление оснований Шиффа
Конденсация карбонильных соединений с первичными аминами приводит к образованию оснований Шиффа, восстановление которых каталитическим гидрированием или комплексными гидридами (обычно NaBH3CN, NaBH4) дает вторичные амины.
Защищенный 5-MeSpd (схема 34 ), полученный исходя из 5-хлорпентанона-2, превращали в 5-азидопентанон-2, вводили в реакцию с N1‑(трет-бутилоксикарбонил)-1,3-диаминопропаном и образовавшееся основание Шиффа восстанавливали с помощью NaBH4. На последней стадии синтеза ди-Вос-азид был с практически количественным выходом восстановлен водородом над Pd/C до искомого производного 5-MeSpd [65].

Схема 34 .
Синтез 1,1-Me2Spd с использованием одновременного восстановления амидо- и азидо-функций (схема 33 ) проходил с очень низким суммарным выходом. Поэтому исходная азидокислота (LII) была в две стадии превращена в азидоальдегид (схема 35 ), который вводили в реакцию с 1,4-диаминобутаном, образовавшееся основание Шиффа и азидогруппу одновременно восстанавливали H2/PtO2 и получали 1,1-Me2Spd с выходом 53%, считая на кислоту (LII) [43].
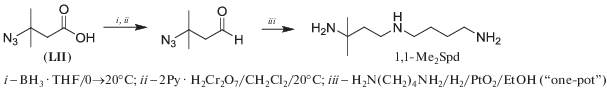
Схема 35 .
Простой и удобный синтез 5-MeSpd (схема 36 ) провели, исходя из 5-азидопентанона-2 [109] через промежуточное основание Шиффа с 1-азидо-3-аминопропаном (синтез азида представлен на схеме 14 ). Полученное основание Шиффа восстанавливали NaBH3CN, а затем азидогруппы восстанавливали каталитическим гидрированием над Pd/C и получали целевой 5-MeSpd с выходом 60%, считая на азидокетон [87].

Схема 36 .
4. ЗАКЛЮЧЕНИЕ
Биохимия полиаминов представляет собой достаточно молодую и быстро развивающуюся область биохимии. Спермин и спермидин присутствуют в милли- и субмиллимолярных концентрациях в клетках всех типов, что a priori определяет множественность их жизненноважных функций, обеспечивающих дифференцировку и нормальный рост клеток. Нарушения метаболизма полиаминов и их внутриклеточного содержания связывают с развитием многих заболеваний, включая онкологические (опухолевые клетки имеют повышенный уровень полиаминов), сердечно-сосудистые, ассоциированные с нарушениями иммунной системы, диабет 2-го типа, синдром Шнайдера-Робинсона и т.п. Модуляторы метаболизма спермина и спермидина имеют определенный терапевтический потенциал, что подтверждается использованием α-дифторметилорнитина для лечения поздних стадий сонной болезни и использованием биологически активных производных полиаминов в сочетании с другими противоопухолевыми препаратами для предотвращения рецидива в послеоперационный период.
Успехи практической полиаминотерапии прямо связаны с развитием наших представлений о клеточных функциях полиаминов и адекватным их описанием на молекулярном уровне. Для этого применяется весь арсенал методов современной биохимии, молекулярной биологии и биоорганической химии. Химические регуляторы метаболизма полиаминов традиционно и достаточно широко используются в подобных исследованиях. Один из алгоритмов превращения спермина и спермидина в их биологически активные производные заключается в создании соответствующих С-метилированных аналогов. Биохимические свойства этих соединений можно регулировать, перемещая метильную группу по скелету полиамина, или, на более тонком уровне, изменяя конфигурацию хирального центра. Эти соединения уже доказали свою состоятельность в качестве инструментов исследования, и с их помощью удалось получить ряд значимых результатов на уровне изолированных ферментов, в культуре клеток, и даже in vivo. С-метилированные аналоги полиаминов имеют безусловный потенциал для изучения существующих и вновь возникающих по мере развития наших знаний “горячих точек” биохимии полиаминов.
Список литературы
Pegg A.E. // J. Biol. Chem. 2016. V. 291. P. 14904-14912.
Igarashi K., Kashiwagi K. // Int. J. Biochem. Cell Biol. 2019. V. 107. P. 104–115.
Miller-Fleming L., Viridiana Olin-Sandoval L., Kate Campbell K., Ralser M. // J. Mol. Biol. 2015. V. 427. P. 3389–3406.
Cohen S.S. A Guide to the Polyamines. N.Y.: Oxford University Press, 1998. P. 595.
Salvi M., Toninello A. // Biochim. Biophys. Acta. 2004. V. 661. P. 113–124.
Berger M.L., Noe Ch.R. // Curr. Top. Med. Chem. 2002. V. 3. P. 51–64.
Nichols C.G., Lee S. // J. Biol. Chem. 2018. V. 293. P. 18779–18788.
Michael A.J. // J. Biol. Chem. 2018. V. 293. P. 18693–18701.
Igarashi K., Kashiwagi K. // J. Biol. Chem. 2018. V. 293. P. 18702–18709.
Park M.H., Wolff E.C. // J. Biol. Chem. 2018. V. 293. P. 18710–18718.
Chattopadhyay M.K., Park M.H., Tabor H. // Proc. Nat. Acad. Sci. U.S.A. 2008. V. 105. P. 6554–6559.
Korhonen V.P., Niiranen K., Halmekyto M., Pietila M., Diegelman P., Parkkinen J.J., Eloranta T., Porter C.W., Alhonen L. Jänne J. // Mol. Pharmacol. 2001. V. 59. P. 231–238.
Mackintosh C.A., Pegg A.E. // Biochem. J. 2000. V. 351. P. 439–447.
Ikeguchi Y., Mackintosh C.A., McCloskey D.E., Pegg A.E. // Biochem. J. 2003. V. 373. P. 885–892.
Nilsson J., Gritli-Linde A., Heby O. // Biochem. J. 2000. V. 352. P. 381–387.
Cason A.L., Ikeguchi Y., Skinner C., Wood T.C., Lubs H.A., Martinez F., Simensen R.J., Stevenson R.E., Pegg A.E., Schwartz C.E. // Eur. J. Hum. Genet. 2003. V. 11. P. 937–944.
Murray-Stewart T., Dunworth M., Foley J.R., Schwartz C.E., Casero R.A. // Med. Sci. (Basel). 2018. V. 6. P. E112.
Kahana C. // Cell. Mol. Life Sci. 2009. V. 66. P. 2479–2488.
Wallace H.M., Fraser A.V. Hughes A. // Biochem. J. 2003. V. 376. P. 1–14.
Casero R.A., Murray Stewart T., Pegg A.E. // Nat. Rev. Cancer. 2018. V. 18. P. 681–695.
Damiani E., Wallace H.M. // Meth. Mol. Biol. 2018. V. 1694. P. 469–488.
Casero R.A., Woster P.M. // J. Med. Chem. 2009. V. 52. P. 4551–4573.
Casero R.A., Marton L.J. // Nat. Rev. Drug Discov. 2007. V. 6. P. 373–390.
Gerner E.W., Bruckheimer E., Cohen A. // J. Biol. Chem. 2018. V. 293. P.18770–18778.
Wallace H.M. // Expert. Opin. Pharmacother. 2007. V. 8. P. 2109–2116.
Goodwin A.C., Destefano Shields C.E., Wu S., Huso D.L., Wu X., Murray-Steward T.R., Hacker-Prietz A., Rabizadeh S., Woster P.M., Sears C.L., Casero R.A., Jr. // Proc. Nat. Acad. Sci. U.S.A. 2011. V. 108. P. 15354–15359.
Hong S.K., Chaturvedi R., Piazuelo M.B., Coburn L.A., Williams C.S., Delgado A.G., Casero R.A., Jr., Schwartz D.A., Wilson K.T. // Inflamm. Bowel Dis. 2010. V. 16. P. 1557–1566.
Chaturvedi R., de Sablet T., Asim M., Piazuelo M.B., Barry D.P., Verriere T.G., Sierra J.C., Hardbower D.M., Delgado A.G., Schneider B.G., Israel D.A., Romero-Gallo J., Nagy T.A., Morgan D.R., Murray-Stewart T., Bravo L.E., Peek R.M., Jr., Fox J.G., Woster P.M., Casero R.A., Jr., Correa P., Wilson K.T. // Oncogene. 2015. V. 34. P. 3429–3440.
Gobert A.P., Wilson K.T. // Trends Microbiol. 2016. V. 24. P. 366–376.
Clement P.M., Henderson C.A., Jenkins Z.A., Smit-McBride Z., Wolff E.C., Hershey J.W., Park M.H., Johansson H.E. // Eur. J. Biochem. 2003. V. 270. P. 254–263.
Nakanishi S., Cleveland J.L. // Amino Acids. 2016. V. 48. P. 2353–2362.
Mathews M.B., Hershey J.W. // Biochem. Biophys. Acta. 2015. V. 1849. P. 836–844.
Lu J., Zhao H.-W., Chen Yu., Wei J.-H., Chen Z.-H., Feng Z.-H., Huang Y., Chen W., Lup J.-H., Fang Y. // Exp. Ther. Med. 2019. V. 17. P. 3741–3747.
Casero R.A., Jr., Pegg A.E. // Biochem. J. 2009. V. 421. P. 323–338.
Hesterberg R.S., Cleveland J.L., Epling-Burnette P.K. // Med. Sci. 2018. V. 6. Pii E22.
Hyvonen M.T., Herzig K.-H., Sinervirta R., Albrecht E., Nordback I., Sand J., Keinanen T.A., Vepsalainen J., Grigorenko N., Khomutov A.R., Kluger B., Janne J., Alhonen L. // Am. J. Pathol. 2006. V. 168. P. 115–122.
Cooper G.J.S., Young A.A., Gamble G.D., Occleshaw C.J., Dissanayake A.M., Cowan B.R., Brunton D.H., Baker J.R., Phillips A.R., Frampton C.M., Poppitt S.D., Doughty R.N. // Diabetologia. 2009. V. 52. P. 715–722.
Meana C., Rubin J.M., Bordallo C., Suarez L., Bordallo J., Sanchez M. // J. Cell Mol. Med. 2016. V. 20. P. 302–312.
Eisenberg T., Abdellatif M., Schoeder S. et al. // Nature Med. 2016. V. 22. P. 1428–1438.
Birholtz L.M., Williams M., Niemand J., Louw A.I., Persson L., Heby O. // Biochem. J. 2011. V. 438. P. 229–244.
Wang B., Pachaiyappan B., Gruber J.D., Schmidt M.G., Zhang Y.M., Woster P.M. // J. Med. Chem. 2016. V. 59. P. 3140–3151.
Schenk T., Chen W.C., Göllner S., Howell L., Jin L., Hebestreit K., Klein H.-U., Popescu A.C., Burnett A., Mills K., Casero R.A., Jr., Marton L., Woster P., Minden M.D., Dugas M., Wang J.C.Y., Dick J.E., Müller-Tidow C., Petrie K., Zelent A. // Nature Med. 2012. V. 18. P. 605–611.
Nagarajan S., Ganem B. // J. Org. Chem. 1986. V. 51. P. 4856–4861.
Nagarajan S., Ganem B., Pegg A.E. // Biochem. J. 1988. V. 254. P. 373–378.
Lakanen J.R., Coward J.K., Pegg A.E. // J. Med. Chem. 1992. V. 35. P. 724–734.
Byers T.L., Lakanen J.R., Coward J.K., Pegg A.E. // Biochem. J. 1994. V. 303. P. 363–368.
Byers T.L., Ganem B., Pegg A.E. // Biochem. J. 1992. V. 287. P. 717–724.
Varnado B.L., Voci L.M., Meyer L.M., Coward J.K. // Bioorg. Chem. 2000. V. 28. P. 395–408.
Nayvelt I., Hyvönen M.T., Alhonen L., Pandya I., Thomas T., Khomutov A.R., Vepsäläinen J., Patel R., Keinänen T.A., Thomas T.J. // Biomacromolecules. 2010. V. 11. P. 97–105.
Григоренко Н.А., Вепсалайнен Й., Ярвинен А., Кейнанен Т.А., Алхонен Л., Янне Ю., Крицын А.М., Хомутов А.Р. // Биоорган. химия. 2004. Т. 30. С. 441–445. (Grigorenko N.A., Vepsalainen J., Jarvinen A., Keinanen T.A., Alhonen L., Janne J., Kritsyn A.M., Khomutov A.R. // Rus. J. Bioorg. Chem. 2004. V. 30. P. 396–399.)
Григоренко Н.А., Вепсалайнен Й., Ярвинен А., Кейнанен Т.А., Алхонен Л., Янне Ю., Хомутов А.Р. // Биоорган. химия. 2005. Т. 31. С. 200–205. (Grigorenko N.A., Vepsalainen J., Jarvinen A., Keinanen T.A., Alhonen L., Janne J., Khomutov A.R. // Rus. J. Bioorg. Chem. 2005. V. 31. P. 183–188.)
Rasanen T.L., Alhonen L., Sinervirta R., Keinanen T., Herzig K-H., Suppola S., Khomutov A.R., Vepsalainen J., Janne J. // J. Biol. Chem. 2002. V. 277. P. 39867–39872.
Alhonen L., Parkkinen J.J., Keinanen T., Sinervirta R., Herzig K.H., Janne J. // Proc. Nat. Acad. Sci. U.S.A. 2000. V. 97. P. 8290–8295.
Хомутов М.А., Хивонен М.Т., Симонян А.Р., Вепсалайнен Й., Алхонен Л., Кочетков С.Н., Кейнанен Т.А. // Биоорган. химия. 2011. Т. 37. С. 253–258. (Khomutov M.A., Hyvönen M.T., Simonian A.R., Vepsäläinen J., Alhonen L., Kochetkov S.N., Keinänen T.A. // Rus. J. Bioorg. Chem. 2011. V. 37. P. 225–230.)
Hyvönen M.T., KeinänenT.A., Khomutov M., Simonian A., Weisell J., Kochetkov S.N., Vepsäläinen J., Alhonen L., Khomutov A.R. // J. Med. Chem. 2011. V. 54. P. 4611–4618.
Hegde S.S., Chandler J., Vetting M.W., Yu M., Blanchard J.B. // Biochemistry. 2007. V. 46. P. 7187–7195.
Montemayor E.J., Hoffman D.W. // Biochemistry. 2008. V. 47. P. 9145–9153.
Khomutov M.A., Hyvönen M.T, Simonian A.R., Weisell J., Vepsäläinen J., Alhonen L., Kochetkov S.N., Keinänen T.A., Khomutov A.R. // Mendeleev Commun. 2018. V. 28. P. 479–481.
Kahana C. // J. Biol. Chem. 2018. V. 293. P. 18730–18735.
Hyvönen M.T., Keinänen T.A., Khomutov M., Simonian A., Vepsäläinen J., Park J.H., Khomutov A.R., Alhonen L., Park M.H. // Amino Acids. 2012. V. 42. P. 685–695.
Даидх С., Хомутов М.А., Симонян А.Р., Кочетков С.Н., Мадхубала Р. // Мол. биол. 2011. Т. 45. С. 673–678.
Phillips M.A. // J. Biol. Chem. 2018. V. 293. 18746–18756.
Hobley L., Kim S.H., Maezato Y., Wyllie S., Fairlamb A.H., Stanley-Wall N.R., Michael A.J. // Cell. 2014. V. 156. P. 844–854.
Kim S.H., Wang Y., Khomutov M., Khomutov A., Fuqua C., Michael A. // ACS Chem. Biol. 2016. V. 11. P. 491–499.
Lebreton L., Jost E., Carboni B., Annat J., Vaultier M., Dutartre P., Renaut P. // J. Med. Chem. 1999. V. 42. P. 4749–4763.
Bergeron R.J., Muller R., Huang G., McManis J.S., Algee S.E., Yao H., Weimar W.R., Wiegand J. // J. Med. Chem. 2001. V. 44. P. 2451–2459.
Jarvinen A.J., Cerrada-Gimenez M., Grigorenko N.A., Khomutov A.R., Vepsalainen J.J., Sinervirta R.M., Keinanen T.A., Alhonen L.I., Janne J.E. // J. Med. Chem. 2006. V. 49. P. 399–406.
Holtta E. // Biochemistry. 1977. V. 16. P. 91–100.
Jarvinen A., Keinanen T.A., Grigorenko N., Khomutov A.R., Uimari A., Vepsalainen J., Narvanen A., Alhonen L., Janne J. // J. Biol. Chem. 2006. V. 281. P. 4589–4595.
Wang Y., Devereux W., Woster P.M., Stewart T.M., Hacker A., Casero R.A., Jr. // Cancer Res. 2001. V. 61. P. 5370–5373.
Hyvonen M.T., Keinanen T.A., Cerrada-Gimenez M., Sinervirta R., Grigorenko N., Khomutov A.R., Vepsalainen J., Alhonen L., Janne J. // J. Biol. Chem. 2007. V. 282. P. 34700–34706.
Keinänen T.A., Grigorenko N., Khomutov A.R., Huang Q., Uimari A., Alhonen L., Hyvönen M.T., Vepsäläinen J. // Bioscience Reports. 2018. V. 38. P. BSR20180527.
Hyvönen M.T., Khomutov M., Petit M., Weisell J., Kochetkov S.N., Alhonen L., Vepsäläinen J., Khomutov A.R., Keinänen T.A. // ACS Chem. Biol. 2015. V. 10. P. 1417–1424.
Хомутов М.А., Хивонен М.Т., Вепсалайнен Й., Алхонен Л., Кейнанен Т.А., Кочетков С.Н., Хомутов А.Р. // Биоорган. химия. 2015. V. 41. P. 612–618. (Khomutov M.A., Keinanen T.A., Hyvonen M.T, Weisell J., Vepsalainen J., Alhonen L., Kochetkov S.N., Khomutov A.R. // Rus. J. Bioorg. Chem. 2015. V. 41. P. 548–553.)
O’Sullivan M.C., Zhou Q. // Bioorg. Med. Chem. Lett. 1995. V. 5. P. 1957–1960.
Khomutov A.R., Shvetsov A.S., Vepsalainen J.J., Kritzyn A.M. // Tetrahedron Lett. 2001. V. 42. P. 2887–2889.
Weisell J., Hyvönen M.T., Häkkinen M.R., Grigorenko N.A., Pietilä M., Lampinen A., Kochetkov S.N., Alhonen L., Vepsäläinen J., Keinänen T.A., Khomutov A.R. // J. Med. Chem. 2010. V. 53. P. 5738–5748.
Хомутов А.Р., Григоренко Н.А., Демин А.В., Вепсалайнен Й., Касеро Р.А., Уостер П.М. // Биоорган. химия. 2005. T. 31. C. 303–311. (Khomutov A.R., Grigorenko N.A., Skuridin S.G., Demin A.V., Vepsalainen J., Casero R.A., Woster P.M. // Rus. J. Bioorg. Chem. 2005. V. 31. P. 271–278.)
Bergeron R.J., Garlich J.R., Stolowich N.J. // J. Org. Chem. 1984. V. 49. P. 2997–3001.
Humora M.J., Quick J. // J. Org. Chem. 1979. V. 41. P. 1166–1168.
Nagarajan S., Ganem B. // J. Org. Chem. 1985. V. 50. P. 5735–5737.
Fukuyama T., Chung-Kuang J., Cheung M. // Tetrahedron Lett. 1995. V. 36. P. 6373–6374.
Favre-Reguillon A., Segat-Dioury F., Nait-Bouda L., Cosma C., Siaugue J.-M., Foos J., Guy A. // Syn. Lett. 2000. V. 6. P. 868–870.
Grigorenko N.A., Khomutov A.R., Keinänen T.A., Järvinen A., Alhonen L., Jänne J., Vepsäläinen J. // Tetrahedron. 2007. V. 63. P. 2257–2262.
Kaur N., Delcros J.-G., Martin B., Phanstiel O. IV // J. Med. Chem. 2005. V. 48. P. 3832–3839.
Khomutov M.A., Chizhov A.O., Kochetkov S.N., Khomutov A.R. // Mendeleev Commun. 2019. in press.
Carboni B., Benalil A., Vaultier M. // J. Org. Chem. 1993. V. 58. P. 3736–3741.
Brand G., Hosseini M.W., Ruppert R. // Tetrahedron Lett. 1994. V. 35. P. 8609–8612.
Krakowiak K.E., Bradshaw J.S. // Synth. Commun. 1996. V. 21. P. 3999–4004.
Iwata M., Kuzuhara H. // Bull. Chem. Pharm. Jpn. 1989. V. 62. P. 1102–1106.
Kashiwagi K., Pahk A.J., Masuko T., Igarashi K., Williams K. // Mol. Pharmacol. 1997. V. 52. P. 701–713.
Bergeron R.J., Weimar W.R., Wu Q., Feng Y., Mc‑Manis J.S. // J. Med. Chem. 1996. V. 39. P. 5257–5266.
Bergeron R.J., Feng Y., Weimar W.R., McManis J.S., Dimova H., Porter C.W., Raisler B., Phanstiel O. // J. Med. Chem. 1997. V. 40. P. 1475–1494.
Saab N.H., West E.E., Bieszk N.C., Preuss C.V., Mank A.R., Casero R.A., Jr., Woster P.M. // J. Med. Chem. 1993. V. 36. P. 2998–3004.
Thoo Lin P.K., Kuksa V.A., Maguire N.M. // Synthesis. 1998. P. 859–866.
Kuksa V.A., Pavlov V.A., Thoo Lin P.K. // Bioorg. Med. Chem. 2002. V. 10. P. 691–697.
Khomutov M., Mikhura I.V., Kochetkov S.N., Khomutov A.R. (неопубликованные данные).
Grigorenko N.A., Vepsalainen J., Jarvinen A., Keinanen T.A., Alhonen L., Janne J., Khomutov A.R. // Mendeleev Commun. 2005. V. 15. P. 142–143.
Григоренко Н.А., Хомутов М.А., Симонян А.Р., Кочетков С.Н., Хомутов А.Р. // Биоорган. химия. 2016. Т. 42. С. 469–474. (Grigorenko N.A., Khomutov M.A., Simonian A.R., Kochetkov S.N., Khomutov A.R. // Russ. J. Bioorg. Chem. 2016. V. 42. P. 423–427.)
Khomutov A.R., Weisell J., Khomutov M.A., Grigorenko N.A., Simonian A.R., Häkkinen M.R., Keinänen T.A., Hyvönen M.T., Alhonen L., Kochetkov S.N., Vepsäläinen J. Methylated polyamines as research tools // Polyamines: Methods and Protocols, Methods in Molecular Biology / Eds. Pegg A.E., Casero R.A. Springer Science+Business Media, LLC. 2011. V. 720. P. 449–461.
Bok Lee Y., Folk J.E. // Bioorg. Med. Chem. 1998. V. 6. P. 253–270.
Edwards M.L., Prakash N.J., Stemerick D.M., Sunkara S.P., Bitonti A.J., Davis G.F., Dumont J.A., Bey P. // J.Med.Chem. 1990. V. 33. P. 1369–1375
Hughes D.L. The Mitsunobu Reaction // Organic Reactions / Ed. Paquette L.A. New York: Wiley, 1992. V. 42. P. 335.
Edwards M.L., Stemerick D.M., McCarthy J.R. // Tetrahedron Lett. 1990. V. 31. P. 3417–3420.
Osby J.O., Ganem B. // Tetrahedron Lett. 1985. V. 26. P. 6413–6416.
Mamos P., Karigiannis G., Athanassopoulos C., Bichta S., Kalpaxis D., Papaioannou D., Sindona G. // Tetrahedron Lett. 1995. V. 36. P. 5187–5190; Karigiannis G., Papaioannou D. // Eur. J. Org. Chem. 2000. V. 10. P. 1841–1863.
Bräse S., Banert K. Organic Azides Syntheses and Applications. John Wiley & Sons Ltd., 2010. P. 507.
Patai S. The Chemistry of Azido Group. Interscience Publishers: John Wiley and Sons, 1971. P. 333.
Vaultier M., Lambert P. H., Carrib R. // Bull. Soc. Chem. Fr. 1986. № 1. P. 83–87.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия