

Биоорганическая химия, 2020, T. 46, № 1, стр. 36-46
Конвергентный синтез галанина крысы и изучение его биологической активности
М. В. Сидорова *, М. Е. Палькеева *, Д. В. Авдеев *, А. С. Молокоедов *, М. В. Овчинников *, А. А. Азьмуко *, Л. И. Серебрякова *, О. М. Веселова *, И. М. Студнева *, О. И. Писаренко *
* ФГБУ “Национальный медицинский исследовательский центр кардиологии” Минздрава России
121552 Москва, 3-я Черепковская ул. 15а, Россия
Поступила в редакцию 11.04.2019
После доработки 19.06.2019
Принята к публикации 26.06.2019
Аннотация
Конвергентным твердофазным синтезом с использованием Fmoc-методологии получен полноразмерный галанин крысы GWTLNSAGYLLGPHAIDNHRSFSDKHGLT-NH2 (G29). Пептидную цепь в ходе синтеза наращивали по одной аминокислоте и фрагментной конденсацией. Фрагменты с С-концевым остатком глицина получали твердофазным методом на 2-хлортритилхлоридной смоле или в растворе. После ВЭЖХ-очистки галанин имел корректную молекулярную массу и чистоту 98%. Кардиопротекторные свойства G29 изучали на модели острого инфаркта миокарда у крыс. Введение G29 уменьшало размеры инфаркта миокарда на 40% и снижало активность маркеров некроза МВ-креатинкиназы и лактатдегидрогеназы в плазме. Пептид G29 улучшал метаболическое состояние сердца – увеличивал содержание АТР, общего фонда адениннуклеотидов, фосфокреатина, общего креатина и снижал уровень лактата по сравнению с контролем. Результаты указывают на возможность использования пептида G29 в качестве препарата для уменьшения реперфузионных повреждений сердца и необходимость изучения механизмов его действия.
ВВЕДЕНИЕ
Нейропептид галанин (G29), состоящий из 29 аминокислотных остатков (а.о.) (29 а.о. – у большинства млекопитающих, 30 а.о. – у человека), синтезируется преимущественно в головном и спинном мозге и, в меньшей степени, в кишечнике, он широко распространен в центральной и периферической нервной системе [1]. Галанин регулирует ряд важных функций организма: запоминания, потребления пищи, засыпания, алкогольной зависимости, невропатической боли, выработки гормонов и др. Этот пептид участвует в центральной регуляции сердечно-сосудистой системы [2, 3], снижает резистентность к инсулину [4] и улучшает поглощение глюкозы в сердце экспериментальных животных на моделях сахарного диабета [5]. В сердце и периферических органах и тканях галанин действует не только через нейрональные механизмы, но и активируя клеточные рецепторы GalR1-3 [4, 5], которые являются потенциальной мишенью для лечения различных заболеваний [6]. Литературные данные [7, 8] позволяют предположить участие галанина в реализации различных механизмов защиты сердца от ишемического и реперфузионного повреждения. Однако до настоящего времени эти механизмы мало изучены и данных о влиянии галанина на сердечно-сосудистую систему немного [9].
Настоящая работа является продолжением наших исследований по поиску и изучению пептидов, обладающих кардиопротекторными свойствами. Ранее нами было показано, что N-концевые фрагменты галанина улучшают восстановление функции изолированного сердца крысы после тотальной ишемии и ограничивают размеры инфаркта миокарда (ИМ) in vivo после региональной ишемии и реперфузии сердца крыс [10]. Эти эффекты обусловлены снижением образования активных форм кислорода, ингибированием апоптоза и связаны с улучшением энергетического состояния сердца [11]. Данная работа посвящена синтезу полноразмерного галанина крысы и изучению его биологической активности.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Выбор стратегии синтеза
Последовательность 29-членного галанина, приведенная на рис. 1, представляется достаточно сложной для ступенчатого твердофазного синтеза (ТФС). При многостадийном ТФС длинных пептидов за счет неколичественных выходов на стадиях деблокирования α-аминогрупп и последующего создания амидных связей образуются “ложные” пептиды с пропуском (делецией) отдельных аминокислотных остатков или их сочетаний. Последовательность галанина содержит ряд трифункциональных аминокислот (таких как Trp, Asp, Ser, Thr, Arg), легко вступающих в побочные реакции в ходе ТФС или постсинтетических процедур. Так, например, при синтезе аспартилпептидов на стадиях отщепления Fmoc-защит действием вторичных аминов происходит образование циклического аспартимида, результатом чего является α→β-перегруппировка амидной связи с образованием изомерного побочного продукта.
Рис. 1.
Аминокислотная последовательность галанина крысы (G29). Стрелками показаны места разбиения на фрагменты.

Образование аспартимида, катализируемое кислотой или основанием, является одной из наиболее серьезных побочных реакций в ТФС пептидов, содержащих аспарагиновую кислоту, и до сих пор не разработана экономически эффективная стратегия ее подавления [12, 13]. Степень протекания транспептидации главным образом определяется конкретной аминокислотной последовательностью. Наиболее подвержены этой побочной реакции последовательности Asp-Gly, Asp-Asn, Asp-Asp [14, 15]. Для подавления транспептидации применяют различные методические приемы (использование объемных, стерически затрудненных защитных групп для β-карбоксильной функции аспарагиновой кислоты, модификации деблокирующего реагента для отщепления Fmoc-защиты, сокращение суммарного времени воздействия основных реагентов), однако полностью исключить эту побочную реакцию до сих пор не удается. В связи с вышесказанным очистка продукта ТФС, содержащего близкие по составу и свойствам примеси, – сложный многостадийный процесс.
Для преодоления перечисленных проблем был разработан конвергентный, или блочный [16–18] синтез полипептидов. В соответствии с этой стратегией последовательность пептида разбивается на относительно короткие фрагменты, которые получают на твердой фазе или в растворе и, после соответствующей очистки, конденсируют на полимерном носителе или в растворе. Этот метод был успешно применен при синтезе ряда полипептидов, насчитывающих от 24 до 100 а.о. [19–21]. В нашей лаборатории этот подход был использован в синтезе последовательности 1-42-β-амилоида [22] и 26-членного фрагмента, соответствующего иммунодоминантной последовательности 197–222 β1-адренорецептора [23]. Сравнительная оценка ступенчатого и фрагментного синтеза этих двух пептидов убедительно показала преимущества последнего подхода [22–24].
В этой связи для получения галанина был выбран комбинированный способ наращивания пептидной цепи: на отдельный участках пошагово, а на других – конденсацией фрагментов. Последовательность пептида была разбита на фрагменты по остаткам глицина (разбиение показано стрелками на рис. 1), соответствующие последовательностям 23–27 (ФI), 9–12 (ФII) и 1–8 (ФIII). На участке 13–22, не содержащем “удобных” для фрагментации (нерацемизующихся при конденсации) аминокислотных остатков, было решено наращивать пептидную цепь ступенчато по одной аминокислоте. В сочетании с Fmoc-защитой α‑аминогруппы для блокирования боковых функций аминокислот использовали But – для гидроксильных групп Ser, Thr, Tyr и карбоксильной группы Asp; Boc – для ε-аминогруппы Lys и индольного кольца Trp; Pbf – для гуанидиновой группы Arg; Trt – для карбоксамидной группы Asn и имидазола His.
Синтез защищенных фрагментов
Синтез фрагментов ФI и ФIII был проведен с использованием Fmoc-методологии на полимерном носителе с 2-хлортритил-хлоридной якорной группой [24], которая обеспечивает получение защищенных пептидных блоков со свободной С‑концевой карбоксильной группой благодаря возможности “мягкого” расщепления якорной сложноэфирной связи (например, действием смеси уксусной кислоты и трифторэтанола в дихлорметане), при котором сохраняются защитные группы боковых цепей аминокислот.
Фрагмент ФII был получен классическими методами пептидной химии в растворе по схеме 1 , исходя из бензилового эфира глицина. Пептидную цепь наращивали с использованием Boc- и Z-производных соответствующих аминокислот последовательным синтезом с использованием избытков смешанных ангидридов [25]. Полупродукты синтеза выделяли из реакционных смесей путем экстракции и промывок с последующей кристаллизацией из подходящих растворителей. Индивидуальность полупродуктов получения фрагмента ФII контролировали с помощью ТСХ. Вос-группу удаляли действием трифторуксусной кислоты, а Z-защиту отщепляли каталитическим гидрогенолизом над 5% палладием на угле (Pd/C). На заключительной стадии синтеза ФII после отщепления Z-группы в пептид вводили Fmoc-защитную группу действием Fmoc-ONSu по известной методике [26].

Схема 1 . Синтез пептидного фрагмента Fmoc-Tyr(But)-Leu-Leu-Gly-OH (ФII).
Индивидуальность полученных фрагментов ФI–ФIII анализировали с помощью ТСХ и ВЭЖХ (после полного деблокирования соответствующих образцов). По данным аналитической ВЭЖХ чистота полученных фрагментов была не менее 90%. Структура пептидных фрагментов ФI–ФIII подтверждена данными 1Н-ЯМР-спектроскопии и масс-спектрометрии. Характеристики фрагментов приведены в эксп. части.
Синтез галанина G29
ТФС G29 проводили с С-конца (см. схему 2 ) с использованием 3-кратных избытков Nα-Fmoc-защищенных аминокислот и пептидных фрагментов на амидном полимере Ринка. На первом этапе синтеза ступенчато присоединяли остатки Thr29 и Leu28, затем к дипептидилполимеру PI присоединяли пептапептид ФI и получали гептапептидилполимер PII. Начиная с Phe22 и до Pro13 (в течение 10 циклов ТФС) синтез вели ступенчато, присоединяя по одной аминокислоте DIC/HOBt-методом. На завершающем этапе к пептидилполимеру РIII последовательно присоединяли блоки ФII и ФIII. Для конденсации пептидных фрагментов мы использовали комплекс F (аддукт N,N '-дициклогексилкарбодиимида и пентафторфенола в мольном соотношении 1 : 3) [21], хорошо зарекомендовавший себя в блочном ТФС [27]. Полноту протекания фрагментных конденсаций контролировали тестом Кайзера c нингидрином [28].

Схема 2 . Конвергентный синтез галанина крысы G29. (Комплекс F – аддукт N,N'-дициклогексилкарбодиимида и пентафторфенола в мольном отношении 1 : 3.)
Деблокирование α-аминогруппы в ходе синтеза G29 проводили 25% раствором 4-MePip в DMF. Выше уже говорилось о том, что последовательность Asp-Asn, присутствующая в молекуле галанина, склонна к образованию аспартимида и последующей α→β-перегруппировке амидной связи в условиях ТФС с использованием Fmoc-методологии [15]. Для минимизации этой побочной реакции, начиная со стадии деблокирования остатка Asn18, для отщепления Fmoc-защиты мы использовали модифицированный деблокирующий реагент – 25% раствор 4-метилпиперидина с добавкой 0.1 М 1-гидроксибензотриазола, как было предложено в работе [29]. В результате конвергентного синтеза, проведенного в соответствии со схемой 2 , был получен пептидилполимер PV, после удаления Nα-Fmoc-защиты целевой продукт был отщеплен от полимера и полностью деблокирован действием трифторуксусной кислоты со специальными добавками. Неочищенный продукт G29 содержал по данным аналитической ВЭЖХ 77.1% основного вещества (рис. 2а). Для дополнительной оценки содержания аспартимидов в неочищенном продукте ТФС галанина G29 образец Nα – свободного пептидилполимера PV был обработан деблокирующей смесью, не содержащей воды. Масс-спектрометрия полученного продукта показала, что, наряду с целевым пептидом G29 (m/z 3165.48), в его состав входит вещество с молекулярной массой на 18 Да меньше (m/z 3147.306), которое может быть побочным аспартимидом (продуктом дегидратации) по остатку аспарагиновой кислоты, участвующей в образовании связи Asp-Asn. Соотношение интенсивностей целевого и побочного продуктов в масс-спектре составило 100 : 4.6 (Δ – 18). Галанин был очищен с помощью ВЭЖХ на колонке с обращенной фазой (рис. 2б). Полученный пептид имел корректную молекулярную массу (по данным масс-спектрометрии) и оказался идентичен коммерческому образцу галанина крысы (GeneCust, Люксембург).
Рис. 2.
Профили ВЭЖХ неочищенного (а) и очищенного (б) галанина крысы. Колонка Kromasil C18-100-5 4.6 × 250 мм, размер частиц сорбента 5 мкм; пептиды элюировали со скоростью 1 мл/мин градиентом буфера Б в буфере А от 20 до 80% за 30 мин, буфер А – 0.05 М KH2PO4, pH 3.0, буфер Б 70% ацетонитрил в буфере А, детекция при 220 нм (условия 1).

Таким образом, использованные нами конвергентный подход и модифицированный реагент для отщепления Fmoc-защит обеспечили получение неочищенного галанина крысы с хорошей чистотой и относительно низким содержанием побочных продуктов транспептидации (менее 2.5%); продукт, как видно на рис. 2а, не содержал трудноотделимых примесей с близкими временами удерживания. Выбранные для фрагментной конденсации пептидные блоки были хорошо растворимы в NMP, реакции образования амидной связи в присутствии комплекса F протекали достаточно быстро (в течение 4 ч) и с высокими выходами.
Оценка биологической активности пептида G29 у крыс in vivo
Нами была использована модель региональной ишемии и реперфузии сердца крыс Вистар. На начальном этапе было изучено дозозависимое влияние внутривенного введения G29 в начале реперфузии (после периода региональной ишемии) на размеры инфаркта миокарда (ИМ). Гистохимический анализ срезов левого желудочка (ЛЖ) сердца в конце реперфузии не выявил различий между экспериментальными группами в зоне риска (ЗР). Величина ЗР/ЛЖ, % в контроле, в группе G29 и растворителя пептида G29 (0.2% DMSO) была близкой и составляла в среднем 41.1 ± 0.9%. Это означает, что повреждение сердца было стандартно моделировано у всех животных. В контроле величина инфаркта миокарда, выраженная отношением ИМ/ЗР, составила 43.0 ± 2.0%. Введение 0.2% DMSO не влияло на этот показатель. Любая из изученных доз G29 достоверно ограничивала размеры ИМ (P < 0.005–0.001 (рис. 3а). В наибольшей степени отношение ИМ/ЗР (в %) снижалось под действием дозы 0.5 мг/кг – до 60% от величины в контроле. В дальнейшем она была использована в качестве оптимальной для изучения влияния пептида на активность маркеров некроза в плазме и метаболическое состояние ЗР.
Рис. 3.
Уменьшение повреждения сердца пептидом G29 у крыс in vivo. (a) Дозозависимое снижение размеров инфаркта миокарда (ИМ/ЗР, %) при внутривенном введении G29 после региональной ишемии. Данные представлены как M ± m для серий из 8 опытов. K – контроль. * P < 0.001 от К, # P < 0.005 от К. (б, в) Влияние внутривенного введения оптимальной дозы G29 (0.5 мг/кг) на активность креатинкиназы-МВ (-МВ- КК) и лактатдегидрогеназы (ЛДГ) в плазме в конце реперфузии. Данные представлены как M ± m для серий из 8 опытов. ИС – исходное состояние; K – контроль; DMSO – введение 0.2% DMSO. * P < 0.05 от К и DMSO.
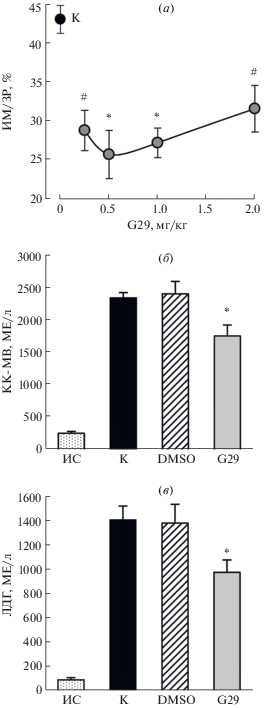
Таблица 1.
Влияние внутривенного введения пептида G29 на метаболическое состояние зоны риска сердца крысы в конце реперфузии
| Исходное состояние | Конец реперфузии | ||
|---|---|---|---|
| контроль | G29, 0.5 мг/кг | ||
| ATP | 21.35 ± 1.18 | 4.73 ± 0.73* | 6.25 ± 0.39* |
| ADP | 5.52 ± 0.43 | 3.38 ± 0.16* | 7.62 ± 0.28*# |
| AMP | 1.36 ± 0.25 | 1.24 ± 0.18 | 2.25 ± 0.18*# |
| ΣAН | 28.24 ± 1.24 | 9.35 ± 0.74* | 16.12 ± 0.43*# |
| ФКр | 23.56 ± 1.78 | 8.18 ± 1.38* | 12.18 ± 0.97*# |
| Кр | 46.43 ± 4.52 | 37.53 ± 4.17 | 40.12 ± 5.05 |
| ΣКр | 69.98 ± 4.27 | 44.91 ± 3.34* | 52.31 ± 4.17* |
| ФКр/Кр | 0.51 ± 0.04 | 0.21 ± 0.02* | 0.30 ± 0.03*# |
| Лактат | 1.15 ± 0.11 | 10.82 ± 0.93* | 2.14 ± 0.17*# |
Активность маркеров некроза креатинкиназы МВ (МВ-КК) и лактатдегидрогеназы (ЛДГ) в плазме крови в исходном состоянии (перед окклюзией ПНА) составляла 270.2 ± 28.5 и 92.8 ± ± 18.5 МЕ/л соответственно. Развитие ИМ в контроле сопровождалось увеличением активности обоих ферментов в конце реперфузии по сравнению с исходным состоянием – до 2350.1 ± ± 80.7 и 1415.0 ± 112.6 МЕ/л для МВ-КК и ЛДГ соответственно (рис. 3б, 3в). Введение оптимальной дозы G29 перед началом реперфузии достоверно снижало активность МВ-КК и ЛДГ на 25 и 30% соответственно, по сравнению с контролем.
Влияние введения пептида G29 на показатели метаболического состояния ЗР в конце реперфузии представлено в табл. 1. Ишемическое и реперфузионное повреждение вызывало значительные изменения в содержании метаболитов энергетического обмена и лактата в сердце. В контроле в конце реперфузии содержание АТP было снижено в 4.5 раза, а общий пул адениннуклеотидов (ΣAH) за счет уменьшения АDP – в 3 раза по сравнению с исходным состоянием. Содержание ФКр снижалось до 35%, что вызывало уменьшение общего креатина (ΣКр) до 64% от исходных значений при практически неизменном уровне Кр. Недостаточное восстановление аэробного обмена в ЗР к окончанию реперфузии сопровождалось активацией гликолиза/гликогенолиза. Это приводило к накоплению лактата, содержание которого увеличивалось почти на порядок по сравнению с исходным значением. Введение пептида G29 увеличивало содержание АТP (P = 0.088) и достоверно АDP и АМP (P < 0.01) по сравнению с контролем. В результате ΣAH был увеличен в 1.7 раза. Одновременно отмечено лучшее восстановление ФКр (на 48% по сравнению с контролем), что сопровождалось сохранением более высокого содержания ΣКр и отношения ФКр/Кр, отражающего фосфорилирование Кр. Эти данные свидетельствуют о более эффективном энергообеспечении ЗР под действием G29, одновременно наблюдалось снижение содержания лактата в ткани сердца до значения, близкого к исходному.
Полученные результаты однозначно указывают на способность пептида G29 уменьшать летальные повреждения кардиомиоцитов при ишемии и реперфузии сердца in vivo. Кардиозащитное действие пептида проявляется в ограничении размеров острого инфаркта, уменьшении активности маркеров некроза в кровотоке и в улучшении энергетического состояния инфарцированного миокарда.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использованы производные L-аминокислот (Novabiochem, Швейцария), DIEA, HOBt, TBTU, TIS, DMF, N-метилпирролидон, дихлорметан и трифторуксусная кислота (Fluka, Швейцария), ацетонитрил и изопропиловый спирт (Panreac, Испания).
Для ТСХ пептидных фрагментов использовали пластинки Kieselgel 60 F254 (Merck, ФРГ) и системы растворителей хлороформ–метанол–АсОН (90 : 8 : 2) (система 1), хлороформ–метанол– АсОН (9 : 1 : 0.5) (система 2), пептиды проявляли в УФ-свете и парами Сl2–бензидин [30].
ВЭЖХ проводили на хроматографах Gilson (Франция) и Knauer (ФРГ). Аналитическую ВЭЖХ галанина выполняли на колонке Kromasil 100-5 C18 с сорбентом С18, размер пор 100 Å, на хроматографе Gilson (Франция). Элюенты: буфер А – 0.05 М КН2РО4, pH 3.0, буфер Б – 70% ацетонитрила в буфере А, элюция градиентом концентрации буфера Б в буфере А от 20 до 80% за 30 мин. Детекция при 220 нм (условия 1). Для аналитической ВЭЖХ пептидных фрагментов использовали буфер А – 0.1% TFA, буфер Б – 80% ацетонитрила в буфере А, градиент от 10 до 70% буфера Б в А (условия 2). Препаративная ВЭЖХ проводилась на колонке Kromasil С18 (30 × 250 мм), размер частиц сорбента 10 мкм. Элюенты: буфер А – 0.1% ТFA, буфер Б – 80% ацетонитрила в буфере А. Элюцию проводили градиентом 0.5% в минуту буфера Б в буфере А от 100% буфера А со скоростью 10 мл/мин. Детекция при длине волны 220 нм. Фракции, соответствующие целевому веществу, объединяли, концентрировали в вакууме и лиофилизовали.
1Н-ЯМР-спектры регистрировали на спектрометре WH-500 Bruker 500 МГц (ФРГ) в DMSO-d6 при 300 K, концентрация пептидов составляла 2–3 мг/мл, химические сдвиги (δ, м.д.) измеряли относительно тетраметилсилана, константы спин-спинового взаимодействия (J) – в герцах. Отнесение сигналов к определенным группам протонов аминокислотных остатков проводилось с помощью метода дифференциального двойного резонанса. Масс-спектры регистрировали на масс-спектрометре Bruker Autoflex speed (Bruker Daltonics Inc., ФРГ), оснащенном твердотельным УФ-лазером с λ 355 нм и рефлектроном, в режиме регистрации положительно заряженных ионов. Для регистрации масс-спектров MALDI использовали стальную мишень MTP 384 ground steel. Энергия лазера подбиралась индивидуально для каждого образца, частота облучения 50 Гц, при регистрации масс-спектра суммировались данные, полученные при 10 последовательных облучениях.
Синтез пептидных фрагментов (общие методики)
Пептидные фрагменты ФI и ФIII получали на 2-хлортритилхлоридной смоле (Iris Biotech, ФРГ), содержащей 1.2 экв. Cl/г. К 3.0 г (3.6 ммоль) смолы прибавляли раствор 3.2 г (10.8 ммоль) Fmoc-Gly-OH в 30 мл DCM, в полученную суспензию приливали 4.25 мл (15.0 ммоль) DIEA и перемешивали 30 мин при 25°С, полимер отфильтровывали, промывали DCM 2 × 30 мл, DMF 2× 30 мл, DCM 2 × 30 мл. Остаточный хлор отщепляли смесью DCM–MeOH–DIEA (32 : 6 : : 2 v/v), промывали DCM 2 × × 30 мл и DMF 2 × × 30 мл. Содержание стартовой аминокислоты составило 0.46 ммоль Fmoc-Gly/г аминоацилполимера. При синтезе фрагментов ФI и ФIII, исходили из 3 г (1.4 ммоль) Fmoc-Gly-аминоацилполимера. Цикл ТФС включал следующие основные стадии: 1) деблокирование α-аминогрупп 25% 4-Ме Pip/DMF в течение 10 мин, 2) промывка DMF, 3) конденсация 4-кратного избытка Fmoc-AA/ DIC/HOBt в смеси DMF/NMP (1 : 1) в течение 2 ч, 4) промывка DMF. Защищенные пептиды отщепляли от полимерного носителя смесью (40 мл) AcOH–трифторэтанол–DCM (1 : 2 : 7) в течение 40 мин. Полимер отфильтровывали, промывали 2 × 30 мл деблокирующей смеси, фильтрат упаривали, продукт осаждали сухим эфиром и после высушивания кристаллизовали из подходящего растворителя.
Фрагмент (ФII) Fmoc-Tyr(But)-Leu-Leu-Gly-OH получали, исходя из 6.74 г (20 ммоль) H-Gly-OBzl ⋅ Tos-OH, методом смешанных ангидридов по приведенной ниже методике.
К охлажденному до –15°С раствору 24.0 ммоль карбоксильного компонента в 80 мл DMF прибавляли 2.66 мл (24 ммоль) NMM и 3.12 мл (24 ммоль) изо-бутилхлорформиата, выдерживали 5 мин и приливали охлажденный до –15°С раствор 20 ммоль аминокомпонента и 2.22 мл (20.0 ммоль) NMM в 30 мл DMF. Реакционную смесь выдерживали 20 мин при –15°С и 1 ч при 0°С. Ход реакции контролировали с помощью ТСХ в системе 1. В реакционную смесь прибавляли раствор KHCO3 до начала слабого выделения углекислого газа и перемешивали 30 мин. Реакционную смесь упаривали, остаток растворяли в 200 мл этилацетата и промывали 2% раствором H2SO4 (3 × 50 мл), водой (3 × 50 мл), 5% раствором NaHCO3 (3 × 50 мл) и водой до нейтральной реакции. Органический слой сушили над безводным сульфатом натрия, осушитель отфильтровывали, фильтрат упаривали. Остаток обрабатывали трифторуксусной кислотой в течение 1 ч, реакционную смесь упаривали, трифторацетат пептида осаждали диэтиловым эфиром, осадок отфильтровывали, промывали на фильтре диэтиловым эфиром, высушивали и вводили в конденсацию с новым карбоксильным компонентом.
К раствору 10.7 г (14.4 ммоль) тетрапептида Z-Tyr(But)-Leu-Leu-Gly-OBzl в 300 мл этанола прибавляли 1.4 г 5% Pd/C и в эту суспензию при перемешивании пропускали ток водорода в течение 2 ч, затем в реакционную смесь приливали 14 мл 1 M NaOH. По окончании реакции катализатор отфильтровывали, фильтрат упаривали. В полученный пептид Н-Tyr(But)-Leu-Leu-Gly-ОН вводили Nα-Fmoc-защиту по описанной методике [26]. Фрагмент ФII кристаллизовали из смеси изо-пропиловый спирт–гексан.
Ниже приведены выходы и характеристики фрагментов ФI–ФIII. Кроме этого образцы полученных фрагментов были полностью деблокированы, после чего их чистоту анализировали с помощью ВЭЖХ. Содержание основного вещества в полученных образцах составило 90–95%.
Fmoc-Ser(But)-Asp(OBut)-Lys(Boc)-His(Trt)-Gly-OH (ФI). Получено 1.3 г (74.0% в расчете на стартовую аминокислоту, присоединенную к полимеру), Rf 0.23 (система 1), 0.62 (система 2). MS, m/z (Iотн, %): 1241.756 (100) [M + Na]+, 1257.777 (38) [M + K]+, 1263.772 (14) [M + 2Na-H]+. Спектр 1H-ЯМР: 1.21 (с, 9H, –Ser(But)), 1.32–1.37 (м, 1 H, γ'CH2, Lys), 1.37–1.42 (м, 19 H, –Asp(OBut ), –Boc, γCH2, Lys), 1.41–1.60 (м, 2 H, δCH2, Lys), 1.64–1.74 (м, 2 H, βCH2, Lys), 2.45 (дд, 1H, J 16.5, 7.3, β'CH2, Asp), 2.69 (дд, 1H, J 16.5, 7.4, β"CH2, Asp), 2.74–2.96 (м, 4 H, εCH2, Lys; βCH2, His), 3.72 (дд, 2 H, J 6.0, 3.0, βCH2, Ser), 3.69 (д, 2 H, J 5.8, αCH2, Gly), 4.11–4.21 (м, 1 H, αCH, Lys), 4.24–4.28 (м, 1 H, αCH, Ser), 4.22–4.27 (м, 3 H, O(CH2), CH, Fmoc), 4.46–4.51 (м, 1 H, αCH, His), 4.60–4.66 (м, 1 H, αCH, Asp), 6.69–6.75 (м, 1 H, εNH, Lys), 6.92–7.05 (м, 5 H, –CH, Trt), 7.16–7.21 (м, 1 H, αNH, Ser), 7.23–7.47 (м, 19 H, CH, Trt, Fmoc; C(4)H, His), 7.59–7.62 (м, 1 H, C2H, His), 7.82 (д, 1 H, J 9.3, αNH, Lys), 7.98 (д, 1 H, J 9.3, αNH, Gly), 8.1 (д, 1 H, J 9.4, αNH, His), 8.23 (т, 1 H, J 5.8, αNH, Asp).
Fmoc-Tyr(But)-Leu-Leu-Gly-OH (ФII). Получено 10.7 г (71.5% в расчете на исходный бензиловый эфир глицина), Rf 0.19 (система 1), 0.50 (система 2). MS, m/z (Iотн, %): 765.471 (100) [M + Na]+, 781.455 (16) [M + K]+, 787.456 (10) [M + 2Na-H]+. Спектр 1H-ЯМР: 0.86 (дд, 12H, J 9.2, 6.8, δ(CH3)2, Leu), 1.18 (с, 9H, But), 1.46–1.53 (м, 4H, βCH2, Leu), 1.59–1.61 (м, 2H, γCH, Leu), 2.71 (т, 1H, J 1.0, β'CH, Tyr), 2.93 (т, 1H, J 1.0, β"CH, Tyr), 3.69–3.74 (м, 2H, αCH2, Gly), 4.16–4.21 (м, 3H, O(CH2), CH, Fmoc), 4.25 (дт, 1H, J 9.1, 7.5, αCH, Tyr), 4.30–4.37 (м, 2H, αCH, Leu), 6.77–6.90 (м, 2H, C (3,5) H, Tyr), 7.12–7.19 (м, 2H, C (2,6) H, Tyr), 7.23–7.34 (м, 2H, CH, Fmoc), 7.37–7.42 (м, 2H, CH, Fmoc), 7.56 (д, 1H, J 3.5, αNH, Tyr), 7.59–7.65 (м, 2H, CH, Fmoc), 7.82–7.90 (м, 3H, CH, Fmoc, αNH, Leu), 8.08 (д, 1H, J 7.7, αNH, Leu), 8.13 (т, 1H, J 5.8, αNH, Gly).
Fmoc-Gly-Trp(Boc)-Thr(But)-Leu-Asn(Trt)-Ser(But)-Ala-Gly-OH (ФIII). Получено 1.7 г (81.8% в расчете на стартовую аминокислоту, присоединенную к полимеру), Rf 0.42 (система 1), 0.69 (система 2). MS, m/z (Iотн, %): 1458.627 (100) [(M–But) + CH3OH + H]+, 1504.030 (27) [M + Na]+. Спектр 1H-ЯМР: 0.88 (дд, 6H, J 20.0, 6.7, δ(CH3)2, Leu), 0.97 (д, 3H, J 5.8, γCH3, Thr), 1.06-1.18 (м, 21H, But; βCH3, Ala), 1.49–1.54 (м, 2H, βCH2, Leu), 1.58 (с, 9H, –Boc), 1.60–1.62 (м, 1H, γCH, Leu), 2.49–2.53 (м, 1H, β'CH, Asn), 2.71–2.78 (м, 1H, β"CH, Asn), 2.91–2.97 (м, 1H, β'CH, Trp), 2.99–3.12 (м, 1H, β"CH, Trp), 3.29–3.36 (м, 1H, β'CH, Ser), 3.38–3.44 (м, 1H, β"CH, Ser), 3.68 (дд, 4H, J 9.0, 6.6, αCH2, Gly), 3.75–3.93 (м, 1H, β CH, Thr), 3.99–4.14 (м, 3H, αCH, Ala; –CH, Fmoc; αCH, Ser), 4.19 (д, 2H, J 5.4, O(CH2), Fmoc), 4.28–4.40 (м, 2H, αCH, Thr; αCH, Leu), 4.60 (дт, 1Н, J 9.3, 7.3, αCH, Asn), 4.78 (дд, 1Н, J 9.0, 6.6, αCH, Trp), 7.05–7.47 (м, 18H, C5H, C6H, Trp; CH, Trt; αNH, Gly), 7.50–7.64 (м, 7H, C2H, Trp; CH, Fmoc; αNH, Ser), 7.66–7.75 (м, 4H, CH, Fmoc; αNH, Leu; C4H, C7H, Trp); 7.79–8.00 (м, 4H, CH, –Fmoc; αNH, Gly; αNH, Thr), 8.01 (д, 1Н, J 9.2, αNH, Ala), 8.13 (д, 1Н, J 9.3, αNH, Trp), 8.32 (т, 1Н, J 5.8, αNH, Asn).
H-Gly-Trp-Thr-Leu-Asn-Ser-Ala-Gly-Tyr-Leu-Leu-Gly-Pro-His-Ala-Ile-Asp-Asn-His-Arg-Ser-Phe-Ser-Asp-Lys-His-Gly-Leu-Thr-NH2 (G29). Синтез галанина крысы проводили с С-конца (см. схему 2 ) исходя из 0.53 г (0.25 ммоль) полимера Ринка с содержанием аминогрупп 0.47 ммоль/г. Цикл ТФС включал следующие стадии: 1) деблокирование α-аминогрупп 25% 4-Ме Pip/DMF (2 × 5 мл в течение 5 мин); начиная со стадии пептидилполимера PIII деблокирование проводили смесью 25% 4-MePip/0.1 М HOBt в DMF (2 раза по 5 мл в течение 5 мин); 2) промывка DMF, 3) конденсация 0.75 ммоль (3-кратного избытка) Fmoc-AA/ DIC/HOBt в 3 мл смеси DMF/NMP (1 : 1) в течение 2 ч; конденсацию 0.75 ммоль фрагментов ФI–ФIII в присутствии 0.75 ммоль комплекса F проводили в 3–4 мл NMP в течение 4 ч; 4) промывка DMF. Заключительное отщепление от носителя и деблокирование галанина осуществляли действием 10 мл смеси TFА–вода–триизопропилсилан–дитиотреитол–1,3-диметоксибензол (80 : 5 : 5 : 5 : 5) в течение 1.5 ч. По окончании реакции полимер отфильтровывали, промывали 2 × × 2 мл деблокирующей смеси, фильтрат упаривали, продукт осаждали сухим диэтиловым эфиром, отфильтровывали, промывали на фильтре эфиром и DCM. Неочищенный продукт ТФС лиофилизовали из 2% АсОН для полного отщепления Trp(Boc) [31]. Было получено 0.52 г неочищенного продукта с чистотой 77.1%, затем пептид очищали методом ВЭЖХ, как описано выше, до 98% чистоты. Получено 0.22 г соединения G29 (28% в расчете на стартовую аминокислоту). G29 MS, m/z: 3163.474 (Мрасчет = 3164.45).
Для дополнительной оценки содержания аспартимидов в неочищенном продукте ТФС G29 50 мг Nα – свободного пептидилполимера PV обрабатывали 3 мл смеси TFА–TIS–дитиотреитол– 1,3-диметоксибензол (85 : 5 : 5 : 5) в течение 1.5 ч. Продукт (16 мг) выделяли, как описано выше. MS, m/z (Iотн, %): 3165.480 (100) [M + H]+, 3147.306 (4.6) [M – H2O]+, 3221.481 (8.0) [M + But], 2690.148 (5.8).
Изучение пептида G29 на модели региональной ишемии и реперфузии сердца у крыс in vivo проведено на самцах крыс Wistar (300–350 г) в соответствии с “Международными рекомендациями по проведению биомедицинских исследований с использованием животных”, принятым Международным советом медицинских научных обществ в Женеве в 1985 г.
У наркотизированных 20% уретаном (1200 мг/кг веса, внутрибрюшинно) животных в условиях торакотомии осуществляли искусственную вентиляцию легких комнатным воздухом с помощью аппарата KTR-5 (Hugo Sacks Electronik, Германия). Яремную вену катетеризировали для окрашивания сердца 1% раствором Эванса в конце опыта. Сонную артерию катетеризировали для регистрации артериального давления. После присоединения артериального катетера к тензометрическому датчику регистрировали систолическое артериальное давление (САД) и ЧСС на полиграфе Biograph-4 (Cанкт-Петербургский госуниверситет аэрокосмического приборостроения). Запись на компьютер выполнена с помощью аналого-цифрового преобразователя USB 6210 (National Instruments, США) и программы в системе LabVIEW 7 (National Instruments, США) [10]. После препарирования животного следовал 30-минутный период стабилизации гемодинамических показателей (исходное состояние), затем окклюзия передней нисходящей коронарной артерии (ПНА) в течение 40 мин, продолжительность последующей реперфузии составляла 60 мин. Пептид G29 вводили внутривенно болюсом в дозах 0.25; 0.5; 1.0 или 2.0 мг/кг веса одновременно с началом реперфузии. В контрольной серии опытов – такой же объем физиологического раствора (0.5 мл).
В отдельной серии опытов было изучено влияние растворителя пептида G29 (0.2% раствора DMSO). В конце опыта для идентификации зоны риска (ЗР) и интактной области миокарда реокклюдировали ПНА и в яремную вену вводили болюсно 2% раствор Эванса (2 мл). Затем вырезали сердце и выделяли левый желудочек (ЛЖ) для последующего определения размеров инфаркта миокарда (ИМ). Замороженный ЛЖ разрезали перпендикулярно длинной оси сердца на 4–5 срезов толщиной около 1.5–2.0 мм, которые затем инкубировали 10 мин в 1% растворе 2,3,5-трифенилтетразолий хлорида в 0.1 М калий фосфатном буфере (рН 7.4 при 37°С). Полученные образцы сканировали, площади ИМ и ЗР определяли методом компьютерной планиметрии, используя программу Imagecal. После этого срезы взвешивали для определения массы ЛЖ. В каждой группе рассчитывали отношения зона риска/вес левого желудочка (ЗР/ЛЖ) и инфаркт миокарда/зона риска (ИМ/ЗР) в %.
Повреждение мембран кардиомиоцитов оценивали по увеличению активности лактатдегидрогеназы (ЛДГ) и МВ-фракции креатинкиназы (MB-КK) в плазме крови. Около 0.5 мл крови собирали в гепаринизированные пробирки из венозного катетера в исходном состоянии (перед окклюзией ПНА) и после 1 ч реперфузии. Активность ферментов в плазме определяли на спектрофотометре Shimadzu UV-1800 (Япония) при λ 340 нм, используя наборы фирмы BioSystems.
В отдельной серии опытов оценивали влияние внутривенного введения оптимальной дозы пептида (0.5 мг/кг) на энергетическое состояние ЗР. По окончании реперфузии ЗР быстро выделяли из ЛЖ и замораживали щипцами Волленбергера, охлажденными в жидком азоте. Замороженную ткань гомогенизировали в холодной 6% HClO4 (10 мл/г ткани) в гомогенизаторе Ultra-Turrax T-25 (IKA-Labortechnik, Германия). Белки осаждали центрифугированием (центрифуга Sorvall RT1, Thermo Fisher Scientific, США) при 2800 g в течение 10 мин при 4°С. Супернатанты нейтрализовали 5 М К2СО3 до рН 7.4. Осадок KСlO4 отделяли центрифугированием в тех же условиях. Безбелковые экстракты хранили при –20°С до определения метаболитов. Сухой вес гомогенизированной ткани определяли после высушивания образцов в течение 1 сут при 110°С. Содержание адениннуклеотидов, фосфокреатина (ФКр), креатина (Кр) и лактата в тканевых экстрактах определяли энзиматическими методами [32], используя спектрофотометр Shimadzu UV-1800 (Япония). Таким же образом оценивали метаболическое состояние ЗР в конце реперфузии в контроле (в опытах с внутривенным введением физиологического раствора) и в исходном состоянии (до окклюзии ПНА).
ЗАКЛЮЧЕНИЕ
Конвергентный подход был успешно применен для получения 29-членного галанина крысы на твердой фазе. Использование очищенных пептидных блоков для наращивания пептидной цепи исключило возможность образования в синтезе трудноотделимых “ложных” пептидов с близкими к целевому пептиду свойствами, благодаря этому пептид G29 был получен с высокими выходом и чистотой. Введение G29 уменьшало размеры инфаркта миокарда на 40% и снижало активность маркеров некроза МВ-креатинкиназы и лактатдегидрогеназы в плазме к концу реперфузии по сравнению с контролем. Пептид G29 (0.5 мг/кг) улучшал метаболическое состояние сердца – увеличивал содержание АТP, общего фонда адениннуклеотидов, фосфокреатина, общего креатина и снижал уровень лактата по сравнению с контролем. Результаты указывают на возможность использования пептида G в качестве препарата для уменьшения реперфузионных повреждений сердца и необходимость изучения механизмов его действия.
Список литературы
Seta Y., Kataoka S., Toyono T., Toyoshima K. // Arch. Histol. Cytol. 2006. V. 69(4). P. 273–280.
Diaz-Cabiale Z., Parrado C., Vela C., Razani H., Covenas R., Fuxe K., Narvaez J.A. // Neuropeptides. 2005. V. 39. P. 185–190.
Abbott S., Pilowsky P. // Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009. V. 296(4). R1019–1026.
He B., Shi M., Zhang L., Li G., Zhang L., Shao H., Li J., Fang P., Ma Y., Shi Q., Sui Y. // Physiol. Behav. 2011. V. 103(3–4). P. 284–289.
Fang P., Sun J., Wang X., Zhang Z., Bo P., Shi M. // Life Sciences. 2013. V. 92. P. 628–632.
Lang R., Gundlach A. L., Kofler B. // Pharmacology & Therapeutics. 2007. V. 115. P. 177–207.
Alston E.N., Parrish D.C., Hasan W., Tharp K., Pahlmeyer L., Habecker B.A. // Neuropeptides. 2011. V. 45(1) P. 33–42.
Kocic I. // J. Pharm. Pharmacol. 1998. V. 50(12). P. 1361–1364.
Wang J., Gareri C., Rockman H.A. // Circulation Research. 2018. V. 123(6). P. 716–735.
Шульженко В.С., Серебрякова Л.И., Студнева И.М., Пелогейкина Ю.А., Веселова О.М., Молокоедов А.С., Овчинников М.В., Палькеева М.Е., Сидорова М.В., Писаренко О.И. // Кардиологический вестник. 2016. Т. 11(3). С 12–21.
Timotin A., Pisarenko O., Sidorova M., Studneva I., Shulzhenko V., Palkeeva M., Serebryakova L., Molokoedov A., Veselova O., Cinato M., Tronchere H., Boall F., Kunduzova O. // Oncotarget. 2017. V. 8(13). P. 21241–21252.
Stathopoulos P., Papas S., Kostidis S., Tsikaris V. // J. Peptide Sci. 2005. V. 11(10). P. 658–664.
Subirόs-Funosas R., El-Faham A., Albericio F. // Tetrahedron. 2011. V. 67(45). P. 8585–8593.
Mergler M., Dick F., Sax B., Staehelin C., Vorherr T. // J. Pept. Sci. 2003. V. 9. P. 518–526.
Behrendt R., Huber S., Martί R., White P. // J. Pept. Sci. 2015. V. 21. P. 680–687.
Weber U., André M. // Hoppe-Seyler Z. Phys. Chem. 1975. V. 356(s1). P. 701–714.
Lloyd-Williams P., Albericio F., Giralt E. // Tetrahedron. 1993. V. 49(48). P. 11065–11133.
Вольпина О.М., Михалева И.И., Иванов В.Т. // Биорганич. химия. 1982. T. 8(1). C. 5–49.
Vasileiou Z., Barlos K., Gatos D. // J. Pept. Sci. 2009. V.15. P. 824–831.
Barlos K., Gatos D. // Biopolymers (Peptide Science). 1999. V. 51. P. 266–278.
Goulas S., Gatos D., Barlos K. // J. Pept. Sci. 2006. V. 12. P. 116–123.
Сидорова М.В., Молокоедов А.С., Овчинников М.В., Беспалова Ж.Д., Бушуев В.Н. // Биоорганич. химия. 1997. Т. 23. № 1. С. 41–50. [Sidorova M.V. Moloko-edov A.S., Ovchinnikov M.V., Bespalova Zh.D., Bushuev V.N. // Russ. J. Bioorgan. Chem. V. 23. № 1. P. 46–55.]
Сидорова М.В., Палькеева М.Е., Азьмуко А А., Овчинников М.В., Молокоедов А.С., Шарф Т.В., Ефремов Е.Е., Голицын С.П. // Биоорганич. химия. 2017. Т. 43(4). С. 339–346. [Sidorova M.V., Palkeeva M.E., Az’muko A.A., Ovchinnikov M.V., Molokoedov A.S., Sharf T.V., Efremov E.E, Golitsyn S.P. // Russ. J. Bioorgan. Chem. 2017. V. 43(4). Р. 351–358.]
Barlos K., Gatos D. // Convergent peptide synthesis // Fmoc Solid Phase Peptide Synthesis. A Practical Approach. Ed. by Chan W.C., White P.D. Oxford Univ. Press, 2001. P. 215–228.
Van Zon A., Beyerman H.C.// Helv. Chim. Acta. 1973. V. 56. P. 1729–1740.
Гершкович А.А., Кибирев В.К. Химический синтез пептидов. Киев: Наукова думка, 1992.
Kovacs J., Kisfaludy L., Ceprini M.Q. // J. Am. Chem. Soc. 1967. V. 89(1). P. 184–184.
Kaiser E., Colescott R.L., Bossinger C.D., Cook P.I. // Anal. Biochem. 1970. V. 34. P. 595–598.
Michels T., Dölling R., Haberkorn U., Mier W. // Org. Lett. 2012. V. 14(20). P. 5218–5221.
Досон Р., Эллиот Д., Эллиот У., Джонс.К. Справочник биохимика. 1991. М.: Мир. С. 391.
Fmoc Solid Phase Peptide Synthesis. A Practical Approach. Ed. Chan W.C., White P.D. Oxford Univ. Press, 2001. P. 65–67.
Bergmeyer H.U. Methods of Еnzymatic Аnalysis. New York: Academic Press, 1974. P. 1464–1467, 1772–1776, 1777–17781, 2127–2131.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия