

Биоорганическая химия, 2020, T. 46, № 2, стр. 158-169
Компьютерное моделирование и синтез потенциальных ингибиторов тирозинкиназы BCR-ABL с мутацией Т315I
А. Н. Федоркевич , О. Л. Шарко , В. В. Шманай
Институт физико-органической химии Национальной академии наук Беларуси,
220072 Минск, ул. Сурганова, 13, Беларусь
Поступила в редакцию 10.10.2019
После доработки 29.10.2019
Принята к публикации 14.11.2019
Аннотация
Методом компьютерного моделирования проведен сравнительный анализ взаимодействия химерного белка BCR-ABL – нормального типа и с мутацией T315I, с известными ингибиторами, а также с предложенными соединениями, потенциально способными к ингибированию мутантного белка. Показано, что предложенные соединения встраиваются в структуру белка с сохранением основных водородных связей и межмолекулярных взаимодействий. Осуществлен синтез двух структур, содержащих пиррольный цикл, которые оказались наиболее перспективными на основании результатов компьютерного моделирования.
ВВЕДЕНИЕ
Тирозинкиназа BСR-ABL – гибридный белок, продукт гибридного гена Bcr-Abl1, формирующегося в результате реципрокной транслокации между хромосомами 9 и 22 (филадельфийская хромосома). Белок BCR-ABL является конститутивно активной тирозинкиназой, ответственной за онкогенную трансформацию клеток онкобелком. Постоянная активность этой тирозинкиназы делает клетку нечувствительной к воздействию факторов роста и вызывает ее избыточную пролиферацию. Формирование белка BCR-ABL провоцирует 95% случаев хронического миелолейкоза и 20–50% случаев острого В-лимфобластного лейкоза взрослых [1].
Хронический миелоидный лейкоз поддается лечению таргетной (целевой) терапией ингибиторами тирозинкиназ: иматинибом, нилотинибом, дазатинибом и др. Терапия значительно улучшает показатели выживаемости. Однако использование данных препаратов со временем приводит к появлению резистентности у пациентов вследствие развития точечных мутаций в BCR-ABL-белке. Появление мутации T315I в тирозинкиназе BCR-ABL приводит к потере ключевой водородной связи между белком и ингибитором и становится критичным для связывания белка с большинством известных ингибиторов. В настоящее время ведется поиск эффективных препаратов, которые способны ингибировать активность белока с наличием мутации T315I [2].
В работе представлены результаты моделирования взаимодействия белка BCR-ABL, в том числе с мутацией T315I, с основными известными ингибиторами тирозинкиназ, а также с предложенными новыми структурами. Были синтезированы перспективные для дальнейшего исследования соединения с наличием фрагмента пиррола в структуре.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Первым специфическим ингибитором тирозинкиназы BCR-ABL, прошедшим клинические испытания и в настоящее время используемым для лечения больных хроническим миелоидным лейкозом, является иматиниб (рис. 1).

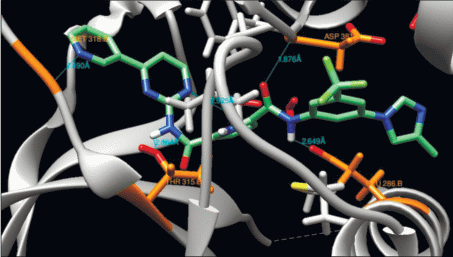
Иматиниб связывается с неактивной конформацией белка [3] и образует при этом 6 водородных связей и большое число ван-дер-ваальсовских взаимодействий. Ключевыми для связывания с ингибитором являются в белке остатки Met-318, Thr-315, Glu-286 , Asp-381 (рис. 2) [4].
Рис. 2.
Иматиниб в структуре комплекса с тирозинкиназой Bcr-Abl: в неактивной конформации “DFG-out” (а) и с T315I-мутацией (б).

Появление мутации T315I ведет к потере одной из основных водородных связей между белком и молекулой иматиниба из-за исчезновения ОН-группы треонина. Также при появлении мутации возникают серьезные cтерические препятствия для встраивания ингибитора в активный центр белка. Иматиниб не встраивается в структуру такого мутантного белка, что критически влияет на его ингибирующую способность (рис. 3).

Основная идея данной работы состоит в том, чтобы сконструировать молекулу ингибитора таким образом, чтобы фрагмент молекулы, который вызывает стерические затруднения при встраивании в структуру белка, был более гибким и менее объемным. Это позволит ингибитору встраиваться в струткуру белка с наличием мутации T315I с сохранением ингибирующей активности. Для этого один из шестичленных циклов в молекуле иматиниба был заменен на пятичленный пирролльный фрагмент, сочленение с пиридин-пиримидиновым фрагментов стало более гибким в случае структуры (2) (рис. 4). Оценку эффективности взаимодействия белка и лиганда проводили с помощью молекулярного докинга в программе AutoDock Vina.
Рис. 4.
Ингибитор (1) в модельном комплексе с тирозинкиназой Abl (водородные связи с Met-318, Thr-315, Glu-286, Asp-381).
Процесс молекулярного докинга может быть разделен на три стадии: подготовка молекулы белка, подготовка молекулы лиганда и расчет эффективности взаимодействия белка и лиганда. Первый шаг предполагает оптимизацию структуры белка. В качестве исходной структуры была взята человеческая тирозинкиназа ABL в комплексе с иматинибом [5]. Структура комплекса получена методом дифракции рентгеновских лучей с разрешением 2.4 Å. Оптимизация была проведена с использованием силового поля AMBER, частичные заряды были получены из AMBER ff99SB, из структуры белка были удалены молекулы растворителя, ковалентно-несвязанные ионы, добавлены недостающие протоны аминокислот, присутствующие при физиологических значениях pH. Структура лигандов оптимизирована путем минимизации энергии структуры в программе ChemBio 3D в силовом поле MMFF94. Результаты расчета энергии взаимодействия белка и предложенных лигандов, в сравнении с известными ингибиторами, представлены в табл. 1.
Таблица 1.
Результаты компьютерного моделирования взаимодействия Bcr-Abl-белка без мутаций и с T315I мутацией с иматинибом, нилотинибом и потенциальными ингибиторами (1) и (2)
| Bcr-Abl тирозинкиназа | ΔG, ккал/моль | |||
|---|---|---|---|---|
| иматиниб | нилотиниб | (1) | (2) | |
| Без мутаций | –9.9 | –10.4 | –11.7 | –11.5 |
| С T315I-мутацией | –* | –* | –10.6 | –10.8 |
Из данных табл. 1 можно сделать вывод, что структуры (1) и (2) имеют более высокое абсолютное значение энергии связывания, чем ингибиторы, взятые для сравнения, и, следовательно, потенциально будут эффективнее взаимодействовать с молекулой белка. Анализ состояния лигандов в комплексе с молекулой белка указывает на сохранение для них основных водородных связей, характерных для иматиниба (рис. 4–7).
Рис. 5.
Ингибитор (1) в активном центре тирозинкиназы BCR-ABL (водородные связи с Met-318, Thr-315, Glu-286, Asp-381). Ингибитор (2) в активном центре тирозинкиназы BCR-ABL (водородные связи с Met-318, Thr-315, Glu-286, Asp-381).

Рис. 6.
Ингибитор (1) в активном центре тирозинкиназы BCR-ABL (показана поверхность потенциальной энергии белка).
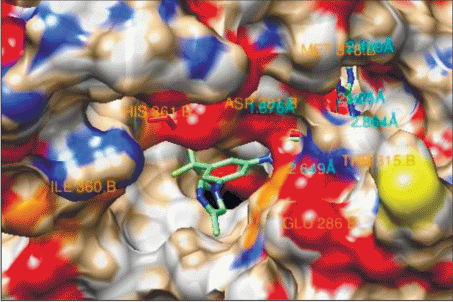
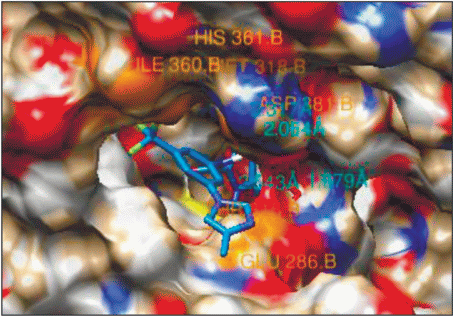
Рис. 7.
Ингибитор (2) в активном центре тирозинкиназы BCR-ABL (показана поверхность потенциальной энергии белка).
В отличие от известных ингибиторов, а именно иматиниба и нилотиниба [6], соединения (1) и (2) не образуют водородные связи с изолейцином-360 и гистидином-361 (рис. 4, 5). Так как данные связи не являются ключевыми [4], можно ожидать, что их отсутствие не будет критично сказываться на ингибирующей способности рассматриваемых соединений.
Результаты молекулярного докинга показывают, что предложенные структуры, в отличие от иматиниба и нилотиниба, способны встраиваться в тирозинкиназу BCR-ABL с мутацией T315I (рис. 8–11).
Рис. 8.
Ингибитор (1) в активном центре тирозинкиназы BCR-ABL с мутацией T315I (водородные связи с Met-318, Glu-286, Asp-381).

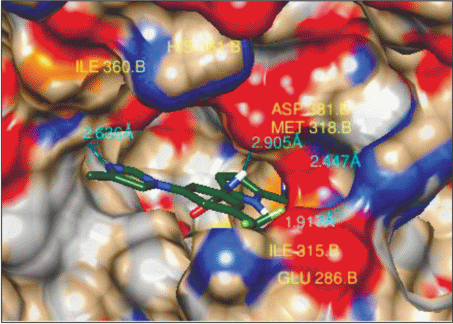
Рис. 9.
Ингибитор (1) в активном центре тирозинкиназы BCR-ABL с мутацией T315I (показана поверхность потенциальной энергии белка).

Рис. 10.
Ингибитор (2) в активном центре тирозинкиназы BCR-ABL с мутацией T315I (водородные связи с Met-318, Glu-286, Asp-381).

Рис. 11.
Ингибитор (2) в активном центре тирозинкиназы BCR-ABL с мутацией T315I (показана поверхность потенциальной энергии белка).
В рамках данной работы осуществлен синтез двух предложенных ингибиторов. Синтез ключевых блоков представлен ниже, на схеме.
Схема 1 .
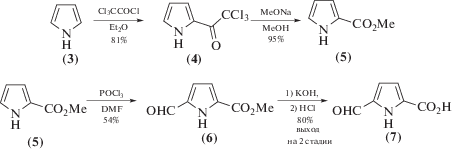
Альдегидокислота (7) синтезирована из коммерчески доступного пиррола (3). Первая стадия предполагает ацилирование пиррола хлорангидридом трихлоруксусной кислоты. Выход соединения (4) оказался высоким (81%), несмотря на сильное обугливание реакционной смеси. Использование эквивалентного по отношению к хлорангидриду количества Et3N для связывания выделяющегося HCl не изменяет данную тенденцию. Реакция соединения (4) с каталитическим количеством метилата натрия в метаноле проходит быстро при комнатной температуре и легко контролируется с помощью ТСХ. Очистка продукта на данной стадии не требуется. Очистка продукта, а также разделение образующихся изомеров обязательны на стадии формилирования соединения (5).
Соединение (5) получено с выходом 95%. В спектре 1Н-ЯМР присутствует синглет при 3.86 м.д. с интегральной интенсивностью, равной трем. Данный сигнал указывает на наличие метильной группы в полученном соединении. В целом, спектральные характеристики полученного соединения совпали с описанными в литературе [8].
Реакция формилирования по Вильсмейеру-Хааку, используемая для получения соединения (6), проходила достаточно легко. Средний выход в 54% на данной стадии можно объяснить побочным образованием 4-региоизомера альдегидоэфира (6). Выход основного 5-изомера значительно зависит от температуры проведения реакции. В данной работе раствор хлориминиевой соли, образующейся при смешивании диметилформамида и POCl3, в CH2Cl2 добавляли при –25°С, далее температуру реакционной смеси медленно доводили до комнатной и затем кипятили полчаса. При –25°С хлориминиевая соль и эфир (5) практически не реагируют, поэтому понижение температуры смешивания реагентов почти не влияет на выход целевого вещества (6). Если нагревание вести не плавно, а быстро нагревать реакционную смесь, то выход снижается до 40%. В 1Н-ЯМР-спектре соединения (6) присутствует сигнал протона альдегидной группы при 9.67 м. д.
Гидролиз метилового эфира (6) проходил в соответствии с описанными методиками [7]. 1Н-ЯМР-спектр показывает отсутствие сигнала метильной группы и наличие уширенного синглета карбоксильной группы при 12.85 м.д. Общий выход кислоты (7) составил 33%.
Соединение (10) получено исходя из коммерчески доступного 3-ацетилпиридина (8) (схема 2 ). Кипячение в DMF-DMA приводит к образованию единственного продукта (9). В его спектре 1Н-ЯМР присутствуют два синглета от двух метильных групп при 2.95 и 3.18 м.д.; также присутствуют протоны при двойной связи при 5.68 и 7.84 м.д. Большая константа спин-спинового взаимодействия, равная 12.5 Гц, указывает на Е-конфигурацию двойной связи [8].
Образование амина (10) происходило путем кипячения в течение 12 ч раствора соединения (9) с 1 эквивалентом гуанидингидрохлорида и 1.1 экв. NaOH в н-бутаноле [9]. Соединение образуется практически чистым и не требует дополнительной очистки после реакции. Его 1Н-ЯМР-спектр совпал с описанным в литературе [9]. Выход соединения (10) в расчете на 3-ацетилпиридин составил 72%.
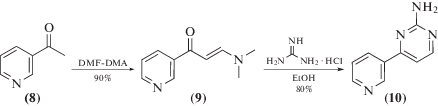
Схема 2.
Синтез ингибиторов (1) и (2) представлен на схемах 3 и 4 .
Соединение (11) получено из амина (10) и альдегидокислоты (7). Умеренный выход на данной стадии (45%) можно объяснить низкой нуклеофильностью аминогруппы соединения (10). Дальнейшее окисление альдегидопиррола (11) в кислоту проходило с достаточно высоким выходом. Конденсация кислоты (12) с коммерчески доступным 3-(3-метил-1Н-пиррол-1-ил)-5-(трифторметил)анилином (13), который используется в синтезе известного ингибитора Bcr-Abl-тирозинкиназы – нилотиниба [10], происходило в стандартных условиях с применением DСС в качестве конденсирующего реагента и бензотриазола в качестве активатора карбоксильной группы. Соединение (1) получено с общим выходом 11% в расчете на пиррол.

Схема 3 .
Конденсация кислоты (7) с амином (10) проходила с достаточно низким выходом ввиду малой нуклеофильности амина (10). Соединение (2) получено с общим выходом 12% исходя из пиррола.
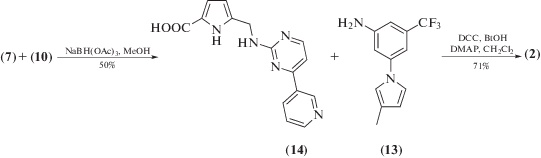
Схема 4 .
Все устойчивые соединения были выделены и охарактеризованы с использованием 1H-, 13С-ЯМР.
Таким образом, в работе методом компьютерного моделирования охарактеризованы структуры предложенных ингибиторов в комплексе с тирозинкиназой BCR-ABL как с наличием мутации T315I так и без нее. Показано, что сохраняются ключевые водородные связи с остатками Met-318, Thr-315, Glu-286, Asp-381. Предложенные соединения синтезированы и охарактеризованы. Результаты исследования биологической активности разработанных новых ингибиторов BCR-ABL-тирозинкиназы будут представлены в отдельном сообщении.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и методы исследования
Исходные вещества. Использованные в ходе работы реактивы и растворители имели квалификацию “ч” и “чда”. Очистку и высушивание растворителей производили в соответствии с рекомендациями [11]. В качестве исходных соединений использовали пиррол, 3-ацетилпиридин и амин (13) фирмы “Acros”.
Методы анализа. Моделирование взаимодействия белка с лигандом осуществляли с использованием программы UCSF Chimera 1.10.2, модуль AutoDock Vina. Кристаллографические данные о целевом белке взяты из базы RCSB Protein Data Bank. Файлы: “Human Abl kinase domain in complex with imatinib” – белок без мутаций в комплексе с иматинибом (ингибитор BCR-ABL-тирозинкиназы первого поколения). “The crustal structure of human abl1 kinase domain T315I mutant in complex with DCC-2036” – структура белка с T315I-мутацией в комплексе с потенциальным ингибитором. Данные файлы были загружены в формате *.pdb.
Оценку индивидуальности синтезируемых веществ и наблюдение за ходом проводимых реакций осуществляли методом ТСХ на пластинках Silufol UV-254. При проявлении использовали ультрафиолетовую лампу с длинной волны 254 нм, 1% раствор KMnO4 в воде, 5% раствор фосфоромолибденовой кислоты в этаноле. В качестве элюента использовали смеси петролейного эфира, этилацетата, а также метанола и хлороформа, метанола и метилена, толуола и ацетона в различных соотношениях. Выделение и очистку индивидуальных веществ осуществляли с помощью перекристаллизации из петролейного эфира, а также методом колоночной хроматографии на силикагеле 60 Merck (70–230 mesh), с использованием в качестве элюента смесей тех же растворителей.
Спектры 1Н-ЯМР синтезированных соединений (δ, м.д.; J, Гц) записывали для растворов в CDCl3 или DMSO-d6 на приборе Bruker AC 500 с рабочей частотой 500 МГц относительно сигнала остаточных протонов в дейтерохлороформе (δ 7.26 м.д.) и DMSO-d6 (2.50 м.д.). Спектры 13С-ЯМР записывали для растворов в CDCl3 или DMSO-d6 на приборе Bruker АС 500 с рабочей частотой 100.6 МГц. Химические сдвиги указаны относительно сигнала CDCl3 (δ 77.00 м.д.) и DMSO-d6 (39.43 м.д.). ИК-спектры снимали на FT-IR-спектрофотометре “PerkinElmer Spectrometer 100” в таблетке с KBr.
(2-Трихлороацетил)-1Н-пиррол (4). К перемешиваемому раствору трихлорацетилхлорида (37.00 г, 0.23 моль) в сухом Et2O (100 мл) добавляли пиррол (12.50 г, 0.18 моль) по каплям за 3 ч при комнатной температуре и далее реакционную смесь перемешивали 1 ч при комнатной температуре и нейтрализовали 20% водным раствором K2CO3 (100 мл) до pH 8–9. Органическую фазу отделяли, водную экстрагировали Et2O (3 × 25 мл). Объединенную органическую фазу промывали последовательно насыщенным водным раствором NaHCO3 и насыщенным водным раствором NaCl, сушили Na2SO4. Осушитель отфильтровывали, растворитель удаляли при пониженном давлении. Продукт (4) очищали методом колоночной хроматографии на силикагеле (ПЭ–EtOAc, 15 : 1–5 : 1) и получали в виде белых кристаллов (34.04 г, 89%). Rf 0.44 (ПЭ–EtOAc, 5 : 1), Тпл 75.5°С. Спектральные характеристики совпали с описанными в литературе [7]. Спектр 1H-ЯМР (CDCl3): 6.34–6.37 (м, 1H), 7.16–7.17 (м, 1 H), 7.38–7.40 (м, 1H), 9.51 (уш. с., 1 H).
Метил-1Н-пиррол-2-карбоксилат (5). В сухом MeOH (64 мл) растворяли Na (0.223 г, 9.7 ммоль) при комнатной температуре. К полученному перемешиваемому раствору добавляли (2-трихлороацетил)-1Н-пиррол (14.700 г, 69.2 ммоль) шестью порциями за 30 мин. Реакционную смесь перемешивали 2 ч при комнатной температуре (контроль ТСХ). Растворитель упаривали, остаток разбавляли Et2O (80 мл). К раствору добавляли 3 М НСl (5 мл). Органическую фазу отделяли, водную экстрагировали Et2O (3 × 15 мл). Объединенную органическую фазу промывали последовательно насыщенным водным раствором NaHCO3 и насыщенным водным раствором NaCl, сушили Na2SO4. Осушитель отфильтровывали, растворитель упаривали при пониженном давлении. Продукт использовали в следующей стадии без дополнительной очистки. Эфир (5) получали в виде белых кристаллов (8.215 г, 95%). Rf 0.31 (ПЭ–EtOAc, 5 : 1). Тпл 73.0°С. Спектральные характеристики совпали с описанными в литературе [6]. Спектр 1H-ЯМР (CDCl3): 3.86 (с, 3H), 6.26–6.28 (м, 1H), 6.91–6.93 (м, 1H), 6.95–6.97 (м, 1H), 9.27 (м, 1 H).
Метил-5-формил-1Н-пирол-2-карбоксилат (6). К охлажденному до 0°С сухому DMF (2.890 г, 39.6 ммоль) добавляли POCl3 (6.070 г, 39.6 ммоль) за 15 мин. Реакционную смесь перемешивали до полной кристаллизации (30 мин). Смесь отогревали до комнатной температуры; полученную иминиевую соль растворяли в сухом CH2Cl2 (20 мл) и охлаждали до –25°С. Раствор метил-1Н-пиррол-2-карбоксилата (5) (3.536 г, 28.3 ммоль) в CH2Cl2 (20 мл) добавляли шприцом за 1 ч при 0°С. Раствор доводили до комнатной температуры и затем кипятили 30 мин при перемешивании. Реакционную смесь охлаждали до комнатной температуры, добавляли насыщенный водный раствора NaHCO3 до pH 8–9 и кипятили 15 мин. Органическую фазу отделяли, водную экстрагировали Et2O (3 × 10 мл). Объединенную органическую фазу промывали насыщенным водным раствором NaCl, сушили Na2SO4. Осушитель отфильтровывали, растворитель удаляли при пониженном давлении. Метил-5-формил-1Н-пирол-2-карбоксилат (6) (2.770 г, 64%) выделяли методом колоночной хроматографии на силикагеле (ПЭ–EtOAc, 15 : 1–3 : 1) в виде белых кристаллов. Rf 0.37 (ПЭ–EtOAc, 65 : 35). Тпл 96.2°С. Также был получен побочный продукт – метил-4-формил-1Н-пирол-2-карбоксилат (0.736 г, 17%). Спектральные характеристики совпали с описанными в литературе [7]. Спектр 1H-ЯМР (CDCl3): 3.92 (с, 3H), 6.94 (м, 2H), 9.67 (с, 1H), 9.88 (уш. с, 1H).
5-Формил-1Н-пирол-2-карбоновая кислота (7). Раствор метилкарбоксилата (6) (1.00 г, 6.53 ммоль) и KOH (0.403 г, 7.18 ммоль) в EtOH (8 мл) и H2O (2 мл) кипятили 3 ч. Растворитель упаривали, осадок растворяли в H2O (8 мл). Раствор подкислили 3 М НСl до pH 4–6, осадок отфильтровали промывали H2O, сушили при 100°С до постоянной массы. Альдегидокислоту (7) получали в виде бледно-желтых кристаллов (0.817 г, 90%). Rf 0.44 (ПЭ–EtOAc, 1 : 1). Тпл 188°С (с разложением). Спектральные характеристики совпали с описанными в литературе [7]. Спектр 1H-ЯМР (DMSO-d6,): 6.82–6.84 (м, 1H), 6.93–6.95 (м, 1H), 9.69 (с, 1H), 12.85 (уш. с., 1H), 13.06 (уш. с., 1H).
(Е)-3-(Диметиламино)-1-фенилпроп-2-ен-1-он (9). Смесь 3-ацетилпиридина (2.428 г, 20 ммоль) и DMF-DMA (4.760 г, 40 ммоль) кипятили при перемешивании до исчезновения 3-ацетилпиридина (контроль ТСХ) около 6 ч. Смесь охлаждали, растворитель упаривали. Осадок перекристаллизовывали из ПЭ. Продукт (9) получали в виде желтых кристаллов (3.115 г, 89%). Rf 0.43 (EtOAc). Тпл 90.2°С. Спектральные характеристики совпали с описанными в литературе [10]. Спектр 1H-ЯМР (CDCl3.): 2.95 ( с., 3Н), 3.18 (с, 3Н), 5.68 (д., J 12.3, 1Н), 7.35 (м, 1Н), 7.84 (д., J 12.3, 1Н), 8.19 (д., J 7.9, 1H), 8.66 (д., J 4.8, 1Н), 9.08 (д., J = 2.2, 1Н).
4-(Пиридин-3-ил)пиримидин-2-иламин (10). Раствор продукта (9) (6.400 г, 36.3 ммоль), гуанидингидрохлорида (3.056 г, 36.3 ммоль) и NaOH (1.595 г, 39.9 ммоль) в н-бутаноле (40 мл) кипятили 12 ч. Раствор охлаждали, осадок отфильтровывали, промывали на фильтре 96% этанолом, затем холодной водой. Полученные кристаллы сушили в вакууме при 40°С. Продукт (10) получали в виде серо-белых кристаллов (5.162 г, 82%). Rf 0.20 (EtOAc). Тпл 188.2°С. Спектральные характеристики совпали с описанными в литературе [10]. Спектр 1H-ЯМР (500 МГц,): 5.21 (с, 2H), 7.11 (д, J 5.2, 1H), 7.45 (дд, J 4.8, 8.0, 1H), 8.35 (д, J 8.0, 1H), 8.44 (д, J 5.2, 1H), 8.75 (д, J 4.8, 1H), 9.24 (д, J 2.2, 1H).
N-(4-(Пиридин-3-ил)пиримилин-2-ил)-5-формил-1Н-пиррол-2-карбоксамид (11). К суспензии соединения (7) (139 мг, 1 ммоль), в 10 мл CH2Cl2 последовательно добавляли, DMAP (122 мг, 1 ммоль), раствор амина (10) (172 мг, 1 ммоль) в 5 мл СH2Cl2. Далее в реакционную смесь всыпали DCC (227 мг, 1.1 ммоль). Реакционную смесь перемешивали 12 ч. Осадок отфильтровали, промыли на фильтре CH2Cl2. К реакционной смеси добавляли насыщенный раствор NaHSO3 (20 мл). Водный слой отделяли, подкислили 2 М HCl до pH 4. Экстрагировали EtOAc (3 × 20 мл). Объединенную органическую фазу промывали насыщенным водным раствором NaCl (1 × 20 мл), сушили Na2SO4. Осушитель отфильтровывали, растворитель упаривали при пониженном давлении. Продукт (11) (132 мг, 45%) выделяли методом колоночной хроматографии на силикагеле (толуол–ацетон, 7 : 3) в виде белых кристаллов. Rf 0.24 (толуол–ацетон, 7 : 3). Тпл 174.3°С. Спектр 1H-ЯМР (DMSO-d6): 7.03–7.05 (м, 1H), 7.06–7.08 (м, 1 H), 7.13 (д, J 5.3, 1H), 7.55 (дд, J 4.7, 8.1, 1H), 8.18 (д, J 8.1, 1H), 8.67 (д, J 5.3, 1H), 8.73 (д, J 4.7, 1H), 9.04 (с, 1H), 9.32 (с, 1H), 11.95 (уш. с., 1H), 12.26 (уш. с., 1H). Спектр 13С-ЯМР (DMSO-d6): 106.55, 107.98, 109.73, 115.28, 122.01, 124.55, 132.91, 135.645, 136.63, 147.32, 150.58, 157.53, 159.47, 160.12, 175.44.
5-((4-(Пиридин-3-ил)пиримидин-2-ил)карбамоил)-1Н-пиррол-2-карбоновая кислота (12). К 2 мл раствора карбоксамида (11) (132 мг, 0.42 ммоль) в ацетоне добавляли суспензию перманганата калия (135 мг, 0.85 ммоль) в 8 мл смеси ацетон–вода (1 : 1) за 1 ч. Раствор перемешивали дополнительно 1 ч при 40°С и еще 1 ч при комнатной температуре. Тиосульфат натрия добавляли порциями до обесцвечивания раствора. Ацетон удаляли при пониженном давлении, водный слой подкисляли до pH 3–4 5% раствором HCl. Полученный осадок фильтровали, промывали водой на фильтре. Белый осадок сушили при пониженном давлении. Продукт (12) (130 мг, 96%) использовали в следующей стадии без дополнительной очистки. Rf 0.19 (MeOH–CHCl3, 1 : 8). Тпл 197.3°С. Спектр 1H-ЯМР (DMSO-d): 7.04–7.05 (м, 1 H), 7.06–7.08 (м, 1H), 7.14 (д, J 5.2, 1H), 7.57 (дд, J 4.7, 8.0, 1H), 8.16 (д, J 8.0, 1H), 8.65 (д, J 5.2, 1H), 8.75 (д, J 4.7, 1H), 9.02 (с, 1H), 10.71 (с, 1H), 11.65 (уш. с., 1H), 12.06 (уш. с., 1H). Спектр 13С-ЯМР (DMSO-d6): 107.55, 110.98, 111.73, 115.08, 121.01, 124.55, 138.91, 139.58, 140.33, 149.47, 154.59, 155.88, 157.43, 160.12, 160.34.
5-(4-Пиридин-3-илпиримидин-2-ил)аминометил-1Н-пиррол-2-карбоновая кислота (14). К суспензии NaBH4 (2.00 г, 53 ммоль) в 22 мл CHCl3 добавляли безводную AcOH (14 мл, 244 ммоль) при 0–5°С за 1 ч. Смесь перемешивали при 0–5°С дополнительно 1.5 ч. Альдегидокислота (7) (3.480 г, 25 ммоль) и раствор амина (10) (4.472 г, 26 ммоль) в CHCl3 (12 мл) были добавлены к образованной суспензии. Смесь перемешивали при 0–5°С 1 ч, затем 12 ч при комнатной температуре. К смеси добавляли H2O (15 мл) и Na2CO3 до pH 9–10. Водную фазу промывали CHCl3 (3 × 10 мл). Далее pH раствора доводили до 4 1 М раствором HCl. Осадок отфильтровывали, промывали H2O на фильтре. Полученные желтые кристаллы сушили в вакууме при комнатной температуре. Выход 50% (3.688 г). Rf 0.23 (MeOH–CHCl3 1 : 8). Тпл 186.2°С. Спектр 1H-ЯМР (DMSO-d6): 2.48 (с, 2Н), 4.52 (м, 1Н), 6.01 (м, 1Н), 6.60 (м, 1Н), 7.23 (д, J 5.16, 1Н), 7.55 (уш. с, 1Н), 7.58 (дд, J 4.94, 8.03, 1Н), 8.39 (д, J 5.00, 1Н), 8.50 (м, 1Н), 8.68 (д, J 5.00, 1Н), 9.25 (м, 1Н), 11.48 (уш. с, 1Н). Спектр 13С-ЯМР (DMSO-d, δ6): 37.70, 106.56, 107.99, 109.71, 115.27, 122.02, 124.56, 132.90, 135.65, 136.64, 147.33, 150.59, 159.47, 162.01, 164.45. ИК-спектр (в KBr, υ, см–1): 3436, 1668.
N2-(3-(4-Метил-1Н-имидазол-1-ил)-5-(трифтрометил)фенил)-N5-(4-пиридин-3-ил)пиримидин-2-ил)-1Н-пиррол-2,5-дикарбоксамид (1). К раствору кислоты (12) (105 мг, 0,34 ммоль) в смеси CH2Cl2–DMF (10 мл, 1 : 1) были последовательно добавлены бензотриазол (BtOH) (50 мг, 0.38 ммоль), DMAP (46 мг, 0.38 ммоль), раствор амина (13) (91 мг, 0.38 ммоль) в 5 мл СH2Cl2. Далее в реакционную смесь всыпали DCC (79 мг, 0.38 ммоль) и перемешивали 16 ч. Осадок отфильтровывали, промывали на фильтре CH2Cl2. Растворитель упаривали до объема в 5 мл при пониженном давлении, остаток выливали в 50 мл воды при перемешивании. Образующийся осадок отфильтровывали. Промывали на фильтре водой, 2 мл 96% EtOH. Кристаллы перекристаллизовывали из изопропанола. Продукт (1) получали в виде белого порошка (132 мг, 73%). Тпл 228.6°С. Rf 0.29 (MeOH–CHCl3, 1 : 4). Спектр 1H-ЯМР (DMSO-d6): 3.34 (с, 3H), 4.68 (с, 2H), 6.06 (м, 1Н), 6.28 (м, 1H), 7.30 (д, J 5.0, 1H), 7.41 (д, J 4.0, 1H), 7.53 (д, J 8.1, 1H), 7.65 (м, 1H), 7.78 (м, 1H), 7.90 (т, J 7.8, 1H), 8.04 (д, J 8.1, 1H), 8.45 (м, 2H), 8.69 (д, J 5.0, 1H), 9.29 (с, 1H), 12.35 (уш. с. 2Н). Спектр 13С ЯМР (DMSO-d6): 13.62, 37.15, 88.03, 108.33, 109.88 110.03, 110.20, 110.38, 112.84, 114.18, 115.36, 122.6, 125.45, 127.55, 128.80, 135.06, 135.96, 137.66, 140.39, 145.55, 149.47, 150.32, 153.77, 157.21, 160.50, 161.68.
N-(3-(4-Метил-1Н-имидазол-1-ил)-5-(трифторметил)фенил)-5-(((4-(пиридин-3-ил)пиримидин-2-ил)амино)метил)-1Н-пиррол-2-карбоксамид (2). Соединение (2) получали из килоты (14) по методике, аналогичной методике получения соединения (1), в виде бледно-желтого порошка (124 мг, 71%). Тпл 204.2°С. Rf 0.35 (MeOH : CHCl3, 1 : 4). Спектр 1H-ЯМР (DMSO-d6): 3.32 (с, 3H), 6.08 (м, 1Н), 6.20 (м, 1H), 7.26 (д, J 4.9 , 1H), 7.39 (д, J 4.0, 1H), 7.33 (д, J 8.0, 1H),7.64 (м, 1H), 7.78 (м, 1H), 7.90 (т, J 8.0, 1H), 8.06 (д, J 8.0, 1H), 8.43 (м, 2H), 8.54 (д, J 5.0, 1H), 9.31 (с, 1H), 11.98 (уш. с. 2Н). Спектр 13С-ЯМР (DMSO-d6): 13.62, 107.06, 108.36, 109.88, 110.04, 110.06, 112.23, 114.18, 114.63, 115.38, 122.06, 122.60, 125.71, 128.80, 129.73, 138.34, 140.33, 144.41, 149.47, 150.72, 154.59, 155.88, 156.90, 157.31, 157.40 158.28.
Список литературы
Tanaka R., Kimura S. // Expert Rev. Anticancer Ther. 2008. V. 8 (9). P. 1387–1398.
Lin Y., Meng Y., Huang L. // JACS. 2014. V. 136. P. 14 753–14 762.
Breccia M., Alimena G. // OncoTargets and Therapy. V. 1. P. 49–58.
Lin Y., Meng Y., Huang L. // Proc. Natl. Acad. Sci. USA. 2013. V. 110. № 5. P. 1664–1669.
Cowan-Jacob S.W., Fendrich G., Floersheimer A. // Acta Crystallogr., Sect. D. 2007. V. 63. P. 80–93.
Jabbour E., El Ahdab S., Cortes J., Kantarjian H. // Expert Opin. Investig. Drugs. 2008. V. 17. № 7. P. 1127–1136.
Schmuck C., Bickert V., Merschky M. // Eur. J. Org. Chem. 2008. P. 324–329.
Zheng R., Zeng X., He H., He J. // Synth. Commun. 2012. V. 1. № 42. P. 1521–1531.
Koroleva E.V., Ignatovich Zh.V., Ignatovich S.V., Gusak K.N. // Russ. J. Org. Chem. 2011. V. 47. № 8. P. 1222−1226.
Deadman B.J., Hopkin M.D., Baxendale I.R. // Org. Biomol. Chem. 2013. Is. 11. P. 1766–1800.
Гордон А., Форд Р. // М.: Мир, 1976.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия