

Биоорганическая химия, 2020, T. 46, № 3, стр. 310-321
Синтез производных флуоресцентного родаминового красителя на основе дигидрохинолина для анализа нуклеиновых кислот методом полимеразной цепной реакции в реальном времени
И. В. Матвиенко 1, 2, В. М. Байрамов 1, Н. А. Парыгина 1, 2, В. Е. Курочкин 3, Я. И. Алексеев 2, 3
1 ФГБНУ Всероссийский научно-исследовательский институт сельскохозяйственной биотехнологии
127550 Москва, ул. Тимирязевская, 42, Россия
2 ООО “Синтол”
127550 Москва, ул. Тимирязевская, 42, Россия
3 ФГБУН Институт аналитического приборостроения РАН
198095 Санкт-Петербург, ул. Ивана Черных, 31/33, Россия
Поступила в редакцию 13.08.2019
После доработки 02.09.2019
Принята к публикации 15.11.2019
Аннотация
Разработана оптимизированная схема синтеза производных ксантенового флуоресцентного красителя на основе дигидрохинолина. Впервые продемонстрирована возможность их использования в качестве эффективных флуорофоров в составе гибридизационных зондов для ПЦР в реальном времени с детекцией на индивидуальном спектральном канале прибора для ПЦР в реальном времени.
ВВЕДЕНИЕ
Благодаря своим хорошим фотохимическим и фотофизическим спектральным характеристикам, а именно высоким коэффициенту экстинкции и квантовому выходу, родаминовые флуоресцентные красители находят широкое применение для визуализации биологических макромолекул [1]. По сравнению с флуоресцентными красителями флуоресцеинового ряда, родаминовые красители более фотостабильны, их спектр флуоресценции не зависит от рН в диапазоне от 4 до 10 [2]. По этим причинам они находят широкое применение не только в биотехнологии для введения в состав белков и нуклеиновых кислот, но также и в медицине для диагностической визуализации живых клеток или живых организмов в доклинических исследованиях [3]. За последние годы большое разнообразие родаминовых красителей было выведено на рынок для конъюгирования с биомолекулами [2]. Тем не менее задача синтеза эффективных красителей, флуоресцирующих в спектральном диапазоне от 610 до 665 нм остается актуальной, так как наиболее распространенные красители, используемые для введения в состав нуклеиновых кислот, белков и других биологических макромолекул, флуоресцируют либо в более коротковолновой области спектра (5-карбоксиродамин 6Ж (5-R6G, λem = 562 нм), тетраметилкарбоксиродамин (5-TAMRA, λem = 583 нм), карбокси-Х-родамин (6-ROX, λem = 610 нм), либо в близкой ИК области (3,3,3',3'-тетраметил-индокарбоцианин (Cy5, λem = 669 нм).
Ранее Liu Jixiang и др. [4] синтезировали флуоресцентные красители, содержащие в своей структуре фрагмент 2,2-диметил-1,2-дигидрохинолина, аннелированного с тиофеном, бензтиофеном или нафталином.
Целью данной работы было разработать метод синтеза красителей родаминовой природы, содержащих аналогичный по структуре аннелированный дигидрорхинолиновый фрагмент с улучшенными спектральными характеристиками, а также изучить его свойства в составе гибридизационных зондов для ПЦР в реальном времени.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В качестве целевой была выбрана структура (V), синтез которой приведен на схеме 1 . Последовательность синтеза соединений I–IV, изображенная на верхней части схемы 1 , описана впервые.
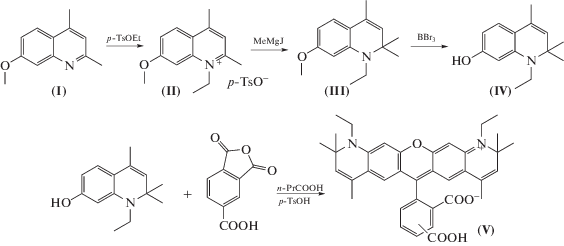
Схема 1 . Синтез целевого красителя (5(6)-карбокси-2-(1,11-диэтил-2,2,4,8,10,10-гексаметил-10,11-дигидро-2Н-пирано[3,2-g:5,6-g']дигидрохинолиниум-6-ил)бензоата, (5/6-Sy630).
При кипячении замещенных хинолинов с этилтозилатом в хлорбензоле были получены соответствующие четвертичные аммониевые соли, которые при обработке избытком реактива Гриньяра давали производные 1-этил-1,2-дигидрохинолина. Метоксигруппу удаляли обработкой трехбромистым бором при комнатной температуре. В связи с тем, что производные 7-гидрокси-1,2-дигидрохинолина чувствительны к действию минеральных кислот и кислот Льюиса, ядро красителя синтезировали кипячением их с тримеллитовым ангидридом в масляной кислоте с добавлением каталитических количеств п-толуолсульфокислоты. Полученную смесь изомеров разделяли при помощи хроматографии.
N-Гидроксисукцинимидные эфиры (VI) получали взаимодействием соответствующих карбоксипроизводных с дисукцинимидилкарбонатом в сухом хлористом метилене с добавлением 1.5 эквивалентов основания Хюнига и 0.5 эквивалентов 4-диметиламинопиридина. Также были синтезированы их пропилазидные производные взаимодействием активированных эфиров (VI) с 3-аминопропилазидом, как изображено на схеме 2 .
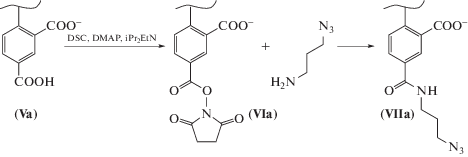
Схема 2 . Получение N-гидроксисукцинимидного эфира и пропилазида красителя 5-карбокси-Sy630.
2-Аминофенилкетоны, такие как 2-аминобензофенон и 2-аминоацетофенон и их замещенные производные являются удобными исходными соединениями для получения хинолинов [5]. 4‑Метокси-2-аминоацетофенон был получен по методу Tsutomu Sugasawa и др. [6] путем взаимодействия м-анизидина с ацетонитрилом в присутствии треххлористого бора и хлорида алюминия и последующим гидролизом полученного 2‑аминофенилкетимина, как это показано на схеме 3 . Синтез хинолинов, основанный на конденсации α-замещенного анилина с кетонами, впервые описан Фридлендером в 1882 г. [7]. В дальнейшем были разработаны многочисленные вариации этого метода с использованием различных катализаторов [8–20].
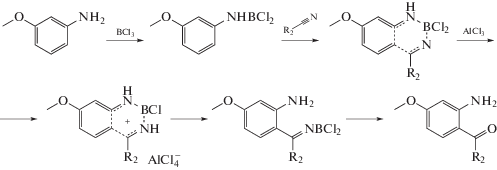
Схема 3 . Получение 4-метокси-2-аминоацетофенона.
Перспективной казалась идея получить и 2,4-диметил-7-метоксихинолин по этому же способу реакцией 2-амино-4-метоксиацетофенона с ацетоном, но по сообщению E. Roberts и E.E. Turner [21] взаимодействие 3,4-дихлор-2-аминоацетофенона с ацетоном протекает лишь при нагревании в запаянном сосуде при 185°С в течение 6 ч. Тогда была получена 2,4-диметил-7-метоксихинолин-3-карбоновая кислота (схема 4 ).

Схема 4 . Получение 2,4-диметил-7-метоксихинолин-3-карбоновой кислоты.
Однако эта кислота весьма устойчива к декарбоксилированию, эффективно удалить карбоксильную группу удается лишь разложением ее серебряной соли. Более рациональным представляется получение 2,4-диметил-7-метоксихинолина путем нагревания с концентрированной серной кислотой продукта конденсации м-анизидина с ацетилацетоном (схема 5 ), в работе [21] так были получены различные хлорпроизводные хинолина.

Схема 5 . Получение 2,4-диметил-7-метоксихинолина.
Спектральные характеристики 5(6)-изомеров карбокси-Sy630 Va и Vb, а также их производных VIa, VIb, VIIa и VIIb приведены в табл. 1. Значения квантовых выходов и коэффициентов экстинкции были получены аналогично [22].
Таблица 1.
Спектральные характеристики изомеров новых флуоресцентных красителей (Va и Vb) и их производных (VIa, VIb, VIIa и VIIb) в системе этанол–вода (1 : 1)
| Название | Обозначение | λex/λem, нм | Квантовый выход, % | ε, М–1 см–1 |
|---|---|---|---|---|
| 5-Карбокси-Sy630 | 5-Sy630 (Va) | 589/615 | 70 ± 8 | 101 900 ± 1100 |
| 6-Карбокси-Sy630 | 6-Sy630 (Vb) | 586/612 | 88 ± 12 | 90 300 ± 4600 |
| 5-Азидопропил-Sy630 | 5-Sy630-N3 (VIIa) | 592/619 | 66 ± 8 | 71 300 ± 2000 |
| 6-Азидопропил-Sy630 | 6-Sy630-N3 (VIIb) | 592/617 | 97 ± 3 | 107 800 ± 3900 |
Синтезированные N-гидроксисукцинимидные производные 5 и 6 изомеров красителя Sу630 (VIa, VIb) были присоединены на 5'-концы олигонуклеотидов, путем конденсации с 5'-термиальной аминогруппой (Aminolink-C6) олиготимидилатов T20. Соответствующие азидные производные (VIIa, VIIb) были присоединены на 5'‑концы олиготимидилатов T20, путем конденсации с 5'‑терминальной алкильной группой (Alkyn). Результаты масс-спектрометрического анализа MALDI-TOF и спектральные характеристики синтезированных олигонуклеотидов приведены в табл. 2 и 3 соответственно.
Таблица 2.
Результаты MALDI-TOF анализа модифицированных олигонуклеотидов
| Олигонуклеотид, 5' → 3' | Обозначение | Молекулярная масса | m/z [M + H]+ |
|---|---|---|---|
| 5-Sy630-Aminolink-C6-t-ttt-ttt-ttt-ttt-ttt-ttt-t | 5-Sy630-Aminolink-C6-T20 | 6775.9 | 6780.1 |
| 6-Sy630-Aminolink-C6-t-ttt-ttt-ttt-ttt-ttt-ttt-t | 6-Sy630-Aminolink-C6-T20 | 6775.9 | 6781.3 |
| 5-Sy630-Alkyn-t-ttt-ttt-ttt-ttt-ttt-ttt-t | 5-Sy630-Alkyn-T20 | 6930.0 | 6942.6 |
| 6-Sy630-Alkyn-t-ttt-ttt-ttt-ttt-ttt-ttt-t | 6-Sy630-Alkyn-T20 | 6930.0 | 6927.7 |
| 5-Sy630-Aminolink-C6-a-gcg-gct-cct-act-tct-gca-ggg-g-BHQ2 | 5-Sy630-Aminolink-C6-Fc | 8382.0 | 8371.6 |
| 6-Sy630-Aminolink-C6-a-gcg-gct-cct-act-tct-gca-ggg-g-BHQ2 | 6-Sy630-Aminolink-C6-Fc | 8382.0 | 8372.1 |
| 5-Sy630-Alkyn-a-gcg-gct-cct-act-tct-gca-ggg-g-BHQ2 | 5-Sy630-Alkyn-Fc | 8536.1 | 8521.2 |
| 6-Sy630-Alkyn-a-gcg-gct-cct-act-tct-gca-ggg-g-BHQ2 | 6-Sy630-Alkyn-Fc | 8536.1 | 8517.1 |
| 6-ROX-ag-cgg-ctc-cta-ctt-ctg-cag-ggg-BHQ2 | FC_Pr_up_ROX | 8310.8 | 8308.8 |
| Cy5-ag-cgg-ctc-cta-ctt-ctg-cag-ggg-BHQ2 | FC_Pr_up_Cy5 | 8300.6 | 8303.8 |
| cac-ata-ttt-aca-gaa-tgg-caa-agg | Fc-up | 7393.8 | 7395.1 |
| ctg-aag-aca-cat-ttt-tac-tcc-caa | Fc-low | 7255.7 | 7256.5 |
Таблица 3.
Спектральные характеристики модифицированных олигонуклеотидов
| Олигонуклеотид, 5' → 3' | Обозначение | λex/λ em, нм |
|---|---|---|
| 5-Sy630-Aminolink-C6-t-ttt-ttt-ttt-ttt-ttt-ttt-t | 5-Sy630-Aminolink-C6-T20 | 602/628 |
| 5-Sy630-Alkyn-t-ttt-ttt-ttt-ttt-ttt-ttt-t | 5-Sy630-Alkyn-T20 | 602/629 |
| 6-Sy630-Aminolink-C6-t-ttt-ttt-ttt-ttt-ttt-ttt-t | 6-Sy630-Aminolink-C6-T20 | 602/625 |
| 6-Sy630-Alkyn-t-ttt-ttt-ttt-ttt-ttt-ttt-t | 6-Sy630-Alkyn-T20 | 601/624 |
| 5-Sy630-Aminolink-C6-a-gcg-gct-cct-act-tct-gca-ggg-g-BHQ2 | 5-Sy630-Aminolink-C6-Fc | 604/629 |
| 6-Sy630-Aminolink-C6-a-gcg-gct-cct-act-tct-gca-ggg-g-BHQ2 | 6-Sy630-Aminolink-C6-Fc | 603/626 |
| 5-Sy630-Alkyn-a-gcg-gct-cct-act-tct-gca-ggg-g-BHQ2 | 5-Sy630-Alkyn-Fc | 602/626 |
| 6-Sy630-Alkyn-a-gcg-gct-cct-act-tct-gca-ggg-g-BHQ2 | 6-Sy630-Alkyn-Fc | 603/626 |
| 6-ROX-ag-cgg-ctc-cta-ctt-ctg-cag-ggg-BHQ2 | FC_Pr_up_ROX | 588/613 |
| Cy5-ag-cgg-ctc-cta-ctt-ctg-cag-ggg-BHQ2 | FC_Pr_up_Cy5 | 648/668 |
Как видно из данных, приведенных в табл. 3, максимумы длин волн эмиссии у 5-ого и 6-ого карбокси изомеров красителя Sy630 отличаются на 4–5 нм, что также характерно для 5-ых и 6-ых карбокси изомеров других красителей ксантенового ряда (FAM, R6G, TAMRA, ROX). Однако, эта разница исчезает при введении 5-ого или 6‑ого карбокси изомера красителя Sy630 в гибридизационные зонды, видимо, из-за влияния гетерогенной по составу, по сравнению олиготимидилатом, последовательности зондов.
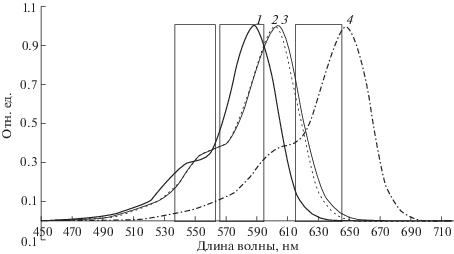
Рис. 1.
Нормализованные спектры возбуждения олиготимидилатов, содержащих на 5'-конце красители 6-ROX (1), 6-Sy630 (2), 5-Sy630 (3) и Cy5 (4). Прямоугольниками выделены спектральные диапазоны пропускания интерференционных светофильтров, использованных для постановки ПЦР-РВ.
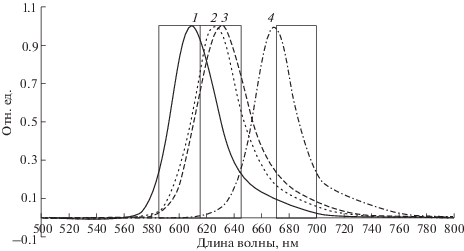
Максимумы спектров поглощения и флуоресценции производных красителя Sy630 в составе олигонуклеотидов имеют промежуточные значения по сравнению с максимумами поглощения и флуоресценции красителей 6-ROX и Сy5 (рис. 1, 2 ). Для обеспечения возможности детектировать новый флуоресцентный краситель Sy630 в индивидуальном спектральном канале, в прибор для ПЦР-РВ АНК-М [23] была установлена пара светофильтров 580 нм (30 нм)/630 нм (30 нм), где 30 нм – значение полной ширины пропускания на половине амплитуды. Для того, чтобы снизить перекрестное свечение сигнала флуоресценции красителя Sy630 в спектральный канал, соответствующий красителю 6-ROX, имеющему в составе гибридизационных зондов (см. табл. 3) характеристики длин волн возбуждения – 587–588 нм и длин волн эмиссии – 613–614 нм, для его детекции использовали не стандартную пару светофильтров с характеристиками возбуждения/эмиссии 550 нм (25 нм)/600 нм (30 нм). Для детекции сигнала флуоресценции красителя Cy5 использовали стандартную пару светофильтров 630 нм (30 нм)/ 685 нм (25 нм).
Рис. 2.
Нормализованные спектры эмиссии олиготимидилатов, содержащих на 5'-конце красители 6-ROX (1), 6-Sy630 (2), 5-Sy630 (3) и Cy5 (4). Прямоугольниками выделены спектральные диапазоны пропускания интерференционных светофильтров, использованных для постановки ПЦР-РВ.
На рис. 3 приведены результаты постановок ПЦР-РВ с одновременной детекцией красителей 6-ROX, 5-Sy630-NHS (VIa), 6-Sy630-NHS (VIb) и Cy5 в составе гибридизационных зондов на индивидуальных спектральных каналах.
Рис. 3.
Результаты ПЦР-РВ (данные нормированы к нулю и по максимуму) серии из четырех 10-кратных разведений ДНК человека с концентрациями 5, 1, 0.2, 0.04 нг/мкл в двух повторах, полученные с использованием гибридизационных зондов. (а) FC_Pr_up_ROX, (R2 = 1.0, E = 97%); (б) 5-Sy630-Aminolink-C6-Fc, (R2 = 1.0, E = 98%) и 6-Sy630- Aminolink-C6-Fc (R2 = 0.999, E = 100%); (в) FC_Pr_up_Cy5 (R2 = 0.997, E=98%).

На рис. 4 приведены результаты постановок ПЦР-РВ с одновременной детекцией красителей 6-ROX, 5-Sy630-N3 (VIIa), 6-Sy630-N3 (VIIb) и Cy5 в составе гибридизационных зондов на индивидуальных спектральных каналах.
Рис. 4.
Результаты ПЦР-РВ (данные нормированы к нулю и по максимуму) серии из четырех 10-кратных разведений ДНК человека с концентрациями 5, 1, 0.2, 0.04 нг/мкл в двух повторах, полученные с использованием гибридизационных зондов. (а) FC_Pr_up_ROX, (R2 = 1.0, E = 97%); (б) 5-Sy630-Alkyn-Fc, (R2 = 0.998, E = 100%) и 6‑Sy630-Alkyn-Fc (R2 = 1.0, E = 97%); (в) FC_Pr_up_Cy5 (R2 = 0.999, E = 101%).

Как видно из приведенных на рис. 3 и 4 данных, системы праймеров и зондов, меченных производными нового флуоресцентного красителя Sy630, обеспечивают высокую (выше 97%) эффективность ПЦР в реальном времени в широком диапазоне концентраций ДНК. Рассчитанные значения R2, близкие к 1 и значения эффективности ПЦР-РВ, близкие к 100% для зондов, в которые красители вводили либо в виде N-гидроксисукцинимидных эфиров, либо в виде азидопропильных производных, свидетельствуют о том, что оба типа производных нового красителя могут быть использованы для введения в состав гибридизационных зондов для ПЦР в реальном времени. При этом стоит отметить, что в силу более высокой стабильности азидопропильных производных они являются более предпочтительными кандидатами для рутинной работы по введению флуоресцентных меток в состав гибридизационных зондов для ПЦР-РВ. При этом наиболее предпочтительным является использование производного 6-изомера азидопропила, так как он имеет большие значения квантового выхода и коэффициента экстинкции (табл. 1), произведение которых пропорционально светимости флуорофора. Подобранная комбинация интерференционных светофильтров для трех соседних спектральных каналов обеспечивает эффективное определение ДНК-мишени на каждом индивидуальном спектральном канале прибора для ПЦР-РВ, хотя не является оптимальной. Для обеспечения наилучшего возбуждения и сбора сигнала флуоресценции для детекции флуоресценции красителя Sy630 необходимо использовать интерференционные светофильтры, с максимумами пропускания, сдвинутыми на 20–30 нм в красную область спектрального диапазона.
Таким образом, впервые продемонстрирована возможность постановки ПЦР в реальном времени с гибридизационными зондами, содержащими производные нового флуоресцентого красителя на основе дигидрохинолина (5(6)-карбокси-2-(1,11-диэтил-2,2,4,8,10,10-гексаметил-10,11-дигидро-2Н-пирано[3,2-g:5,6-g']дигидрохинолиниум-6-ил)бензоата – 5/6-карбокси-Sy630. Продемонстрирована возможность использования таких зондов в мультиплексной ПЦР-РВ с детекцией на индивидуальном спектральном канале прибора для ПЦР-РВ. Это позволяет увеличить количество одновременно выявляемых в одной пробирке ДНК и/или РНК мишеней на одну, по сравнению со стандартным набором флуоресцентных красителей и соответствующих им комбинаций интерференционных светофильтров широко используемых сегодня в приборах для ПЦР-РВ для постановки мультиплексной ПЦР в реальном времени.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали ацетонитрил (ч.д.а) и бензол (х.ч.) перегнанные над Р2О5, хлороформ (х.ч.), метил-трет-бутиловый эфир (ч.д.а) и хлористый метилен (х.ч.) над СаН2, диизопропилэтиламин 99% (Fisher Scientific) перегоняли над КОН, ацетилацетон (ч.д.а.) (Черкасского завода химреактивов им. XXV съезда КПСС) перегоняли в вакууме с дефлегматором. Остальные реактивы использовали без дополнительной очистки: этил п-толуолсульфонат 98%, м-анизидин 99%, циклопентанон 99+%, 1 М раствор треххлористого бора в хлористом метилене, трибромид бора 99% (Acros Organics); тетрагидрофуран dry (max 0.0075% water) (Panreac); магний в гранулах 12–50 меш 99.8%, метилацетоацетат 99% (Alfa Aesar); 4-диметиламинопиридин 99+% (Fluka); фосфор(V) оксид (ч.), натрий сернокислый б/водн. (х.ч.), п-толуолсульфокислота моногидрат (ч.), этилацетат (х.ч.), гексан (х.ч.), диэтиловый эфир (ч.д.а) (Химмед); соляная кислота (х.ч.), гидроксид натрия (ч.д.а.), масляная кислота (имп.), серная кислота (х.ч.), бихромат калия (х.ч.), метилйодид (имп.) (Вектон); хлорбензол (ч.д.а.) (Экос-1); хлорид аммония (х.ч.) (Реахим); тримеллитовый ангидрид 97%, N,N'-дисукцинимидилкарбонат 95+%, азид натрия 99.5+%, 3‑бромопропиламина гидробромид 98%, диметилсульфоксид USP, трис-[(1-бензил-1Н-1,2,3-триазол-4-ил)метил]амин 97%, меди(II) сульфат пентагидрат 98%, трис(2-карбоксиэтил)фосфина гидрохлорид (Sigma-Aldrich); метанол 99.9% (Lab-Scan); N,N,N,N',N'-тетраметил-О-(N-сукцинимидил)мочевины тетрафторборат (Hangzhou Dayangchem); остальные реактивы отечественного производства марки “х.ч”.
ТСХ выполняли на пластинках Kieselgel 60 (Merck, Германия) в системах, приведенных ниже.
Олигонуклеотиды, использованные в работе, синтезировали и очищали как описано ранее [22]. Последовательности праймеров и зондов Fc-up, Fc-low, 5-Sy630-Aminolink-C6-Fc, 6-Sy630-Aminolink-C6-Fc, 5-Sy630-Alkyn-Fc, 6-Sy630-Alkyn-Fc, FC_Pr_up_ROX, FC_Pr_up_Cy5 были подобраны к Fc-фрагменту гена иммуноглобулина G человека (FCGR3B, Sequence ID: NM_001271036.1).
В работе использовали магнитную мешалку с подогревом (Heildolph MR 3001 K, Германия), роторный испаритель (Buchi Rotavapor R200, Швейцария), синтезатор ДНК (ASM-2000, Биоссет, Россия), препаративный хроматограф (Reveleris Prep Grace, США), термостат (Циклотемп-303, Россия), шейкер для пробирок (Eppendorf Mixer 5432, Германия), вакуумный испаритель (Univapo 150 ECH, Германия), микроцентрифугу-встряхиватель (Циклотемп-901, Россия). Спектры поглощения красителей и их конъюгатов с олигонуклеотидами регистрировали на спектрофотометре Nanodrop ND-1000 (ThermoFisher Scientific, США). Спектры возбуждения и флуоресценции измеряли на спектрофлуориметре PerkinElmer LS55 (Perkin Elmer, США) со следующими параметрами: скорость сканирования 500 нм/мин, ширина щели при измерении возбуждения – 15 нм, при измерении эмиссии – 10 нм. Спектральный диапазон возбуждения – от 450 до 700 нм; эмиссии – от 500 до 750 нм. Спектры ЯМР записывали на приборе DRX500 (Bruker Daltonics, Германия), снимали в Институте органической химии им. Н.Д. Зелинского РАН (г. Москва). Масс-спектры MALDI-TOF снимались на приборе Microflex LRF (Bruker Daltonics, Германия) в центре коллективного пользования научным оборудованием ФГБНУ ВНИИСБ “Биотехнология”. ПЦР в реальном времени проводили на приборе АНК-48 (Институт аналитического приборостроения РАН, Россия) по следующей циклограмме: 95°С – 5 мин, затем 49 циклов: 60°С – 40 с, 95°С – 15 с. Полученные данные автоматически обрабатывали с помощью программного обеспечения АНК-Shell 1.0.5.100.
2,4-Диметил-7-метоксихинолин (I). Ацетилацетон 45 мл (43.875 г, 0.438 моль, 1.1 экв.) и м‑анизидин 45 мл (1 экв., 0.398 моль, 49.06 г) кипятили с обратным холодильником в течение 2.5 ч, нагревая на масляной бане при 140°С. Реакционную массу упаривали в вакууме при 70°С. Полученное масло при перемешивании и охлаждении на ледяной бане выливали в 250 мл концентрированной серной кислоты. Затем реакционную массу нагревали на кипящей водяной бане 1.5 ч. Выливали в 1 л воды, охлаждали на водяной бане и приливали раствор 135 г бихромата калия в 1 л воды. Охлаждали на ледяной бане. Выпавший осадок бихромата хинолина отфильтровывали, тщательно промывали на фильтре водой и растворяли в 900 мл 12% раствора гидроксида натрия. Перемешивали до полного растворения осадка, экстрагировали 4 × 200 мл диэтилового эфира. Экстракты сушили над безводным сульфатом натрия и упаривали в вакууме. Получено 38.0 г вещества в виде бесцветного масла, при стоянии в холодильнике (на 4°С) медленно кристаллизующегося в виде крупных призм. Выход 2,4-диметил-7-метоксихинолина 51%. Rf = 0.85 (метанол–хлороформ 1 : 3), m/z найдено: 188.165, рассчитано: 187.1. ЯМР 1Н в СDCl3 (500 МГц) δ (м.д.): 7.73 (1H, d, J = 9.1, С5Н), 7.30 (1Н, d, J = 2.4, С8Н), 7.06 (1H, dd, J = 9.1, 2.4, C6H), 6.90 (1H, s, C3H), 3.88 (3Н, s, ОСН3), 2.61 (3Н, s, С2–CH3), 2.53 (3Н, s, С4–CH3). ЯМР 13С в СDCl3 (126 МГц) δ (м.д.): 159.86 (C2), 158.41 (C7), 148.97 (C8a), 143.50 (C4), 124.16 (C5), 120.95 (C4a), 120.20 (C3), 117.56 (C6), 106.81 (C8), 54.87 (OCH3), 24.60 (C2–CH3), 17.95 (C4–CH3).
1-Этил 2,4-диметил-7-метокси-хинолиния тозилат (IIa). 2,4-Диметил-7-метоксихинолин 38.0 г (0.203 моля) и этилтозилат 38 мл (1.1 экв., 0.223 моля, 44.7 г) кипятили с обратным холодильником в 115 мл хлорбензола в течение 20 часов. При охлаждении реакционной массы выпадали кристаллы. Осадок отфильтровывали, промывали на фильтре петролейным эфиром, сушили на воздухе. Фильтрат упаривали в вакууме масляного насоса. Полученное масло при стоянии кристаллизовалось. Получено 72.236 г вещества в виде кремовых кристаллов. Выход 1-этил 2,4-диметил-7-метокси-хинолиния тозилата 92%. Rf = 0.27 (метанол–хлороформ 1 : 3), m/z найдено: 215.969, рассчитано: 216.3. ЯМР 1Н в СDCl3 (500 МГц) δ (м.д.): 8.08 (1H, d, J = 9.2, С5Н), 7.64 (2Н, d, J = 8.0, Tos(2' + 6')), 7.58 (1Н, d, J = 1.9, С8Н), 7.52 (1H, s, C3H), 7.40 (1H, dd, J = 9.2, 2.1, C6H), 6.98 (2H, d, J = 7.9, Tos(3' + 5')), 5.09 (2H, q, J = 7.2, CH2CH3), 4.10 (3Н, s, ОСН3), 3.04 (3Н, s, С2–CH3), 2.81 (3Н, s, С4–CH3), 2.25 (3H, s, Tos(CH3), 1.58 (3H, t, J = 7.3, CH2CH3).
2,2,4-Триметил-1-этил-7-метокси-1,2-дигидрохинолин (IIIa). К раствору 9.438 г (24.36 ммоль) 1-этил-2,4-диметил-7-метокси-хинолиния тозилата в 100 мл абсолютного тетрагидрофурана прибавляли по каплям в темноте, в атмосфере аргона реактив Гриньяра, полученный из 2.43 г (4 экв., 100 ммоль) магния и 6.23 мл (100 ммоль, 14.194 г) йодистого метила в 100 мл метилтретбутилового эфира. Перемешивали в темноте в течение 2 суток. При охлаждении на ледяной бане добавляли 150 мл насыщенного раствора хлорида аммония. Осадок отфильтровывали, промывали небольшим количеством воды и метилтретбутилового эфира. Фильтрат разделяли в делительной воронке. Водную фазу экстрагировали 2 × 100 мл диэтилового эфира. Объединенную органическую фазу сушили над безводным сульфатом натрия и упарили в вакууме. Полученный продукт очищали с помощью флэш-хроматографии на силикагеле. Элюировали смесью гексан–хлороформ 9 : 1. Получено 2.742 г вещества в виде масла. Выход 2,2,4-триметил-1-этил-7-метокси-1,2-дигидрохинолина 49%. m/z найдено: 231.168, рассчитано: 231.2. ЯМР 1Н в СDCl3 (500 МГц) δ (м.д.): 6.97 (1H, d, J = 8.3, С5Н), 6.14 (1H, dd, J = 8.3, 2.3, C6H), 6.08 (1Н, d, J = 2.3, С8Н), 5.10 (1H, s, C3H), 3.79 (3Н, s, ОСН3), 3.32 (2H, q, J = 7.0, N–CH2CH3), 1.97 (3Н, s, С4–CH3), 1.32 (6Н, s, С2–(CH3)2), 1.22 (3H, t, J = 7.0, N–CH2CH3). ЯМР 13С в СDCl3 (126 МГц) δ (м.д.): 160.69 (C7), 145.11 (C8a), 127.48 (C4), 127.12 (C3), 124.59 (C5), 116.73 (C4a), 98.91 (C6), 97.65 (C8), 57.02 (C2), 55.19 (OCH3), 38.32 (N-CH2CH3), 28.80 (C2–(CH3)2), 18.90 (C4–CH3), 14.39 (N–CH2CH3).
2,2,4-Триметил-1-этил-7-гидрокси-1,2-дигидрохинолин (IVa). 2,2,4-Триметил-1-этил-7-метокси-1,2-дигидрохинолин 2.655 г (11.5 ммоль) растворяли в 20 мл сухого хлороформа и в темноте при охлаждении на ледяной бане, в атмосфере аргона добавляли по каплям 2.22 мл (5.762 г, 23 ммоль, 2 экв.) трехбромистого бора в 10 мл сухого хлороформа. Выдерживали 1 ч при –3°С, затем 1 ч 40 мин при комнатной температуре. При охлаждении льдом добавляли 22.5 мл 12% раствора гидроксида натрия. Отделяли на делительной воронке нижний слой. Водную фазу подкисляли, экстрагировали хлороформом. Объединенные экстракты сушили безводным сульфатом натрия и пропускали через слой силикагеля, элюировали хлороформом и упаривали в вакууме. Получено 1.106 г масла, темнеющего на воздухе. Выход 2,2,4-триметил-1-этил-7-гидрокси-1,2-дигидрохинолина 44%. m/z найдено: 217.230, рассчитано: 217.1. ЯМР 1Н в СDCl3 (500 МГц) δ (м.д.): 6.87 (1H, d, C5H), 6.03 (2H, m, C6H + C8H), 5.05 (1H, s, C3H), 3.27 (2H, q, NCH2), 1.92 (3H, s, C4–CH3), 1.29 (6H, s, C2–CH3)2), 1.18 (3H, t, CH2CH3).
5(6)-Карбокси-Sy630 4(5)-карбокси-2-(1,11-диэтил-2,2,4,8,10,10-гексаметил-10,11-дигидро-2Н-пирано[3,2-g:5,6-g']дихинолин-1-иум-6-ил)бензоат (Va) и (Vb). К 3.63 г (16.7 ммоль, 2 экв.) 2,2,4-триметил-1-этил-7-гидрокси-1,2-дигидрохинолина в 130 мл масляной кислоты добавляли 60 мг (0.32 ммоль) гидрата п-толуолсульфокислоты и 1.603 г (8.35 ммоль, 1 экв.) тримеллитового ангидрида. Кипятили в темноте, в атмосфере аргона с обратным холодильником 24 ч. Упаривали в вакууме (при 80°С), заливали гексаном, на следующий день гексан отбрасывали. Предварительно очищали хроматографией на нейтральной окиси алюминия. Элюировали изопропанолом с 0–15% воды с 5% триэтиламина. Фракции, содержащие 5-й изомер, упаривали и растворяли остаток в 5 мл сухого хлористого метилена. Приливали по каплям, при перемешивании на магнитной мешалке, к 100 мл абсолютного диэтилового эфира. Осадок отфильтровывали, промывали эфиром, сушили в вакууме. Получено после сушки в вакууме 0.265 г чистого 5-карбокси изомера, остальные фракции, содержащие продукт, дополнительно очищали хроматографией на силикагеле. Элюировали 0–20% метанола в хлороформе с 1% триэтиламина. 6‑карбоксиизомер (Vb) выходил при 10% метанола, 5-карбоксиизомер (Va) – при 15–20%. Получено 0.796 г 6-карбокси-Sy630 в виде фиолетовой пены. Выход 6-карбокси-Sy630 16%. Rf = 0.24, (метанол–хлороформ 1 : 4). Масс-спектр: m/z, найдено: 591.315, рассчитано: 590.3, квантовый выход = = 0.77 (в системе этанол–вода 1 : 1), коэффициент экстинкции (при максимуме поглощения) = 9 × × 104 М–1 см–1, максимум длины волны поглощения = 584 нм (в системе этанол–вода 1 : 1), максимум длины волны флуоресценции = 609 нм (в системе этанол–вода 1 : 1). ЯМР 1Н триэтиламмониевой соли в СDCl3 (500 МГц) δ (м.д.): 8.23 (1H, d, J = 7.9), 7.98 (1H, d, J = 8.1), 7.80 (1H, s), 6.37 (2H, s), 6.24 (2H, s), 5.09 (2H, s), 3.36 (4H, q, J = 6.9, 2*NCH2CH3), 3.01 (6H, q, J = 7.3, N(CH2CH3)3), 1.63 (6H, s, C4–CH3 + C8CH3), 1.31 (12H, s, 2*C2–CH3 + 2*C11–CH3), 1.24 (15H, m, 2*NCH2CH3 + N(CH2CH3)3). Получено 1.199 г 5-карбокси-Sy630 в виде фиолетового порошка. Выход 5-карбокси-Sy630 24%. Rf = 0.59 (метанол–хлороформ 1 : 9 + 5% триэтиламина), m/z Масс-спектр: m/z, найдено: 591.334, рассчитано: 590.3, квантовый выход = 0.50 (в системе этанол–вода 1 : 1), коэффициент экстинкции (при максимуме поглощения) = 1.007 × 105 М–1 см–1, максимум длины волны поглощения = 585 нм (в системе этанол–вода 1 : 1), максимум длины волны флуоресценции = 612 нм (в системе этанол–вода 1 : 1). ЯМР 1Н триэтиламмониевой соли в СDCl3 (500 МГц) δ (м.д.): 8.66 (1H, s), 8.31 (1H, dd, J = = 7.9, 1.3), 7.16 (1H, d, J = 7.8), 6.31 (2H, s), 6.25 (2H, s), 5.08 (2H, s), 3.35 (4H, q, J = 6.9, 2*NCH2CH3), 3.05 (6H, q, J = 7.2, N(CH2CH3)3), 1.61 (6H, s, C4–CH3 + C8CH3), 1.24–1.32 (27H, m, 2*C2–CH3 + 2*C11–CH3 + 2*NCH2CH3 + + N(CH2CH3)3).
5-Sy630-NHS 5-карбокси-2-(1,11-диэтил-2,2,4,8,10,10-гексаметил-10,11-дигидро-2Н-пирано[3,2-g:5,6-g’]дихинолин-1-иум-6-ил)бензоата N-гидроксисукцинимидный эфир (VIa). 5-Карбокси-Sy630 95 мг (0.161 ммоль) растворяли в 9.5 мл сухого хлористого метилена, добавляли 42 мкл (31 мг, 0.242 ммоль, 1.5 экв.) диизопропилэтиламина, 50 мг (0.193 ммоль, 1.2 экв.) дисукцинимидилкарбоната и 10 мг (0.08 ммоль, 0.5 экв.) 4-диметиламинопиридина. Перемешивали при комнатной температуре 2 ч, затем добавляли еще 25 мг дисукцинимидилкарбоната и перемешивали еще 1 ч. Выливали в 10 мл 0.6 М раствора соляной кислоты и отделяли нижний слой в делительной воронке. Органическую фазу сушили безводным сульфатом натрия, упаривали в вакууме, полученное вещество растворяли в 2 мл сухого хлористого метилена и добавляли по каплям при перемешивании к 30 мл абсолютизированного метилтретбутилового эфира. Отфильтровывали, промывали метилтретбутиловым эфиром, сушили в вакууме. Выход соединения (VIa) составил 43 мг (39%), фиолетовый порошок. Rf = 0.75, (метанол–хлороформ 1 : 3), m/z найдено: 688.1, рассчитано: 687.3 максимум длины волны поглощения = 592 нм (в системе этанол–вода 1 : 1), максимум длины волны флуоресценции = 641 нм (в системе этанол–вода 1 : 1). ЯМР 1Н в СDCl3 (500 МГц) δ (м.д.): 9.08 (1H, s), 8.41 (1H, d, J = 7.7), 7.40 (1H, d, J = 7.7), 6.61 (2H, s), 6.58 (2H, s), 5.44 (2H, s), 3.62 (4H, d, J = 6.5), 2.98 (4H, s), 1.76 (6H, s), 1.48 (12H, s), 1.38 (6H, не разрешившийся триплет).
6-Sy630-NHS 4-Карбокси-2-(1,11-диэтил-2,2,4,8,10,10-гексаметил-10,11-дигидро-2Н-пирано[3,2-g:5,6-g']дихинолин-1-иум-6-ил)бензоата N-гидроксисукцинимидный эфир (VIb). 6-Карбокси-Sy630 227 мг (0.38 ммоль) растворяли в 22 мл сухого хлористого метилена, добавляли 100 мкл (75 мг, 0.576 ммоль, 1.5 экв.) диизопропилэтиламина, 117 мг (0.456 ммоль, 1.2 экв.) дисукцинимидилкарбоната и 23 мг (0.19 ммоль, 0.5 экв.) 4-диметиламинопиридина. Перемешивали при комнатной температуре 1 ч, затем добавляли еще 12 мг дисукцинимидилкарбоната и перемешивали еще 1.5 ч. Выливали в 25 мл насыщенного раствора дигидрофосфата калия, встряхивали в делительной воронке. Органическую фазу отделяли, сушили безводным сульфатом натрия, упаривали в вакууме. Очищали хроматографией на обращенной фазе, элюировали системой вода–ацетонитрил. Продукт собирали при 40% ацетонитрила. Фракции, содержащие продукт, упаривали до половины объема и экстрагировали хлористым метиленом, сушили безводным сульфатом натрия и упаривали в вакууме. Выход соединения (VIb) составил 67 мг (26%) фиолетовая пена. Rf = 0.72, (метанол–хлороформ 1 : 3), m/z найдено: 688, рассчитано: 687.3, максимум длины волны поглощения = 603 нм (в воде). ЯМР 1Н в СDCl3 (400 МГц) δ (м.д.): 8.51 (1H, d, J = 8.2), 8.44 (1H,dd, J = 8.2, 1.6), 7.99 (1H, d, J =1.5), 6.64 (2H, s), 6.60 (2H, s), 5.46 (2H, s), 3.61–3.64 (4H, m), 2.94 (4H, s), 1.77 (6H, s), 1.48 (12H, d), 1.39 (6H, t, J = 7.0).
5-Sy630-N3 5-((3-Азидопропил)карбамоил)-2-(1,11-диэтил-2,2,4,8,10,10-гексаметил-10,11-дигидро-2H-пирано[3,2-g:5,6-g']дихинолин-1-иум-6-ил)бензоат (VIIa). 0.265 г 5-Карбокси-Sy630 (0.45 ммоль) растворяли в 10 мл безводного хлористого метилена, прибавляли при перемешивании 0.162 г (0.54 ммоль, 1.2 экв.) TSTU, затем 94 мкл (0.54 ммоль, 1.2 экв, 70 мг) диизопропилэтиламина и 27 мг (0.22 ммоль, 0.5 экв.) DMAP. Через 1 ч 20 мин реакционную смесь выливали при перемешивании в 100 мл 1%-ного раствора соляной кислоты. Экстрагировали 2 × 50 мл хлороформа, сушили безводным сульфатом натрия и упаривали в вакууме. Остаток растворяли в 10 мл безводного хлористого метилена, добавляли 68 мкл (0.675 ммоль, 1.5 экв.) аминопропилазида и 118 мкл (87 мг, 0.675 ммоль, 1.5 экв.) диизопропилэтиламина. Перемешивали 20 мин и упаривали в вакууме. Очищали хроматографией на силикагеле, элюировали 0–5% метанола в хлороформе, затем 6–8% метанола в хлороформе с 1% триэтиламина. Упаривали в вакууме и сушили в вакууме над Р2О5. Выход соединения (VIIa) 212 мг (70%), фиолетовый порошок. Rf = 0.54, (метанол–хлороформ 1 : 4), m/z найдено: 673.423, рассчитано: 672.3, квантовый выход = 0.74 (в системе этанол–вода 1 : 1), коэффициент экстинкции (при максимуме поглощения) = 1.286 × 105 М–1 см–1, максимум длины волны поглощения = 596 нм (в воде), 590 нм (в системе этанол–вода 1 : 1), максимум длины волны флуоресценции = 620 нм (в системе этанол–вода 1 : 1). ЯМР 1Н в СDCl3 (500 МГц) δ (м.д.): 8.62 (1H, s, C4'H), 8.19 (1H, d, J = 7.8, C7'H), 7.58 (1H, t, J = 5.6, CONH), 7.20 (1H, d, J = 7.8, C6'H), 6.72 (2H, s, C12H + C13H), 6.50 (2H, s, C5H + C7H), 5.32 (2H, s, C3H + C9H), 3.61 (2H, dd, J = 12.6, 6.4, CONHCH2), 3.53 (4H, m, N1CH2CH3 + N11CH2CH3), 3.45 (2H, t, J = 6.7, CH2N3), 1.97 (2H, qt, J = 6.7, CONHCH2CH2), 1.72 (6H, s, C4–CH3 + C7–CH3), 1.42 (12H, d, J = 5.3, C2–2*CH3 + C10–2*CH3), 1.34 (6H, m, N1CH2CH3 + N11CH2CH3).
6-Sy630-N3 4-((3-Азидопропил)карбамоил)-2-(1,11-диэтил-2,2,4,8,10,10-гексаметил-10,11-дигидро-2H-пирано[3,2-g:5,6-g']дихинолин-1-иум-6-ил)бензоат (VIIb). 6-Sy630-NHS 66 мг (0.096 ммоль) растворяли в 10 мл сухого хлористого метилена, добавляли 19 мг (0.192 ммоль, 2 экв.) 3-аминопропилазида. Перемешивали 4 ч при комнатной температуре, разбавляли 10 мл хлористого метилена, промывали 2 × 10 мл 0.1% раствора трифторуксусной кислоты, 2 × 10 мл раствора гидрокарбоната натрия (насыщенный раствор–вода 1 : 1), 20 мл насыщенного раствора хлорида натрия. Очищали хроматографией на силикагеле. Элюировали 5% метанола в хлороформе с 1% триэтиламина. Выход соединения (VIIb) 41 мг (64%), сине-фиолетовый порошок. Rf = 0.27, (метанол–хлороформ 1 : 10 с 5% триэтиламина), m/z найдено: 673.248, рассчитано: 672.3.
ЯМР 1Н в СDCl3 (500 МГц) δ (м.д.): 8.24 (1H, d, J = 8.1), 8.11 (1H, d, J = 8.0), 7.75 (2H, s), 6.66 (2H, s), 6.53 (2H, s), 5.35 (2H, s), 3.58 (4H, неразрешившийся q, J = 6.8, 2*CH2N), 3.50 (2H, dt, J = 15.8, 7.9, CH2NHCO), 2.36 (2H, t, J = 6.9, CH2N3), 1.92 (2H, qt, J = 6.8, –CH2–), 1.66 (6H, s, 2*CH3), 1.44 (12H, s, 4*CH3), 1.33 (6H, t, J = 6.9, 2*CH2CH3). На основании спектра HMBC и HSQC удалось соотнести ЯМР 13С и подтвердить структуру каждого изомера. ЯМР 13С в СDCl3 (126 МГц) δ (м.д.): 166.93 (COO–), 165.61 (CONH), 157.46 (2C, F), 157.29 (2), 151.54 (2C, D), 136.27 (6), 136.10 (1), 133.02 (3), 131.29 (2C, X), 131.18 (1), 128.45 (5), 127.79 (4), 125.41 (2C, E), 123.04 (2C, A), 122.29 (2C, H), 113.63 (2C, C), 94.90 (2C, B), 59.42 (2C, 2*C(CH3)2), 48.96 (a), 39.56 (2C, 2*NCH2CH3), 37.27 (c), 29.22, 29.05 (4C, 4*CH3), 28.16 (b), 17.86 (2*CH3), 12.76 (2*CH2CH3).
2-Амино-4-метоксиацетофенон (VIII). В приборе, снабженном хлоркальциевой трубкой, к 25 мл 1 М раствора треххлористого бора в хлористом метилене при охлаждении льдом по каплям добавляли раствор м-анизидина 2.777 мл (24.72 ммоль, 3.044 г) в 20 мл сухого бензола. Затем добавляли ацетонитрил 2.58 мл (2 экв. = 49.44 ммоль, 3.03 г) и хлорид алюминия 3.626 г (1.1 экв. = 27.19 ммоль). Кипятили 2 ч с обратным холодильником, затем отгоняли с дефлегматором хлористый метилен (пока температура не достигала 80°С). Кипятили еще 5 ч. Добавляли еще 1.29 мл (1 экв.) ацетонитрила и продолжали кипятить 8 ч. Затем при охлаждении льдом добавляли 90 мл 2 М раствора соляной кислоты. Кипятили 1 ч 15 мин с обратным холодильником, охлаждали, отделяли органический слой, водный подщелачивали 101 мл 2 М раствора гидроксида натрия до рН 3. Охлаждали до 3°С, выпавший осадок отфильтровывали, фильтрат экстрагировали 2 × 50 мл хлористого метилена, сушили над безводным сульфатом натрия и упаривали в вакууме. Остаток объединяли с полученным осадком и перекристаллизовывали из водного этанола. Получено 1.078 г вещества в виде блестящих бесцветных призм. Выход 2-амино-4-метоксиацетофенона 27%.
ЯМР 1Н в CDCl3 (500 МГц) δ (м.д.): 7.64 (1H, d, J = 9.0, C6H), 7.30 (1.5H, b, NH2), 6.23 (1H, d, J = 2.3, C3H), 6.13 (1H, dd, J = 9.0, 2.5, C5H), 3.73 (3H, s, OCH3), 2.41 (3H, s, COCH3).
Метиловый эфир 2,4-диметил-7-метоксихинолин-3-карбоновой кислоты (IX). Смешивали 0.772 г (4.67 ммоль) 2-амино-4-метоксиацетофенона, 0.605 мл (1.2 экв. = 5.6 ммоль, 0.651 г) метилацетоацетата и 0.804 г (1 экв.) п-толуолсульфокислоты. Медленно нагревали до 100°С на масляной бане и выдерживали при этой температуре полчаса. Охлаждали, добавили 14 мл воды и нейтрализовали 1.56 мл 12% раствора гидроксида натрия, перемешивали 5 мин, образовавшийся осадок отфильтровывали, промывали на фильтре 3 раза водой. Очищали флэш-хроматографией на силикагеле. Элюировали в системе 2–50% этилацетата в гексане. Вещество выходило при 25–30% этилацетата. Получено 0.664 г в виде бледно-желтой массы. Выход метилового эфира 2,4-диметил-7-метоксихинолин-3-карбоновой кислоты 64%. Rf = 0.73 (метанол–хлороформ 1 : 9). ЯМР 1Н в CDCl3 (500 МГц) δ (м.д.): 7.82 (1H, d, J = 9.2, С5Н), 7.30 (1Н, s, С8Н), 7.13 (1H, dd, J = 9.1, 2.5, C6H), 3.96 (3H, s, C7–OCH3), 3.92 (3H, s, COO–CH3), 2.65 (3H, s, C2–CH3), 2.59 (3H, s, C4–CH3). ЯМР 13C в CDCl3 (126 МГц) δ (м.д.): 170.04 (СОО), 161.36 (С7), 155.08 (С8а), 149.34 (С2), 141.90 (С4), 125.90 (С3), 125.39 (С4а), 120.81 (С5), 119.38 (С6), 107.54 (С8), 55.69 (С7–ОСН3), 52.54 (СООСН3), 24.07 (С2–СН3), 15.94 (С4–СН3).
2,4-Диметил-7-метоксихинолин-3-карбоновая кислота (X). К 0.66 г (3 ммоль) метилового эфира 2,4-диметил-7-метоксихинолин-3-карбоновой кислоты добавляли 8 мл 2 М раствора гидроксида натрия (5.5 экв., 16.5 ммоль), кипятили с обратным холодильником в течение 15 ч. Добавили 12 мл воды и 2.66 мл 6 М раствора соляной кислоты. Выдерживали ночь в холодильнике, отфильтровали осадок, промывали на фильтре водой, сушили в вакууме над Р2О5. Получено 0.436 г вещества в виде бледно-желтых кристаллов. Выход 2,4-диметил-7-метоксихинолин-3-карбоновой кислоты 70%. ЯМР 1Н в DMSO-d6 (500 МГц) δ (м.д.): 8.27 (1H, d, J = 9.4, С5Н), 7.62 (1Н, d, J = 2.3, С8Н), 7.47 (1H, dd, J = 9.3, 2.4, C6H), 3.96 (3H, s, OCH3), 2.83 (3H, s, C2–CH3), 2.79 (3H, s, C4–CH3).
N-гидроксисукцинимидные эфиры 5- и 6-карбокси изомеров Sy630 вводили в состав 5'-аминосодержащих олигонуклеотидов по следующей общей методике. К 200 мкл 5 мкМ раствора олигонуклеотида, содержащего аминогруппу на 5'‑конце в 0.1 М водном растворе гидрокарбоната натрия (рН 8) добавляли 5-ти кратный избыток N‑гидроксисукцинимидного эфира красителя (20 мкл раствора с концентрацией 10 мг/мл в ДМСО). Полученную смесь перемешивали на шейкере со скоростью 1400 об./мин при температуре 25°С в течение 12 часов. Модифицированные олигонуклеотиды очищали методом электрофореза в денатурирующем полиакриламидном геле с последующей очисткой обращенно-фазовой ВЭЖХ. Азидные производные 5- и 6-карбокси изомеров Sy630 вводили в состав 5'-алкинсодержащих олигонуклеотидов по методике, описанной в [22]. ПЦР в реальном времени проводили по следующей циклограмме: денатурация: 95°С – 5 мин, затем 49 циклов: 60°С – 40 с, 95°С – 15 с. Рабочие концентрации праймеров были 5 мкМ, зондов – 2.5 мкМ. В реакцию брали 5 единиц фермента ДНК-полимеразы SynTaq с ингибирующими активность фермента антителами (ООО “Синтол”, E-039-1000).
Список литературы
Johnson I.D. Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies. 11th ed. Life Technologies. Corp., 2010.
Valeur B., Berberan-Santos M.N. Molecular Fluorescence: Principles and Applications. 2nd ed. Wiley-VCH. Weinheim. Germany, 2013.
(a) Farzan V.M., Ulashchik E.A., Martynenko-Makaev Yu.V., Kvach M.V., Aparin I.L., Brylev V.A., Prikazchikova T.A., Maklakova S.Yu., Majouga A.G., Ustinov A.V., Shipulin G.A., Shmanai V.V., Korshun V.A., Zatsepin T.S. // Bioconjugate Chem. 2017. 28 (10). P. 2599.
Liu Jixiang, Diwu Zhenjun, Leung Wai-Yee, Lu Yixin, Patch B., Haugland R.P. // Tetrahedron Lett. 2003. 44. P. 4355.
(a) Elderfield R.C. Heterocyclic Compounds. New York. N.Y. 1961. 45. P. 60.
Sugasawa Tsutomu, Toyoda Tatsuo, Adachi Makoto, Sasakura Kazuyuki // J. Am. Chem. Soc. 1978. 100. P. 4842.
Friedlander P. // Ber. Dtsch. Chem. Ges. 1882. 15. P. 2572–2575.
Wang Guan-Wu, Jia Cheng-Sheng, Dong Ya-Wei // Tetrahedron Lett. 2006. 47. P. 1059.
Jia Cheng-Sheng, Ze Zhang, Tu Shu-Jiang, Wang Guan-Wu // Org. Biomol. Chem. 2006. 4. P. 104.
Selvam N.P., Saravanan C., Muralidharan D., Perumal P.T. // J. Heterocyclic Chem. 2006. 43. P. 1379.
Narasimhulu M., Reddy T.S., Mahesh K.C., Prabhakar P., Rao Ch.B., Venkateswarlu Y. // J. Mol. Catal. A: Chem. 2007. 266. P. 114.
De S.K., Gibbs R.A. // Tetrahedron Lett. 2005. 46. P. 1647.
Wu Jie, Zhang Liang, Diao Tian-Ning // Synlett. 2005. P. 2653.
Kumar S., Saini A., Sandhu J.S. // Synth. Commun. 2007. 37. P. 4071.
Bose D.S., Kumar R.K. // Tetrahedron Lett. 2006. 47. P. 813.
Varala R., Enugala R., Adapa S.R. // Synthesis. 2006. P. 3825.
Arumugam P., Karthikeyan G., Atchudan R., Muralidharan D., Perumal P.T. // Chem. Lett. 2005. 34. P. 314.
Zhang L., Wu J. // Adv. Synth. Catal. 2007. 349. P. 1047.
Zolfigol M.A. Salehi P., Ghaderi A., Shiri M. // Catal. Commun. 2007. 8. P. 1214.
Wu Jie, Xia Hong-Guang, Gao Ke // Org. Biomol. Chem. 2006. 4. P. 126.
Roberts E., Turner E.E. // J. Chem. Soc. 1927. P. 1832.
Натыров А.Н., Власова Н.А., Матвиенко И.В., Волков Е.М., Байрамов В.М., Курочкин В.Е., Алексеев Я.И. // Биоорг. хим. 2018. 44. 5. С. 570–580. [Natyrov A.N., Vlasova N.A., Matvienko I.V., Volkov E.M., Bajramov V.M., Kurochkin V.E., Alekseev Y.I. // Rus. J. Bioorg. Chem. 2018. 44. 5. P. 562–571.]
Алексеев Я.И., Белов Ю.В., Варламов Д.А., Коновалов С.В., Курочкин В.Е., Маракушин Н.Ф., Петров А.И., Петряков А.О., Румянцев Д.А., Скоблилов Е.Ю., Соколов В.Н., Фесенко В.А., Чернышев А.В. // Научное приборостроение. 2006. 16. 3. С. 132–136.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия