

Биоорганическая химия, 2020, T. 46, № 5, стр. 486-504
Физико-химические процессы – вероятные родоначальники жизни
С. П. Коваленко *
Институт микробиологии НАН Беларуси
220141 Минск, ул. акад. В.Ф. Купревича, 2, Республика Беларусь
* E-mail: spkovalenko2008@mail.ru
Поступила в редакцию 31.12.2019
После доработки 17.02.2020
Принята к публикации 20.02.2020
Аннотация
Проанализированы материальные и энергетические ресурсы, способные обеспечить абиотические синтезы веществ, из которых сформировались предбиологические структуры. Показано, что основными исходными веществами могли быть одноуглеродные соединения, в том числе формальдегид и цианистый водород. В качестве возможных энергетических ресурсов рассматриваются вулканическая активность и фотохимическое действие солнечных УФ-лучей. В результате реакций между исходными веществами и фоторедукции их продуктов в серосодержащих водоемах синтезировались аминокислоты, липиды, углеводы и нуклеотиды. Циклы увлажнения/высушивания водоемов благоприятствовали полимеризации мономеров (аминокислот и нуклеотидов). Среди возникших олигонуклеотидов могли оказаться рибозимы. Некоторые из них катализировали процессы удлинения олигонуклеотидов. Удлиненные РНК-подобные структуры приобретали новые функции, в том числе способность катализировать репликацию РНК. Слои из липидоподобных пептидов и жирных кислот исполняли роль простейших мембран, образуя протоклетки. Детали описанных процессов обсуждаются.
ВВЕДЕНИЕ
Человечество всегда интересовалось, как появились на Земле живые существа, создавая множество мифов. Первая научная теория происхождения жизни предложена русским биохимиком А.И. Опариным [1] еще в первой четверти прошлого века. Согласно ей из неорганических веществ в “первичном бульоне” синтезировались белковые молекулы, сформировавшие фазово-обособленные коллоидные структуры (коацерваты), которые эволюционировали под действием естественного отбора, в результате чего образовались живые клетки. Однако в то время не существовало достаточной экспериментальной базы для строго научных исследований происхождения жизни на Земле.
Наш век ознаменовался бурной интенсификацией исследований этой проблемы. Она была подготовлена величайшими достижениями второй половины двадцатого века: открытием структуры и функций ДНК и РНК, расшифровкой генетического кода, прочтением геномов многих живых существ, развитием компьютерной техники, созданием огромных баз данных о строении белков и нуклеиновых кислот филогенетически различных организмов, миниатюризацией исследовательских процессов, открытием ферментативных свойств РНК, разработкой методов искусственной эволюции нуклеиновых кислот и селекции активных рибозимов, а также большим прогрессом в области геохимии, геофизики, космохимии и других смежных наук.
В результате этих успехов идеи Ч. Дарвина относительно “маленького теплого пруда” и А.И. Опарина об “обособлении из первичного бульона предбиологических структур” стали наполняться конкретным содержанием на базе надежных экспериментальных данных.
УФ-излучение Солнца инициировало в атмосфере и наземных водоемах фотохимические процессы, в результате которых неорганические соединения стали источниками “сырья” для синтезов большого числа органических веществ. Из них образовались аминокислоты, сахара, липиды и нуклеотиды.
Расчеты показывают достаточность суммы магматических, атмосферных и космических ресурсов для поддержания непрерывного абиотического синтеза важнейших биомолекул и сборки функциональных структур.
На русском языке проблема происхождения жизни освещается недостаточно. Со времени издания книг К.Ю. Еськова [2], Е.В. Кунина [3] и М.А. Никитина [4] прошли годы, и за это время накопилось много новых важных результатов. Например, в лаборатории Дж. Сазерленда создана система, в которой на основе всего четырех исходных веществ в результате окислительно-восстановительных процессов, инициируемых УФ-облучением, могут синтезироваться главные компоненты живых организмов [5–7]. Группе ученых во главе с Ф. Холлиджером удалось осуществить самокопирование всей нуклеотидной последовательности рибозима (правда, пока только по частям) [8] и тем самым продемонстрировать принципиальную возможность рибозимного синтеза самокопирующейся молекулы РНК. В лаборатории Дж. Шостака добились неферментативного матричного синтеза цепочек ДНК длиной до 25 нуклеотидов в модельных протоклетках [9]. Учитывая, что многие активные рибозимы имеют примерно такую же или даже меньшую длину, можно считать, что проблема их абиотического копирования решена. Дж. Шостак [10] и Р. Кришнамурти [11, 12] практически одновременно экспериментально доказали возможность формирования однородных по углеводным компонентам молекул РНК и ДНК из образовавшейся абиогенно смеси нуклеотидов с различными углеводными компонентами их молекул.
Данный обзор представляет собой попытку восполнить вышеуказанный пробел в публикациях на русском языке по проблеме происхождения жизни, а также выделить из огромного научного материала, накопленного при ее исследовании, важнейшие факты и связать их в правдоподобную систему процессов, возможно, способствовавших возникновению предбиологических структур, обладающих потенциалом для превращения в живые организмы.
РЕСУРСЫ, НЕОБХОДИМЫЕ ДЛЯ ЗАРОЖДЕНИЯ ЖИЗНИ
Для возникновения сложной системы, которую представляют собой живые существа, необходимы, как минимум, три условия:
1. Наличие разнообразных минеральных и органических веществ и их постоянный обильный приток.
2. Жидкость, в которой они могли бы растворяться, стекать в одно место и там накапливаться. Это условие одновременно накладывает ограничения на температурный диапазон и размеры участков их накопления.
3. Источники энергии для их химических преобразований.
Согласно современным геологическим и космологическим представлениям (см. [2, 13–16] и ссылки в них), Земля сформировалась 4.56 млрд лет назад из газопылевого облака. Химический состав таких облаков достаточно изучен. Водорода в них на порядок больше, чем гелия, и в 1000 раз больше, чем кислорода. Кислорода явно не хватает на окисление всех химических элементов, поскольку атомарное соотношение O : C : N : Si : Mg : : Fe : S (самых распространенных элементов, исключая водород и инертные газы) равняется 100 : 60 : 20 : 10 : 10 : 10 : 5 [17]. Один только углерод мог бы связать весь кислород в CO2, не говоря уж о многократно превосходящем его водороде. При формировании планеты химические элементы вступали в жесткую конкуренцию за кислород. Поэтому он оказался полностью связанным в окислах, вероятно, с низшей степенью окисления, а многие металлы остались неокисленными. Образовались также простейшие соединения: гидриды, карбиды, нитриды, сульфиды, фосфиды, галогениды и др.
Первые полмиллиарда лет (катархейский эон) практически не оставили надежных данных, позволяющих оценить температурный режим, наличие и состав атмосферы, гидросферы, соотношение поверхностей суши и океана и т.п. Многие геологи считают, что Земля изначально оказалась обделенной водой и другими летучими веществами из-за “выдувания” их солнечным ветром, не имела гидросферы и атмосферы и лишь “поздняя тяжелая бомбардировка” метеоритами, кометами и астероидами (о времени и интенсивности которой судят по ее следам, сохранившимся на Луне) восполнила этот дефицит [13–15]. Хотя подсчитанная столь ничтожная доля воды в общей массе Земли (0.023%) [14] вполне могла бы быть ее собственной. Однако для нашей темы важен конечный результат: так или иначе, Земля получила ресурсы, необходимые для зарождения жизни.
В силу ряда причин (в том числе – столкновения с крупным небесным телом [16, 18], породившего Луну) Земля пережила период полного расплавления своей поверхности, превращения ее в “океан магмы” с температурой выше 2000°С. Атмосфера образовалась за счет выделения из этого океана газов (а при этой температуре газами становились многие твердые в привычных для нас условиях вещества, например, ZnS, ZnCl2 и FeCl2 с т. кип. ниже 1200°С). Неокисленные металлы (железо, никель и др.) как более тяжелые перемещались к центру, образовав ядро планеты. Одновременно всплывали легкие силикатные породы, содержащие алюминий, магний и двухвалентное железо, а также несовместимые с магмой KREEP-базальты (богатые K – калием, REE – редкоземельными элементами, P – фосфором) [2, 14, 16]. Они сформировали земную кору, обладающую сильными восстановительными свойствами. Именно из таких пород, сохранившихся до сих пор на Луне благодаря отсутствию там воды и атмосферы, состоит ее поверхность, сформированная, как считают [16, 18], из выбитых с Земли материалов.
После остывания земной коры на ней из водяных паров конденсировалась вода вместе с растворенными в ней продуктами дегазации магмы и атмосферных фотохимических процессов. Контакт жидкой воды с породами земной коры положил начало процессам эрозии. Наиболее важным в создании ресурсов для абиотических синтезов был процесс окисления минералов ортопироксена и оливина до трехвалентного состояния железа и одновременного восстановления воды до водорода ([16] и ссылки в ней).
Водород восстанавливал растворенную в воде углекислоту до муравьиной кислоты, формальдегида, метанола и далее до метана. Водный раствор подщелачивался при этом до pH 9–11 [16]. Этот процесс называется серпентинизацией и рассматривается как один из важнейших поставщиков органических веществ.
В настоящее время такой процесс осуществляется только при излияниях вулканической лавы в воду и в местах разломов тектонических плит на океанском дне, например в регионе Lost City в Атлантическом океане. Там и до сих пор наблюдаются сильные потоки водорода, метана и других газов, что послужило основанием для гипотезы о возникновении жизни в таких гидротермальных источниках. Рассмотрение этой гипотезы не является целью данного обзора.
Ведь на молодой Земле серпентинизация происходила повсеместно, и все водоемы первоначально обладали щелочными свойствами. Растворение в них летучих окислов (углерода, серы, фосфора и др.) снижало значение pH до различных уровней, так что по этому показателю ассортимент водоемов был широким. Геохимики проводят аналогию между древними озерами и современной гидротермальной системой вулкана Kverkfjőll (Исландия), где расположено множество водоемов с различными величинами рН (2–7.5) и температуры (0–95°С) [19]. Так возникло большое разнообразие стартовых условий для начала абиотических синтезов на поверхности древней Земли.
Интересно, что процесс серпентинизации приводит также к образованию естественных сплавов железа и никеля, обладающих каталитическими свойствами [16]. Это доказано как обнаружением таких сплавов (таенита, камацита и аваруита) в местах, где происходит серпентинизация, так и в лабораторных имитациях этого процесса [20]:
Образовавшиеся железоникелевые сплавы катализируют восстановление водородом CO2, а другой продукт серпентинизации, магнетит (Fe3O4) – восстановление N2 до NН3 [16].
Процесс дегазации магмы продолжается до сих пор (по-видимому, с гораздо меньшей интенсивностью), проявляясь в вулканической деятельности. Сжатые огромным давлением горячие газы проникают через капиллярные поры и микротрещины в грунтовые воды. Накапливаясь там и нагревая их до кипения, они создают фумаролы и гейзеры, т.е. выбросы пара и жидкой воды вместе с растворенными веществами на земную поверхность, образуя неглубокие водоемы. П. Риммер и О. Шортл приводят химический состав этих газов [19]. Он включает как неорганические (SO2, H2S, CO, HCN), так и органические вещества: метан, ацетилен, диацетилен, цианоацетилен, акрилонитрил, формальдегид и др.
Перечисленные вещества образуются в магме в результате сложных химических процессов с участием свободных радикалов, подробно описанных авторами [19]. Обращает на себя внимание высокое содержание органических веществ, ацетилена и диацетилена, суммарно превышающее даже содержание SO2 или CO.
Авторы с сожалением отмечают, что в описанных ими процессах образуется мало цианамида, необходимого для синтеза нуклеиновых оснований. Его концентрацию в водоеме они оценивают ниже 10 мкМ, хотя рассчитанные ими концентрации других веществ довольно высоки, например, для цианоацетилена и HCN около 0.1 М, акрилонитрила и ацетилена – миллимолярные [19]. В опытах, имитирующих абиотические процессы, используются и значительно меньшие концентрации, например, 10–4 М [21] при синтезе пептидов и даже микромолярные при фосфорилировании альдолов амидотрифосфатом ([22] и ссылки в ней). Так что еще задолго до насыщения гидротермального источника извергаемые вещества могли сразу реагировать между собой и накапливаться в форме более устойчивых продуктов.
Например, HCN с солями железа образует гексацианоферраты, с серосодержащими газами роданиды, с формальдегидом гликолонитрил, а в присутствии NH3 – предшественник глицина 2‑аминоацетонитрил (по реакции Штрекера). При медленном гидролизе HCN образует еще более медленно гидролизуемый формамид (полупериод его гидролиза равен 199 лет при 25°С и pH 7 [23]). Цианоацетилен HC≡C–CN наполовину гидролизуется через 10 дней при 30°С [23] до более стабильного к гидролизу цианоацетальдегида, который в результате реакции Штрекера превращается в предшественника аспарагиновой кислоты, ее нитрил. Ацетилен и диацетилен вступают во взаимодействие с водой, хотя и не так легко, как цианоацетилен. Ацетилен в присутствии ионов ртути превращается в предшественника жирных кислот – ацетальдегид (реакция Кучерова). Недавно были открыты более распространенные катализаторы этой реакции – соли цинка и меди [24].
Скорости реакций, конечно, были невелики, но они, как и концентрации веществ в водоеме, многократно возрастали при испарении воды. Скорее всего, полного высыхания не происходило, так как в водоеме всегда находилось небольшое количество формамида. Наверняка оставался хотя бы мультимикронный слой раствора реагентов в этом полярном высококипящем растворителе, и все реакции продолжались в нем.
Гидротермальные источники (фумаролы и гейзеры) выносят на поверхность, кроме газов, множество летучих веществ, захватываемых водяным паром или горячей водой, – оксиды, сульфиды и/или галогениды различных элементов (K, Na, Zn, Fe, Mn, B, P, Si и др.). А.Я. Мулкиджанян и др. [25, 26] установили минеральный состав конденсатов паров гидротермального поля на Камчатке. Важнейшие показатели: преобладание ионов калия над ионами натрия, высокое содержание Zn, Mn, P по сравнению с морской водой. Этот факт позволил авторам высказать идею, что в таких местах могли возникнуть первые клетки, минеральный состав которых наиболее похож на состав конденсатов паров фумарол и гейзеров.
Еще ранее автор гипотезы “цинкового мира” А.Я. Мулкиджанян [27] отмечал важную роль сульфида цинка для первоначальных абиотических синтезов. Этот минерал довольно распространен на Земле и обладает уникальными каталитическими способностями. При УФ-облучении в водной среде он восстанавливает CO2 до муравьиной кислоты, азот – до аммиака. Он способен накапливать энергию УФ-лучей, а затем отдавать ее в темноте (фосфоресцировать). Кроме того, он, как и другие минералы-полупроводники, перехватывает энергию возбужденного состояния, вызванного облучением, с молекул адсорбированных веществ за пикосекунды, в то время как фоторазрушение аденозинмонофосфата (AMP) происходит лишь через 20 микросекунд. Таким образом, ZnS мог служить надежным убежищем от УФ-лучей для адсорбированных нуклеотидов и РНК (см. [27] и ссылки в ней).
Помимо описанных локальных источников материальных ресурсов наземные водоемы снабжались ими за счет глобальных процессов, происходящих в атмосфере. Атмосфера практически не содержала кислорода, а потому над Землей отсутствовал озоновый слой, задерживающий УФ-лучи. Поэтому в атмосфере и на поверхности Земли интенсивно происходили фотохимические реакции. Атмосфера состояла, в основном, из азота, водяного пара и углекислого газа с примесью выделяемых из недр CO, CH4, H2S, COS, SO2 и других газов. Энергия УФ-лучей, грозовых разрядов и частых падений метеоритов порождала радикальные процессы, в результате которых образуются органические вещества (формальдегид и в меньшем количестве гликолевый альдегид) и продукты азотфиксации (HCN и окислы азота, преимущественно NO). Продуктивность этих процессов представлена в табл. 1.
Таблица 1.
Потоки продуктов дегазации магмы и атмосферных фотохимических процессов*
| Вещество | Глобальные потоки | Суточные потоки на 1 км2 | Ссылка | |||
|---|---|---|---|---|---|---|
| кг/год | моль/год | см–2 с–1 | г | моль | ||
| НСНО | 3 × 109 | 1011 | – | 16.1 | 0.537 | [28] |
| HCN | 7.21 × 107 | 2.67 × 109 | 107 | 0.39 | 0.014 | [29] |
| NO | 4.2 × 109 | 1.4 × 1011 | – | 22.6 | 0.752 | [30] |
| NН3 из NO | 6.12 × 108 | 3.6 × 1010 | – | 3.3 | 0.193 | [30]** |
| NН3 из магмы | 1010 | 7.14 × 1011 | – | 53.7 | 3.837 | Рассчитано по [23] |
| SО2 | >1.71 × 1010 | >2.67 × 1011 | (1–3) × 109 | >91.9 | >1.435 | [31] |
| Н2S | >2.82 × 109 | >8.28 × 1010 | (3.1–77) × 108 | >15.1 | >0.445 | [31] |
* Жирным шрифтом выделены данные авторов, остальные цифры рассчитаны нами по нижнему уровню. ** Рассчитано по данным авторов о конечной производительности суммы процессов, приводящих к преобразованию NO в аммиак. В океане, как считали авторы, за их счет создавалась очень низкая концентрация NН3 (3.6–70 мкМ). Мы же относим эти данные к небольшим наземным водоемам, где происходили такие же процессы.
Вместе с дождями органические соединения выпадали на поверхность и стекали в водоемы, в том числе те, о которых шла речь выше. Окислы азота восстанавливались соединениями двухвалентного железа до NH3, что, по мнению Д. Саммерса и С. Чанга [30], было главным источником аммиака. Поскольку авторы рассматривали именно процессы образования и накопления аммиака в океане, их вывод был довольно пессимистичен в плане обеспечения им всех необходимых абиотических синтезов: рассчитанная концентрация NH3 оказалась слишком низкой. Но в малых водоемах, где эти процессы также происходили, ситуация была более благоприятной.
Н. Китадаи и С. Маруяма (см. [23] и ссылки в ней) высказали предположение, что азот дегазировался из магмы преимущественно в виде аммиака в первые полмиллиарда лет существования Земли. И его годовой поток в атмосферу Земли, согласно расчетам, составлял 1013 г азота. Аммиак в больших количествах мог образовываться также при фоторедукции нитрилов, о чем сказано ниже.
Источники фосфора были разнообразны и довольно обильны, но “проблема фосфора” заключается в плохой растворимости солей основной кислотной его формы – ортофосфата – со многими металлами. Фосфиты примерно в 1000 раз более растворимы и более реактивны [23]. Их главный источник – гидролиз фосфидов, вроде шрайберзита (Fe3P). В присутствии аммиака при этом гидролизе образуются водорастворимые амиды и диамиды фосфорной и фосфористой кислот [32]. Авторы оценивают продукцию этих веществ в 1015–1019 моль за первый миллиард лет существования нашей планеты. Более точная оценка, к сожалению, невозможна из-за неполноты данных о содержании фосфидов на древней Земле.
Табл. 1 показывает, что только за счет фотохимических процессов в атмосфере за 100 тысяч лет (геологически небольшой срок) накопилось бы столько органических веществ, сколько содержит вся современная биосфера (6 × 1014 кг). Но абиотические синтезы биомолекул не ждали этого накопления, они начались еще с образования атмосферы.
Космос тоже обильно поставлял различные вещества в составе метеоритов, особенно во время “поздней тяжелой бомбардировки” (4 млрд лет назад или ранее). Общее количество органических веществ, полученных нашей планетой извне в тот период (за ~100 млн лет), составляет согласно расчетам 1016−1018 кг [33]. Это существенный вклад, несмотря на то, что большая часть космической органики нерастворима в воде и вряд ли участвовала в абиотических процессах. В метеоритах обнаружено более 170 различных органических веществ, в том числе все азотистые основания, необходимые для РНК и ДНК, 12 аминокислот, входящих в молекулы белков, и многие жирные кислоты [23]. Кроме того, некоторые исследователи [34] считают существенными каталитические активности метеоритов и выделяемую ими при падении энергию, инициирующую химические процессы в атмосфере.
Хотя дары Космоса могли в некоторых процессах облегчить формирование биоструктур, нельзя исключать, что такие же ресурсы существовали на молодой Земле. Например, важный источник фосфора шрайберзит, долго считавшийся исключительно “космическим пришельцем”, российские и израильские геологи недавно обнаружили в древних рудных минералах, доказав его земное происхождение [35]. Следует также учесть, что, несмотря на разнообразие веществ, поставленных метеоритами, они не могли выстроить непрерывную слаженную цепь абиотических синтезов всех необходимых для жизни компонентов. Эту последовательность процессов обеспечили ресурсы нашей родной планеты.
“АБИОТИЧЕСКИЙ ПИТОМНИК”
Большая заслуга лаборатории Дж. Сазерленда (Великобритания) не только в том, что ее сотрудники нашли “обходной” путь синтеза пиримидиновых нуклеотидов и исследовали пути возможных абиотических синтезов аминокислот, углеводов, липидов и нуклеотидов. Они еще создали весьма удачную модель, имитирующую первичный “метаболизм”, обеспечивающий непрерывную поставку практически всех “строительных блоков” для синтеза будущих биологических структур. Эта модель представляет собой систему, в которой действует окислительно-восстановительный цикл на основе катализатора – металла с переменной валентностью (железа в составе комплексного иона, гексацианоферрата), восстановителя – сульфитных ионов (в более ранних работах H2S) и источника энергии – УФ-излучения, инициирующего работу цикла [5].
Как уже говорилось выше, основными продуктами атмосферных фотохимических процессов являются формальдегид и HCN, реагирующие затем между собой с образованием гликолонитрила. Он и стал исходным веществом для последующих превращений, начинающихся с его фоторедукции до гликолевого альдегида с отщеплением аммиака.
Первоначально возникшее щелочное озеро при выпадении дождей и/или проникновении магматических газов насыщалось HCN, образующим с ионами Fe2+ комплексные ионы [Fe(CN)6]4–. Одновременно происходило насыщение его сернистым газом и, возможно, фосфорной или фосфористой кислотой (за счет гидролиза шрайберзита и выброса вулканами триметафосфата), что приводило к снижению pH до нейтрального уровня и даже слабого подкисления. Формальдегид образовывал аддукт с бисульфитами в результате реакции [5]:
Именно эту ситуацию имитировали в лаборатории Сазерленда [5, 6]. При pH 6.5 реакционную смесь (NaНSO3, K4[Fe(CN)6], НO–СН2–CN и Na2HPO4) облучали ртутной лампой. Первыми продуктами фоторедукции стали гликолевый альдегид (за счет восстановления нитрильной группы до альдегидной) и аммиак, который тут же вступал с гликолонитрилом в реакцию Штрекера с образованием нитрила глицина. Гликолевый альдегид реагировал с HCN, превращаясь в глицеронитрил. Образуется также немного (от 4 до 8%) ацетальдегида (за счет восстановления не только нитрильной, но и гидроксильной группы гликолонитрила). Часть ацетальдегида тоже вступила в реакцию Штрекера, образовав нитрил аланина [5, 6]. А часть аммиака и HCN атаковала гликолевый альдегид, превратив его в нитрил серина. Так начал функционировать абиотический питомник, изображенный на рис. 1.
Рис. 1.
Первоначальные абиотические синтезы. В розовом поле изображены синтезы аминокислот, в зеленом – нуклеотидов, в сером – углеводов, в желтом – липидов. Голубое поле показывает вещества, образующиеся в атмосфере, коричневое – вещества из недр Земли. Процессы фоторедукции отмечены значком hν.
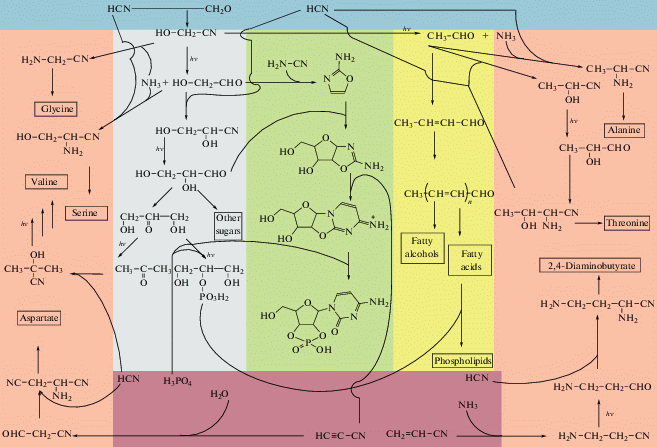
С новыми порциями формальдегида и HCN описанный процесс возобновлялся. Но так как уже возникли первые продукты фоторедукции, они тоже становились исходными веществами для дальнейших реакций, показанных на рис. 1.
Следует обратить внимание, что с каждым циклом количество альдегидных групп и выделившегося аммиака постоянно растет. Альдегиды не убывают в реакциях с HCN: превратившись в гидроксинитрилы, они тут же регенерируются после фоторедукции. Предел этого нарастания определяется количеством HCN, поступающим из недр и атмосферы, точнее – той частью HCN, которая расходуется на реакцию с альдегидами. Ведь часть этого вещества расходуется на образование роданидов и гексацианоферратов. Зато альдегиды могут возникать при гидратации ацетиленов и фоторедукции других нитрилов (цианоацетилена и акрилонитрила). Так что указанные на рис. 1 процессы лимитирует лишь HCN.
Что касается аммиака, то его количество может нарастать постоянно. Все аминонитрилы, предшественники аминокислот, хоть и медленно (в течение нескольких лет) гидролизуются до аминокислот, высвобождая NH3. Этот гидролиз может сильно ускорить каталитическое действие формальдегида [36], а также реакции аминонитрилов с тиолами [21, 37] и реакция Бюхерера-Бергса, о которой будет сказано далее. А вот фоторедукционный процесс, описанный здесь, α‑аминонитрилы не восстанавливает в отличие от β-аминонитрилов [38] и α-гидроксинитрилов [5]. Кстати, способность нитрильных групп β-аминонитрилов подвергаться фоторедукции открывает короткий путь к абиотическому синтезу диаминокислот. Так, из акрилонитрила может образоваться 2,4-диаминомасляная кислота (см. рис. 1). Она могла быть предшественницей других, более сложных положительно заряженных аминокислот в первичном генетическом коде.
При фоторедукции pH реакционной смеси повышается за счет высвобождения аммиака, который, хоть и расходуется на другие продукты, но образует аминонитрилы, обладающие тоже основными свойствами. Впрочем, это повышение временное, так как происходит постоянный приток кислотных газов, и pH в абиотическом питомнике постоянно колеблется между почти нейтральным и слабощелочным (по-видимому, до значений 9–10). В периоды подщелачивания создаются благоприятные условия для гомоальдолизации ацетальдегида [7]. Основным ее продуктом является кротоновый альдегид. В постепенно убывающих концентрациях образуются гексадиеналь, октатриеналь и еще более длинные ненасыщенные альдегиды [7]. При восстановлении их (никелевым катализатором, в качестве которого мог бы выступить упомянутый выше аваруит) образуется смесь насыщенных жирных спиртов, альдегидов и кислот [7].
Продукт фоторедукции глицеронитрила глицеральдегид изомеризуется в более устойчивую форму трехуглеродного сахара – дигидроксиацетон. Его фоторедукция приводит к двум продуктам – ацетону и глицерину [38]. Ацетон вовлекается в абиотический синтез аминокислот (давая валин), глицерин – в синтез липидов [38]. Глицерин в растворе фосфатов и мочевины превращается в 1,2-циклофосфат, гидролиз которого дает смесь глицерин-1-фосфата и глицерин-2-фосфата. Эти же эфиры могли синтезироваться и другими путями – при взаимодействии глицерина со шрайберзитом [39] или в формамидном растворе [40] (после испарения воды). Реакция фосфатных эфиров глицерина с жирными кислотами приводит к образованию фосфолипидов (см. [7] и ссылки в ней).
Хотя процессы абиотических синтезов всех нуклеиновых оснований и рибозы в достаточной степени изучены, они малопригодны для возникновения РНК из-за проблем в их связывании между собой (см. обзоры [12, 23]). Дж. Сазерленд и его коллеги решили эти проблемы, открыв “обходной” путь синтеза нуклеотидов [41]. Ключевое звено в цепи этих процессов – реакция гликолевого альдегида с цианамидом. Она образует 2-аминооксазол, который при реакции с глицеральдегидом дает арабино-аминооксазолин – продукт, состоящий из арабинозного пятичленного кольца, сопряженного с другим кольцом, предшественником цитозина (см. рис. 1, зеленое поле). Если гликолевый альдегид находится в избытке (что вполне возможно из-за ограниченности количества цианамида), он реагирует с 2-аминооксазолом (вместо глицеринового альдегида), давая начало синтезу тетрозонуклеотидов [42], возможных предшественников трео-НК (ТНК).
Реакция с цианоацетиленом завершает построение цитозинового кольца. Гидролиз образовавшегося продукта в присутствии фосфатов и мочевины приводит к преобразованию арабинозного кольца в рибозное, благодаря замыканию другого пятичленного цикла (рис. 1, зеленое поле). Продукт – 2',3'-циклофосфат цитидина – является активированной формой натурального нуклеотида. УФ-облучение частично переводит его в 2',3'-циклофосфат уридина. Таким образом, была решена проблема возможности присоединения рибозы к пиримидинам и проблема его региоспецифичности.
Однако эта изящная схема столкнулась с рядом серьезных трудностей:
1) недостаточность ресурсов цианамида на ранней Земле,
2) необходимость определенного порядка реакций, не допускающего взаимодействия цианамида и глицеринового альдегида с образованием побочных продуктов,
3) невозможность получения этим же путем пуриновых нуклеотидов.
Дальнейшие усилия были направлены на поиск путей преодоления этих трудностей.
Одной из таких попыток была концепция “химии потоков”. Согласно ей многие важные этапы добиологической химии могли осуществляться не в стационарных водоемах вроде упомянутого питомника, а в местах встречи потоков различного химического состава [5]. При этом большое значение приобретает порядок, в котором встречаются разные потоки, если их несколько. Это – уязвимое место концепции, так как здесь просматривается аналогия с экспериментами, имитирующими отдельные этапы абиотических процессов в строго определенном порядке. Однако при небольшом числе потоков вероятность “нужного” порядка слияния потоков еще достаточно высока. Поскольку гидротермальных регионов множество, то в каких-нибудь из них могла сложиться благоприятная ситуация.
Авторы попытались преодолеть первую и вторую трудность чисто гипотетически. Экспериментально был установлен только факт, что 2‑аминотиазол способен избирательно осаждать в разное время гликолевый и глицериновый альдегиды в виде аминалей [43]. Текущий из питомника поток (с 2-аминотиазолом) через несколько часов оставляет после себя сначала участок русла с осадком аминаля гликолевого альдегида, а ниже по течению – осадок аминаля глицеральдегида.
Цианамид действительно мог возникнуть в больших количествах в местах образования гексацианоферратов, где преобладающие минералы содержали кальций. Гексацианоферраты кальция при температурах 700–1000°С разлагаются с образованием кальцийцианамида.
Дальнейшие рассуждения на основе “химии потоков” содержат слишком много допущений. Предполагается, что осажденный в каком-то месте гликолевый альдегид должен прибыть с другим потоком на место залегания кальцийцианамида, причем в виде аддукта с бисульфитом. Количество бисульфита не должно быть выше эквивалентного количества CaCN2, иначе лишний бисульфит встроится в цикл образующегося 2-аминооксазола и помешает его дальнейшим превращениям. Если это условие выполнено, нерастворимый сульфит кальция освободит цианамид, который прореагирует с гликолевым альдегидом. Затем поток должен встретить осадок глицеральдегида. И так далее по рис. 1. Беда только в том, что для синтеза осадителя нужен цианамид, которого не должно быть в первом потоке.
Третью трудность (а, возможно, и все остальные) М. Поунер и сотрудники [44] пытались преодолеть, используя вместо дефицитного цианамида тиоциановую кислоту. Роданиды накопились в питомнике, стоит только довести pH до 4, и начнется их реакция с гликолевым альдегидом, образуя оксазол-2-тион (авторы называют его 2‑тиооксазолом). Он так же легко соединяется с глицеральдегидом, как и его 2-аминоаналог. Вряд ли необходим поток из нейтрального питомника в водоем с низким значением pH. Ведь возможны периодические интенсификации выбросов вулканических кислотных газов, подкисляющих водоем до pH 4. Тем более что для сдвига pH от 7 до 4 требуется всего 10–4 М концентрация кислоты.
Дальнейшие превращения несколько отличаются от вышеописанных, но суть их та же – формирование нуклеинового основания, в данном случае пуринового (в две стадии: сначала пятичленного, затем шестичленного цикла). На этом пути есть развилка, благодаря которой часть продукта сворачивает на путь синтеза пиримидиновых нуклеозидов. Конечный этап обоих путей идентичен – гидролиз в присутствии фосфатов и мочевины с образованием 2',3'-циклофосфатов нуклеозидов. К сожалению, образуются не канонические пуриновые нуклеотиды, а их 8-оксопроизводные. Но в литературе такие производные часто упоминались как вполне приемлемые заменители канонических, пока Дж. Шостак [45] не доказал их непригодность для участия в неферментативной репликации РНК. Кроме того, среди продуктов имеются нуклеотиды арабинозного, ксилозного и ликсозного ряда, хотя рибозные и арабинозные нуклеотиды преобладают.
Другой путь конструирования пуриновых нуклеозидов (FaPy-путь) предложила группа, руководимая Т. Кереллом [46]. На основании литературных данных они решили, что три- и тетрааминопиримидины могли быть синтезированы в абиотических условиях. Авторы теоретически обосновали и экспериментально подтвердили, что в определенных условиях лишь аминогруппа в позиции 5 этих пиримидинов может быть ацилирована муравьиной кислотой. Полученные формиламидопиримидины при рибозилировании любой аминогруппы, соседней с формилированной аминогруппой, могли дать один и тот же продукт (из-за симметричности молекулы). Так была решена проблема региоспецифичности рибозилирования. Последующее замыкание имидазольного цикла произвело пуриновые нуклеозиды. Однако образовалась смесь различных стереоизомеров. Пиранозная форма рибозы преобладала над канонической фуранозной. И хотя β-аномеры возникали чаще, чем α-аномеры, выход канонического аденозина никогда не превышал 20%. Зато этот путь привел к каноническим пуриновым нуклеозидам.
В следующей статье [47] авторы развили свою гипотезу на основе концепции “химии потоков” в сочетании с циклами увлажнения/высушивания. Они изобразили 4 потока, сливающиеся в определенном порядке и затем впадающие в озеро. В результате последовательного взаимодействия всех компонентов образуются пуриновые нуклеозиды. Циклы увлажнения/высушивания обеспечивают также разделение и очистку продуктов.
Все-таки М. Поунеру позже удалось найти путь синтеза канонических пуриновых оснований в составе нуклеозидов [48] благодаря включению в реакционную смесь сероводорода, который, несомненно, присутствовал в предбиотических водоемах. Вместо 8-оксопуриновых нуклеозидов он получил 8-меркаптопуриновые, преимущественно арабинозные нуклеозиды. УФ-облучение удаляло серу, образуя канонические гетероциклы. Так работы [44] и [48] решили проблему абиотического синтеза без потребности в дефицитном цианамиде полного набора нуклеозидов. Но – арабинозного ряда. Вопрос, как возникли канонические пуриновые рибонуклеотиды, остается пока открытым.
М. Поунер, тем не менее, считает результаты своих экспериментов большим успехом, поскольку арабино-НК (АНК) считается одной из возможных предбиотических информационных полимеров. Авторы отмечают ее большую, чем у РНК, устойчивость к гидролизу [49], способность образовывать подобные рибозимам структуры [50] и дуплексы с РНК [49, 51], транскрибироваться с ДНК-матриц [52], служить матрицей для обратной транскрипции ДНК [52].
Ксенонуклеиновые кислоты, у которых и гетероциклические основания, и соединяющие их “мостики” отличаются от природных, исследуются давно. Интерес к ним обусловлен трудностями, обнаруженными при попытках лабораторного осуществления абиотического синтеза и самокопирования цепочек РНК. Целью является поиск более легкого (а, значит, более правдоподобного) сценария спонтанного возникновения самокопирующегося полимера. И такие полимеры вполне могли образоваться: ведь в опытах, наряду с производными рибозы, получаются производные других пентоз и даже тетроз [42], которые тоже могут превратиться в соответствующие нуклеотиды. Как будет рассмотрено далее, из смеси нуклеотидов различных углеводных рядов сначала синтезируются гетерогенные по сахару олигомеры, а затем, после серии репликаций, формируются гомогенные – например, рибозные. Следовательно, различные олигомеры сосуществовали, эволюционировали, взаимодействовали и конкурировали между собой. А Природа отобрала наиболее эффективные.
ВОЗНИКНОВЕНИЕ БИОПОЛИМЕРОВ И ФУНКЦИОНАЛЬНЫХ СТРУКТУР
Биополимеры – это продукты конденсации более простых веществ с отщеплением молекул воды. В водной среде их образование и существование термодинамически и кинетически невыгодно и при установлении равновесия неминуемо приведет к их гидролизу на составные части. Абиотический синтез их возможен либо при нагревании в (почти) безводной среде, например, в периоды высыхания водоемов, особенно в окружении гидрофобных молекул (например, липидов), либо при химической активации их составляющих частей конденсирующими агентами при затрате энергии. Первый вариант предпочтительнее как более естественный. Однако экспериментально установлено, что скорость (а, значит, и наблюдаемая в опыте степень) олигомеризации невысока, если мономеры выступают в обычной, неактивированной форме. Это обстоятельство затрудняет исследование дальнейших процессов усложнения олигомеров, формирования ими функциональных структур, взаимодействия с другими биомолекулами. Экспериментаторы ведь не могут ждать многие годы результатов опыта с натуральными формами реагентов. Поэтому они используют мономеры в активированной форме, не всегда такой, которая могла существовать на ранней Земле. Однако одновременно продолжаются исследования первого варианта, о чем будет сказано позже.
Первоначально в опытах по абиотическому синтезу РНК пытались использовать АТP и другие нуклеозидтрифосфаты, естественные активированные нуклеотиды. Однако в отсутствие белковых ферментов или рибозимов они довольно инертны (см., например, [53]).
Для неферментативного синтеза олигонуклеотидов часто используют нуклеотиды, к фосфатной группе которых присоединена более легко отщепляемая группа. Наиболее активными в этом отношении считаются 2-аминоимидазолиды нуклеотидов [54]. Как показали А. Фаренбах и др. [55], 2-аминоимидазол образуется в системе Дж. Сазерленда одновременно с 2-аминооксазолом при реакции цианамида с гликолевым альдегидом в присутствии аммиака с хорошим выходом (41%). Поэтому его можно считать предбиотическим агентом. Но самостоятельно присоединиться к нуклеотиду он не может. Этому мог способствовать конденсирующий агент, образующийся в гексацианоферратном цикле при действии УФ-лучей – метилизоцианат [56]. Или, как считает А. Фаренбах, хлорциан – тоже возможный продукт УФ-облучения ионов цианида и хлора в водной или газовой среде [57].
Имидазол и его производные применялись Л. Оргелом в синтезе олигонуклеотидов еще с 60-х годов. Удалось даже осуществить неферментативный матричный синтез, получив 14‑звенный олигонуклеотид [58]. Опыты проводили с естественными нуклеотидами, то есть имеющими D-конфигурацию. И, разумеется, с однородными, например, только с рибонуклеотидами.
Но на древней Земле в абиотических синтезах образовывалась сложная смесь нуклеотидов. Они могли содержать разные гетероциклы, не только аденин, гуанин, цитозин и урацил. Соединяющие их мостики тоже могли иметь различную природу. Если в их состав входили углеводы, то они имели бы и L-, и D-конфигурацию. Мы не можем здесь обсуждать все возможные варианты, остановимся лишь на упомянутых выше.
Четыре перечисленных гетероцикла наиболее устойчивы к действию УФ-лучей, поэтому преобладали в абиотических условиях. Кроме того, в опытах Дж. Сазерленда они образуются из простых исходных веществ и не в качестве целевых продуктов, а сразу в составе сложной молекулы, объединяющей углеводную и гетероциклическую часть. А вот углеводная составляющая может быть представлена шестью видами моносахаридов. Преобладающими углеводами, как сказано выше, являются три: арабиноза, рибоза и треоза. Все они выступают в фуранозной форме (согласно пути Поунера-Сазерленда). Если же абиотический синтез нуклеотидов происходил путем, описанным Т. Кереллом, то преобладать должна пиранозная форма.
Прежде всего, рассмотрим проблему рацематов. Ведь только однородные по пространственной конфигурации мономеры могут сформировать полимер правильной спиральной формы. Молекулы РНК, синтезированные из рацемических смесей нуклеотидов, даже имеющие одинаковую их последовательность, совершенно не похожи друг на друга. Их пространственная конфигурация различна из-за хаотичности конфигурации отдельных звеньев. Они не смогут образовать ни правильной спирали, ни структур с какой-либо функциональной активностью. Видимо, и жизнь вряд ли бы возникла, если бы не нашлись естественные факторы, обеспечивающие хиральную чистоту полинуклеотидных цепей.
Оказалось, что в этом могли помочь минералы, в частности, монтмориллонит (глина, продукт водной эрозии вулканического пепла). Дж. Феррис и П. Джоши в серии работ [59–61] показали, что в его присутствии из смеси L- и D-нуклеозидфосфоимидазолидов образуются практически полностью гомохиральные олигонуклеотиды длиной до 11 звеньев. Причем с увеличением длины олигомера возрастает степень его хиральной чистоты. Уже начиная с пентамеров, хиральная чистота продуктов превышает 97%. Таким образом, получаются и чистые L-, и чистые D-олигонуклеотиды. Причина такого селектирующего действия монтмориллонита пока не выяснена, считается, что она обусловлена особенностями кристаллической структуры этого минерала. Не исследована и селектирующая способность монтмориллонита в отношении нуклеотидов, активированных другим способом, а также содержащих разные сахара в их пиранозной или фуранозной формах. А такие исследования в свете вышесказанного очень актуальны.
Важным селектирующим фактором могли быть УФ-лучи. М.А. Никитин в своей книге [4] отмечает, что они могли способствовать отбору, во-первых, самых устойчивых к ним азотистых гетероциклов, склонных образовывать комплементарные пары, т.е. Аde, Gua, Cyt и Ura. Во-вторых, они благоприятствовали накоплению олигонуклеотидов, состоящих из нуклеотидов одной хиральности. Гомохиральные олигонуклеотиды к УФ-лучам более устойчивы, так как расположение гетероциклов в их спиральных молекулах “стопкой” обеспечивает их взаимную защиту за счет стэкинг-взаимодействия. Длинные олигомеры также более устойчивы по сравнению с короткими, поскольку в них больше взаимодействующих гетероциклов. А молекулы РНК, содержащие двуспиральные участки (шпильки), более устойчивы благодаря комплементарному спариванию.
По указанным причинам популяция олигонуклеотидов, в основном, состояла из смеси гомохиральных L- или D-форм. От случайно образовавшихся гетерохиральных цепочек ее очищали УФ‑лучи. Олигонуклеотиды различной хиральности неизбежно должны были длительное время сосуществовать. Предпринимаются попытки теоретически обосновать и экспериментально продемонстрировать возможность абиотического формирования хирально однородных биополимеров путем постепенного накопления небольших избытков одних энантиомеров над противоположными в последовательной серии репликаций. Однако, на наш взгляд, на фоне постоянно текущего абиотического синтеза рацематов такой подход не решает проблему хиральной чистоты.
Не паллиативное, а кардинальное решение этой проблемы (и не только!) произвела Природа, когда среди хирально чистых D-рибозимов появился такой, который оказался способным избирательно распознавать молекулы D-рибозы (именно фуранозной формы!) или ее фосфорилированных производных и присоединять их к основаниям РНК. Это обеспечило интенсивный синтез D-рибонуклеотидов и столь же быстрое накопление полимерных D-РНК. Не создавшие подобного рибозима L-нуклеотиды синтезировались в абиогенных процессах гораздо медленнее и были постепенно вытеснены, как и все ксенонуклеотиды и их полимеры.
Такой D-рибозоспецифичный рибозим экспериментально не получен, но вполне возможен, так как описаны рибозимы, распознающие энантиомерные формы аминокислот и присоединяющие предпочтительно L-аминокислоты к тРНК (см. [62] и ссылки в ней).
На основании своих работ по аминоацилированию молекул тРНК К. Тамура высказал предположение, что сразу после сформирования мира РНК, основанного на D-рибозе, могла произойти селекция L-аминокислот посредством аминоацилирования тРНК [63]. В его экспериментах D‑аминокислоты присоединялись к молекулам тРНК D‑рибозимами в 4 раза медленнее, чем L-аминокислоты. Как выяснилось путем стереометрических измерений [64], затруднения были вызваны пространственной ориентацией D-аминокислот в активном центре рибозима. Таким образом, аминоацилирующие D-рибозимы в процессе формирования генетического кода решили проблему гомохиральности белковых молекул.
Проблема неоднородности мономеров нуклеиновых кислот решена недавними экспериментами Дж. Шостака [10] и Р. Кришнамурти [11, 12]. Дж. Шостак установил, что при неферментативном синтезе олигонуклеотидов на матрицах, составленных из смеси арабино-, рибо- и дезоксирибонуклеотидов, скорость присоединения рибонуклеотидов к праймерам выше, чем их дезокси- и арабино-аналогов. Причем, последние уступают рибонуклеотидам в 15–30 раз. Кроме того, после их присоединения дальнейшее расширение праймера замедляется примерно в 300 раз. Причину авторы видят в особенностях пространственной структуры углеводной части нуклеотидов. Следствием этого является постепенное обогащение синтезируемого полимера рибонуклеотидами в серии репликаций и образования в конечном итоге гомогенной молекулы РНК. А гомогенные РНК-матрицы при копировании тем более произведут гомогенное “потомство”.
С. Бхоумик и Р. Кришнамурти [11] обнаружили, что гетерогенные олигонуклеотиды, составленные из смеси треонуклеотидов и рибонуклеотидов, предпочтительно образуют дуплексы с гомогенными комплементарными цепочками РНК, ТНК или ДНК. Следовательно, они могут служить матрицами для неферментативного лигирования гомогенных нуклеотидных последовательностей, что после нескольких репликаций приведет к образованию полностью гомогенных молекул РНК, ТНК или ДНК. Таким образом, оба неферментативных процесса репродукции нуклеотидных последовательностей способны создать гомогенные цепочки РНК и ДНК из гетерогенной смеси нуклеотидов. Поскольку гомогенные цепочки ТНК тоже образуются в эксперименте, этот вывод, вероятно, справедлив и для ксено-НК.
При всех достоинствах имидазола и его производных, они сами нуждаются в активации для присоединения к фосфатным группам нуклеотидов. Этого недостатка лишены амиды фосфорной кислоты: они легко получаются реакцией NH3 с циклическим триметафосфатом (вулканическим продуктом), со шрайберзитом или с фосфитами [32, 65]. Как установлено в работах Р. Кришнамурти [22, 32, 65], моно- и диамид фосфорной кислоты в нейтральных разбавленных водных растворах (даже в микромолярных концентрациях) активно фосфорилируют альдолы, нуклеозиды, нуклеотиды и нуклеозиддифосфаты. Получены также короткие олигомеры [65]. Наиболее вероятно, что именно они были конденсирующими агентами в абиотических синтезах олигонуклеотидов и пептидов [65].
Среди коротких гомохиральных олигонуклеотидов часто могли возникать такие пары, у которых несколько звеньев на 3'-концах комплементарны. Тогда они, спарившись этими концами, окажутся друг для друга взаимными праймерами. Такой комплекс вполне может быть неферментативно достроен в обе стороны до полной двойной спирали уже большей длины при наличии активированных нуклеотидов.
Несмотря на скромные размеры молекул абиотически возникающих гомохиральных олигонуклеотидов, они могут представлять собой функциональные структуры. Например, рибозим, состоящий всего из 5 звеньев, получен в лаборатории М. Яруса [66]. Он оказался способным катализировать присоединение активированной аминокислоты (аминоациладенилата) к рибозе другого короткого олигонуклеотида сложноэфирной связью. Более того, он еще и катализирует образование пептидной связи между этой аминокислотой и другой активированной аминокислотой, выполняя функцию рибосомы. В экспериментах обнаружено образование пентапептидов с участием этого самого маленького из известных рибозимов [67]. Благодаря небольшой длине этого рибозима вероятность его абиотического синтеза очень велика: возможных комбинаций последовательности нуклеотидов всего 5120. По подсчетам М. Яруса масса такого количества пентануклеотидов равна 4.6 × 10–17 г – ничтожное количество в сравнении с тем, которое может быть накоплено в относительно небольшие сроки в водоемах вокруг даже одного из наземных термальных источников. Так что этот рибозим мог быть распространен на ранней Земле повсеместно.
Выявлены небольшие рибозимы и с другой функцией – лигазной, т.е. со способностью “сшивать” довольно короткие олигонуклеотиды, подобные тем, что получаются синтезом на монтмориллоните или путем неферментативной достройки взаимных праймеров. В любой популяции РНК такие рибозимы спонтанно могли появиться, поскольку часто бывают очень короткими (порядка 20–30 нт), для лигазной активности достаточно даже 7 звеньев [68].
Появление длинных цепей РНК привело к образованию рибозимов с разнообразными функциями. Первостепенное значение имели бы рибозимы, обеспечивающие создание точных копий молекул РНК. Селекцией in vitro в лаборатории Ф. Холлиджера создан рибозим-полимераза РНК, лучший на данный момент с точки зрения точности копирования. Он способен удлинять короткую РНК-затравку на 95 звеньев за сутки [69]. С его помощью размножили настоящую функциональную молекулу РНК – другой рибозим, известный под названием Mini-hammerhead. Этот рибозим состоит всего из 24 нт, но обладает эндонуклеазной активностью: он умеет разрезать РНК в одном строго определенном месте. Рибозим tC19Z скопировал рибозим Mini-hammerhead, сохранив его ферментативную активность. Тем самым впервые была продемонстрирована репликация функциональной молекулы РНК при помощи рибозима-полимеразы. Другой рибозим Ф. Холлиджера (tC9Y) смог скопировать за 60 часов при 17°C цепочку РНК длиной в 206 нт, т.е. длиннее себя [70].
Рибозим-полимеразу с более широким диапазоном действия и большей скоростью копирования удалось получить Дж. Джойсу [71]. С помощью рибозима 24-3 были амплифицированы два РНК-аптамера, РНК-лигаза и даже дрожжевая фенилаланиновая тРНК. Рибозим преодолел все шпильки и петли этих матриц, хотя они обычно являются серьезным барьером для полимераз. Кроме того, этот рибозим оказался активной обратной транскриптазой, присоединяя к праймеру любой из 4 дезоксирибонуклеотидов примерно за 1 мин и доводя длину цепочки ДНК до 32 нт [72].
Недавно Ф. Холиджеру удалось осуществить с помощью рибозима репликацию его каталитически активной молекулы [8]. Правда, не в целом виде, а по частям.
Рибозим получен методом искусственной эволюции. Он состоит из двух субъединиц, одна из которых каталитически неактивна, но как-то помогает другой субъединице. Каталитически активную субъединицу рибозима разделили на 5 фрагментов. В качестве строительного материала использовали не мономеры, а все 64 возможных триплета. Ранее было установлено, что они успешно расплетают двуспиральные участки молекулы рибозима, образуя с ними комплементарные пары. Рибозим действовал как лигаза, сшивая триплеты, причем в обоих направлениях, 5'−3' и 3'−5', и не нуждался в праймерах.
После синтеза всех пяти фрагментов рибозим смог связать их в две молекулы, объединив соседние фрагменты. Для этого пришлось в среду добавить короткие олигонуклеотиды, комплементарные пограничным последовательностям объединяемых фрагментов (они могли спонтанно возникать в предбиологических условиях как обрывки больших молекул при гидролизе). Интересно, что полученные таким путем продукты спонтанно образовали агрегат за счет нековалентных связей. И этот агрегат, будучи соединенным с неактивной субъединицей рибозима, воспроизвел комплементарную последовательность одного из первоначальных фрагментов. К сожалению, это еще не строгое доказательство саморепликации РНК в абиотических условиях, но вселяет надежды, что она возможна.
Идея раскручивания матричной РНК с помощью коротких олигонуклеотидов успешно использована лабораторией другого лауреата Нобелевской премии – Дж. Шостака [73]. Он продолжает начатые Л. Оргелом [58] исследования неферментативного копирования молекул РНК. Одно из главных затруднений на этом пути – быстрое восстановление дуплекса РНК после теплового разъединения цепочек. Но добавление в реакционную смесь гекса- или октамеров нуклеотидов, захватывающих одну из цепочек с образованием с ней комплементарных комплексов, открывает вторую цепочку для матричного неферментативного синтеза олигонуклеотидов. Авторы подчеркивают, что такие “захватчики” вполне могли быть в изобилии в “мире РНК”. Они могли быть либо продуктами гидролиза длинных РНК, либо недостроенными копиями при их синтезе. В любом случае они оказывались комплементарными какому-либо участку молекул РНК, “проживающих” в данном микроокружении.
При попытках воспроизведения абиотического синтеза другого жизненно важного полимера (белка) возникают те же проблемы, что и при синтезе олигонуклеотидов. Нагревание аминокислот в безводной среде тоже приводит к образованию лишь коротких цепочек, хиральная чистота не обеспечивается, минералы оказывают каталитическое действие, но для получения длинных полипептидов необходимо использовать активированные формы аминокислот.
Одной из них являются N-карбоксиаминокислоты, циклические смешанные ангидриды аминокислот и угольной кислоты. Дж. Феррис и Л. Оргел [74] из них получили 55-мерные пептиды при адсорбции на поверхности минералов гидроксиапатита и иллита, служивших катализаторами. N-Карбоксиаминокислоты образуются, например, реакцией аминокислот с одним из вулканических газов – сернистым карбонилом (COS) [75].
Другой путь их абиотического синтеза, длительно и тщательно изучавшийся французскими химиками из университета Монпелье, – через реакцию Бюхерера-Бергса. Как указывалось выше, первым этапом абиотического синтеза аминокислот является образование аминонитрилов. Они поглощают CO2 из насыщенных им водных растворов и через ряд внутримолекулярных реакций превращаются в N-карбамоиламинокислоты [36]. Продукты этого процесса отщепляют аммиак и циклизуются, хотя и довольно медленно (в течение месяца при 80°С) [76]. Это приводит к N‑карбоксиаминокислотам.
Пептиды образуются также при взаимодействии аминокислот с циклическим триметафосфатом [77], а еще лучше с продуктом его аммонолиза – диамидом фосфорной кислоты [32, 65]. Однако наиболее вероятным путем их абиотического синтеза представляется следующий.
Как сказано выше, при недостатке аммиака реакция альдегидов с HCN приводит к образованию α-гидрокси- и α-аминокислот. В опытах Р. Кришнамурти [78–80] показано, что в эквимолярной смеси глицина и молочной кислоты при нагревании до 65°С и выше образуются олигомеры до 10 звеньев, содержащие остатки как молочной кислоты, так и глицина. Причем в растворах одного лишь глицина олигомеров не наблюдается. Если подвергать эту смесь циклам увлажнения/высушивания, то процент включения глицина в олигомеры увеличивается с каждым циклом. То же происходит и при замене глицина на аланин или на смесь этих аминокислот. Замена молочной кислоты на гликолевую улучшает условия образования сополимеров, называемых депсипептидами.
Сложные эфиры образуются и гидролизуются легче, чем пептиды. Видимо, этим объясняется и вовлечение гидроксикислотами аминокислот в реакцию сополимеризации, и последующее увеличение доли аминокислот в сополимере. После нескольких циклов увлажнения/высушивания аминокислоты в сополимерах уже образуют непрерывные последовательности. В конечном итоге вся цепочка становится аминокислотной.
Совершенствование структуры биополимеров для лучшего выполнения ими жизненно важных функций немыслимо без эволюции, подобной Дарвинской (хотя биополимеры – это еще не живые существа). Ведь если структура способна самокопироваться с неизбежными при этом ошибками (“мутациями”), то она обязательно создаст популяцию дочерних структур, из которых “выживут” и продолжат свой “род” при ограниченности ресурсов лишь наиболее успешные. А этот процесс обладает всеми чертами Дарвинской эволюции.
Пока речь шла об отдельных молекулах биополимеров, для их эволюции достаточно было той степени компартментализации, которую предоставляли поры минералов. В них и протекала эволюция предбиотических сообществ полимерных молекул, названных Е.В. Куниным “реплицирующимися ансамблями эгоистичных кооператоров” [3].
Каждый ансамбль постепенно усложнялся благодаря тому, что отдельные его компоненты приобретали различные функции, полезные для выживания и более быстрого роста и развития всего ансамбля. Помимо первичной специализации лигаз и полимераз, необходимой для самовоспроизведения ансамблей молекул РНК, возникали рибозимы, например, ускоряющие фосфорилирование нуклеозидов(тидов) до нуклеозидтрифосфатов любым агентом, даже диамидом фосфорной кислоты. Ведь, несмотря на его высокую активность, скорость катализируемого фосфорилирования значительно выше.
Такой ансамбль более интенсивно потреблял мономеры из окружающего водоема, отбирая их у ансамблей соседних пор, и обгонял последних в росте. В другом ансамбле мог образоваться рибозим, катализирующий какую-либо реакцию абиотического синтеза нуклеотидов. Он тоже получал преимущество и разрастался быстрее. Погодные условия (затопление, перемешивание воды сильным ветром и т.п.) могли способствовать обмену частью рибозимов между ансамблями (нечто подобное горизонтальному переносу генов). Так мог возникнуть новый ансамбль, обладающий обоими полезными свойствами.
По мере усложнения ансамблей для сохранения всех полезных компонентов возникла необходимость более надежного их обособления. Это особенно стало важным в период формирования генетического кода. В водной среде компартментализация возможна благодаря водонепроницаемой оболочке. Такую оболочку на заре жизни могли создать, как считают, молекулы жирных кислот. Обладая полярной группой –СООН и неполярным углеводородным “хвостом”, они формируют мицеллы или пузырьки, состоящие из двух слоев. Обращенная к воде сторона каждого слоя образована полярными группами, внутренняя – неполярными нитями, которые удерживаются рядом ван-дер-ваальсовыми силами. Из таких пузырьков, помещая в них полимерные молекулы, формируют искусственные протоклетки. Мономеры и другие небольшие молекулы могут поступать внутрь пузырька и служить “строительным материалом” для копирования полимеров.
К. Адамала и Дж. Шостак [81] доказали, что внутри таких протоклеток происходит неферментативный матричный синтез олигонуклеотидов из рибонуклеотидов, активированных имидазолом. Они легко проникали через двухслойную мембрану (в отличие от АТP и других нуклеозидтрифосфатов). К цепочкам матриц были прикреплены небольшие комплементарные участки олигонуклеотидов, служившие праймерами. Через 2–3 сут большинство матричных молекул превращались в дуплексы. То есть праймеры были полностью достроены мономерами до комплементарных матрицам цепочек.
В недавней работе этой же лаборатории [9] продемонстрировано неферментативное копирование внутри протоклеток последовательности ДНК длиной в 25 нт, то есть достаточной для транскрипции активных рибозимов. Правда, в качестве мономеров использовались не естественные активированные дезоксирибонуклеотиды, а немного измененные. У них вместо гидроксила в 3'-позиции была аминогруппа. С естественными мономерами удавалось получать только 5-звенные копии. Здесь наблюдался тот же эффект, что и с заменой гидроксила на аминогруппу при фосфорилировании нуклеозидов: более высокая реактивность аминогруппы. Сами авторы признают, что не видят пока какой-либо возможности объяснить предбиотическое появление аминосодержащих нуклеотидных аналогов. Однако не исключено, что такие аминированные нуклеотиды могут возникать, хотя бы в небольших количествах, при высоких концентрациях NH3 (тогда глицеральдегид может прореагировать с 2-аминооксазолом в виде аддукта с аммиаком, как гликолевый альдегид в опыте А. Фаренбаха и др. [55]) или при других, еще не найденных условиях.
Протоклетки, образованные даже столь простыми веществами как жирные кислоты, способны обеспечить обособление и неферментативное копирование молекул НК. Конечно, прочность таких протоклеток невелика: при встряхивании или перемешивании окружающей среды они распадаются и могут терять свое содержимое. В кислой или щелочной среде, а также в соленой воде они также разрушаются. Значительно прочнее мембраны, в состав которых входят фосфолипиды. Возможность абиотического синтеза фосфолипидов показана Дж. Сазерлендом и др. [7]. Они этерифицировали 2-фосфоглицерин карбоновыми кислотами, содержащими 8–10 атомов углерода. Моноэфиры не образовывали пузырьков. Диэфиры и их смеси с моноэфирами формировали пузырьки – липосомы, стабильные при различных значениях pH и температуры. Они были способны удерживать внутри себя моно- и тринуклеотиды в течение, по меньшей мере, двух недель.
Мембраны, состоящие из смеси компонентов с разными полярными группами, обладают большей стабильностью и проницаемостью. Наличие хотя бы небольших количеств фосфолипидов закрепляет молекулы жирных кислот в мембране, снижая скорость их десорбции, а также препятствует разрушению мембран двухвалентными катионами [82].
Фосфолипиды образуют и более сложные, многослойные структуры [83]. Считая их идеальной средой для полимеризации нуклеотидов, Д. Димер провел серию опытов, имитирующих циклы увлажнения/высушивания, используя пузырьки из фосфолипидов с включенными в них неактивированными нуклеотидами (AMP и UMP). Водная суспензия таких пузырьков наносилась небольшими объемами в открытые сосудики, помещенные на медленно вращающийся подогреваемый до 85°С алюминиевый диск. Диск находился в камере, продуваемой CO2 для создания бескислородных условий и удаления паров воды. В одном месте на пути вращения диска помещалось устройство для дозированного добавления воды в сосудики, где вода к этому моменту испарилась, а фосфолипиды образовали тонкие слои с расположенными между ними нуклеотидами. После увлажнения в сосудиках вновь возникали липидные пузырьки с включенными в них мономерами и олигомерами. Появление олигомеров было доказано гель-электрофорезом, наличием гиперхромного эффекта и электрофорезом через наноотверстия (диаметр 1.5 нм), пропускающие лишь по одной молекуле олигонуклеотида. Выявлено образование олигомеров длиной в несколько десятков нуклеотидов (иногда – до 100), количество которых возрастало с каждым циклом увлажнения/высушивания, но затем выходило на плато из-за уравновешивания процессов полимеризации и гидролиза. Параллельные опыты с poly(A) и poly(U) показали, что после каждого цикла гидролизуется около 8% олигомеров.
В более поздних публикациях [84, 85] Д. Димер развивает мысль о естественном отборе в циклах увлажнения/высушивания протоклеток, содержащих разные полимеры. Например, биополимер, стабилизирующий мембрану аналогично современным цитоскелетным белкам, обеспечил бы протоклеткам преимущество, защитив их от механического “стригущего” действия, вызванного турбулентностью. К сожалению, автор не указывает ни природу такого полимера, ни его абиотическое происхождение, ни способы его воспроизведения, чтобы протоклетка смогла передать часть его молекул своим дочерним протоклеткам.
А между тем такого рода полимеры на заре жизни существовали – липидоподобные пептиды. Благодаря вышеописанным процессам полимеризации аминокислот они появились довольно рано (ранее фосфолипидов) и синтезировались в достаточных количествах.
Самыми распространенными аминокислотами были глицин и аланин, а следующими (с большим отрывом) – валин, аспарагиновая и глутаминовая кислоты и серин. Это доказано как изучением содержания аминокислот в метеоритах, так и модельными опытами. Поэтому короткие пептиды (~10 а.о.) по чисто статистическим причинам содержали только один аминокислотный остаток с гидрофильной группой, а остальные были гидрофобными. Таким образом, их молекулы представляли собой цепочки с гидрофильной “головой” и гидрофобным “хвостом” (если полярная аминокислота была в середине цепочки, то с двумя хвостами). Они, как и жирные кислоты, способны формировать в водной среде двухслойные структуры. Такие пептиды способны к самосборке, образуя амилоидные волокна, трубочки, лентообразные пленки и пузырьки, что подробно описано в обзоре первооткрывателя этого явления С. Чжана [86], нашедшего для них массу практических применений в медицине.
Экспериментально установлено [87, 88], что липидоподобные пептиды самопроизвольно образуют нанотрубки и пузырьки диаметром 30–50 нм с толщиной стенок, равной двукратной длине молекулы пептида. Таким образом, эти молекулы ориентированы перпендикулярно стенкам трубочек и пузырьков, образуя двухслойную структуру. Толщина стенок примерно одинакова с толщиной фосфолипидных мембран современных организмов. Прочность стенок выше, чем у двухслойных мембран, образуемых жирными кислотами, так как пептидные цепочки имеют гораздо больший ассортимент межмолекулярных связей, чем гидрофобные “хвосты” молекул жирных кислот. Кстати, в современных клетках цитоскелет, т.е. основу их структуры, определяющую форму клеток, образуют белки, а не липиды.
Скорее всего, на заре жизни пептидные структуры и жирные кислоты образовывали комплексные мембраны, в которых они взаимно дополняли друг друга. Липидные компоненты обеспечивали надежную изоляцию содержимого протоклетки от внешней среды, препятствуя его утечке, а пептидные компоненты придавали мембране прочность без утери эластичности и, возможно, содействовали транспорту субстратов сквозь мембрану.
Разумеется, описанные протоклетки нельзя назвать организмами, даже примитивными. Они не представляли собой единое целое, в котором все компоненты взаимосвязаны и взаимозависимы. Молекулы РНК в них размножались за счет поступающих мономеров, количество остальных компонентов увеличивалось лишь путем захвата новых молекул, производимых абиотическим питомником. Протоклетки росли, увеличиваясь в размерах, затем разделялись случайным образом на неравные части, и не весь ассортимент их содержимого (в том числе, и молекул РНК) мог оказаться в каждой дочерней протоклетке. Те из них, которые получили неполный комплект РНК, могли утратить способность самокопирования и попросту погибнуть, став “пищей” для других протоклеток.
Иными словами, генотип протоклетки (если можно так назвать комплекс молекул РНК в ней) оказывал влияние лишь на часть ее фенотипа – набор самих молекул РНК, появление новых рибозимов и повышение их активности. На мембраны генотип не производил полезного для них действия. Возможно, единственным исключением могло быть появление среди молекул РНК рибозима, катализирующего синтез фосфолипидов (предположение Дж. Шостака [89]), которые, как сказано выше, стабилизируют мембрану.
Лишь после возникновения кодируемого синтеза белковых молекул генотип протоклеток начал постепенно расширять сферу своего влияния, производя и полезный для них строительный материал, и самих “строителей” – разнообразные белковые ферменты. И, по-видимому, только с этого этапа протоклетки можно считать примитивными организмами, а их эволюцию – подлинно Дарвинской. Но процессы, приведшие к возникновению генетического кода – это материал для отдельной большой статьи.
ЗАКЛЮЧЕНИЕ
Первым процессом, ведущим к происхождению жизни на Земле, можно с уверенностью считать фотохимический синтез формальдегида и HCN в только что образовавшейся атмосфере. Она, как утверждают геологи, была в катархейском эоне намного плотнее нынешней, ее давление достигало 100 бар, как на Венере [90]. Какой бы высокой ни была температура земной поверхности, в атмосфере конденсировались микрокапельки воды (очевидно, на высоте гораздо большей, чем сейчас), растворяющие в себе продукты фотохимических реакций и вещества, извергаемые океаном магмы. Можно даже предположить, что образовавшийся аэрозоль представлял собой множество предбиотических микрореакторов ([91] и ссылки в ней). В этих микрореакторах не могли не происходить процессы, описанные выше для наземных водоемов. Абиотический питомник мог существовать в таком микродисперсном виде и, по крайней мере, самые первые стадии изображенных на рис. 1 процессов могли в нем происходить и накапливать гликолонитрил, формамид и другие органические соединения. В микрокапельках происходили также циклы увлажнения/высушивания (второй процесс, важный для зарождения жизни), хотя полному высушиванию могло препятствовать наличие высококипящих жидкостей.
Неизвестно, какова была судьба продуктов атмосферных процессов до образования земной коры и охлаждения ее до температуры конденсации воды. Многие из них довольно устойчивы к УФ-радиации. Кроме того, в атмосферу поступали в виде паров соли цинка (ZnS, а также ZnCl2, который с H2S образует ZnS). ZnS в микрокапельках образовывал микрокристаллики, которые защищали адсорбированные на них продукты от УФ-лучей. Поэтому не исключено, что в наземных условиях абиотические синтезы начались не с нуля, что могло ускорить зарождение жизни.
На земной поверхности при контакте с жидкой водой начался третий процесс (серпентинизация), и продолжились уже в новых условиях первые два. Все это достаточно подробно описано в основном тексте. Отметим только, что в этот период очень возросло в атмосфере содержание метана за счет серпентинизации. А значит – интенсифицировались фотохимические процессы в атмосфере, продуцирующие формальдегид и HCN. Поэтому нелогичным выглядит пересмотр объема продукции HCN (ранее оцениваемого как примерно равный объему продукции формальдегида) в сторону уменьшения [29], объясняемый якобы небольшим содержанием метана в тогдашней атмосфере. Тем более что HCN образуется и другими абиотическими путями. Так что цифры для HCN, приведенные в табл. 1, следует считать сильно заниженными.
Абиотических питомников было множество. Небольших водоемов и сейчас много вокруг каждого вулкана. А в те времена неокрепшей и не столь толстой земной коры вулканическая деятельность была, вероятно, многократно интенсивнее. И в атмосферных микрореакторах, и уже в наземных, гораздо обильнее, чем сейчас, снабжаемых субстратами как из атмосферы, так и из недр, быстро накопилось много органических веществ. При продуктивности абиотических процессов, указанной в табл. 1, на 1 км2 вулканического региона ежесуточно может образоваться 16 г формальдегида, исходного органического вещества для всех синтезов абиотического питомника. Всего около 1 г весят 1019 молекул РНК длиной в 100–200 нт, потенциальных рибозимов-РНК-полимераз. В опытах по искусственной эволюции нуклеиновых кислот используют 1014–1015 молекул (в десятки–сотни тысяч раз меньше) и добиваются успеха. Так что и для успешной естественной эволюции продуктивности абиотических процессов было вполне достаточно.
Хотя конденсации мономеров способствуют циклы увлажнения/высушивания, без конденсирующих агентов вряд ли могло обойтись. Из всех используемых конденсирующих агентов наиболее естественными и не зависящими от других являются амиды фосфорной кислоты [32, 65]. Они активно фосфорилируют нуклеозиды и нуклеотиды даже в микромолярных концентрациях. Но и эти процессы нуждались в появлении рибозимов, которые бы ускоряли их, давая селективное преимущество вырабатывающим их предбиологическим ансамблям. Такие рибозимы не исследовались, но они вполне возможны, так как известен подобный рибозим, катализирующий трифосфорилирование нуклеозидов триметафосфатом [92]. Так что эксперименты, вероятно, дадут положительный результат.
Пока нет консенсуса в вопросе первого информационного полимера. Многие склонны считать, что существовала некая пред-РНК. Вполне логично считать таковой ТНК. Ведь для абиотического синтеза треонуклеотидов не требуется глицеральдегид: доказано, что гликолевый альдегид способен реагировать с 2-аминооксазолом (а возможно, и с оксазол-2-тионом), формируя пятичленное кольцо [42]. Кстати, этим снимается проблема разделения по времени гликолевого и глицеринового альдегидов. Обеспечивается и гомогенность нуклеотидов: ведь если в реакции их синтеза (например, на начальных этапах абиогенеза) был вовлечен только гликолевый альдегид, пентозных нуклеотидов не возникало. ТНК гораздо устойчивее к гидролизу благодаря отсутствию в углеводном кольце свободных гидроксилов. Единственный недостаток: ее молекула менее гибка, чем молекула РНК, что может ограничивать каталитические возможности ТНК. Поэтому она впоследствии была вытеснена РНК, особенно на стадии формирования генетического кода: РНК лучше выполняла адапторные функции.
Образование протоклеток обычно обусловливают инкапсулированием молекул РНК внутрь пузырьков, образованных жирными кислотами. При этом в большинстве работ не рассматривается роль пептидов как структурной основы для протоклеточных мембран. И это несмотря на более высокую вероятность абиотического синтеза липидообразных пептидов, чем молекул жирных кислот достаточной длины (10 и более углеродных атомов), и намного большую прочность пузырьков, образованных пептидами. Заслуживает внимания также недавно высказанная идея [93], что в создании мембран могли участвовать полиэфиры, легко возникающие при конденсации α‑гидроксикислот. Для выяснения абиотической роли пептидов и полиэфиров (или даже депсипептидов) необходимы такие же эксперименты, как проводимые до сих пор с протоклетками, образованными только жирными кислотами.
Описанные процессы реально происходят и, по-видимому, могли при благоприятном сочетании, сложившемся хотя бы в одном из многих миллиардов небольших водоемов, привести к возникновению примитивных организмов, из которых путем эволюции произошло все живое. Однако вряд ли стоит считать их единственно возможными родоначальниками жизни. Еще много остается незаданных Природе вопросов относительно тайны происхождения жизни.
Список литературы
Опарин А.И. // Происхождение жизни. М.: Московский рабочий. 1924.
Еськов К.Ю. // Удивительная палеонтология: История Земли и жизни на ней. М.: ЭНАС, 2008. 312 с.
Кунин Е.В. // Логика случая. О природе и происхождении биологической эволюции. М.: Центрполиграф. 2014. 760 с.
Никитин М.А. // Происхождение жизни. От туманности до клетки. М.: Альпина нон-фикшн. 2016. 542 с.
Ritson D.J., Battilocchio C., Ley S.V., Sutherland J.D. // Nat. Communs. 2018. V. 9. 1821.
Xu J., Ritson D.J., Ranjan S., Todd Z.R., Sasselov D.D., Sutherland J.D. // Chem. Communs. 2018. V. 54. P. 5566–5569.
Bonfio C., Caumes C., Duffy C.D., Patel B.H., Percivalle C., Tsanakopoulou M., Sutherland J.D. // J. Amer. Chem. Soc. 2019. V. 141. P. 3934–3939.
Attwater J., Raguram A., Morgunov A.S., Gianni E., Holliger P. // eLife. 2018. V. 7. e35 255.
O’Flaherty D.K., Zhou L., Szostak J.W. // J. Amer. Chem. Soc. 2019. V. 141. P. 10 481–10 488.
Kim S.C., Zhou L., Zhang W., O’Flaherty D.K., Rondo-Brovetto V., Szostak J.W. // J. Amer. Chem. Soc. 2020. V. 142. P. 2317–2326.
Bhowmik S., Krishnamurthy R. // Nat. Chem. 2019. V. 11. P. 1009–1018.
Yadav M., Kumar R., Krishnamurthy R. // Chem. Rev. 2020. V. 120. https://doi.org/10.1021/acs.chemrev.9b00546
Maruyama S., Ebisuzaki T. // Geosci. Front. 2017. V. 8. P. 253–274.
Ebisuzaki T., Maruyama S. // Geosci. Front. 2017. V. 8. P. 275–298.
Maruyama S., Ikoma M., Genda H., Hirose K., Yokoyama T., Santosh M. // Geosci. Front. 2013. V. 4. P. 141–165.
Preiner M., Xavier J.C., Sousa F.L., Zimorski V., Neubeck A., Lang S.Q., Greenwell H.C., Kleinermanns K., Tuysuz H., McCollom T.M., Holm N.G., Martin W.F. // Life. 2018. V. 8. 41.
Легкоступов М.С. // К вопросу о модели образования планетных систем звезд. М.: ИПМ им. М.В. Келдыша. 2018. 33 с.
Canup R.M., Asphaug E. // Nature. 2001. V. 412. P. 708–712.
Rimmer P.B., Shorttle O. // Life. 2019. V. 9. 12.
McCollom T.M. // Proc. Natl. Acad. Sci. USA. 2016. V. 113. P. 13965–13970.
Vallée Y., Shalayel I., Ly K.D., Rao K.V.R., de Paëpe G., Märker K., Milet A. // Int. J. Dev. Biol. 2017. V. 61. P. 471–478.
Karki M., Gibard C., Bhowmik S., Krishnamurthy R. // Life. 2017. V. 7. 32.
Kitadai N., Maruyama S. // Geosci. Front. 2018. V. 9. P. 1117–1153.
Yang L., Xiao F., Zhang Q., Xu C., Dai B. // Chem. Select. 2019. V. 4. P. 7096–7101.
Mulkidjanian A.Y., Bychkov A.Y., Dibrova D.V., Galperin M.Y., Koonin E.V. // Proc. Natl. Acad. Sci. USA. 2012. V. 109. P. E821–E830.
Диброва Д.В., Гальперин М.Ю., Кунин Е.В., Мулкиджанян А.Я. // Биохимия. 2015. Т. 80. С. 590–611.
Mulkidjanian A.Y. // Biol. Direct. 2009. V. 4. P. 26.
Pinto J.P., Gladstone G.R., Yung Y.L. // Science. 1980. V. 210. P. 183–185.
Tian F., Kasting J., Zahnle K. // Earth Planet. Sci. Lett. 2011. V. 308. P. 417–423.
Summers D.P., Chang S. // Nature. 1993. V. 365. P. 630–633.
Ranjan S., Todd Z.R., Sutherland J.D., Sasselov D.D. // Astrobiology. 2018. V. 18. P. 1023–1040.
Gibard C., Gorrell I.B., Jiménez E.I., Kee T.P., Pasek M.A., Krishnamurthy R. // Angew. Chem. Int. Ed. 2019. V. 58. P. 8151–8155.
Botta O., Bada J.L. // Surv. Geophys. 2002. V. 23. P. 411–467.
Saladino R., Botta L., Di Mauro E. // Life, 2018. V. 8. P. 6.
Britvin S.N., Murashko M.N., Vapnik Y., Polekhovsky Y.S., Krivovichev S.V. // Sci. Rep. 2015. V. 5. P. 8355.
Taillades J., Beuzelin I., Garrel L., Tabacik V., Bied C., Commeyras A. // Orig. Life Evol. Biosph. 1998. V. 28. P. 61–77.
Shalayel I., Coulibaly S., Ly K.D., Milet A., Vallée Y. // Life. 2018. V. 8. P. 47.
Patel B.H., Percivalle C., Ritson D.J., Duffy C.D., Sutherland J.D. // Nat. Chem. 2015. V. 7. P. 301–307.
Pasek M.A., Harnmeijer J.P., Buick R., Gull M., Atlas Z. // Proc. Natl. Acad. Sci. USA. 2013. V. 110. P. 10 089–10 094.
Gull M., Cafferty B.J., Hud N.V., Pasek M.A. // Life. 2017. V. 7. P. 29.
Powner M.W., Gerland B., Sutherland J.D. // Nature. 2009. V. 459. P. 239–242.
Islam S., Aguilar J.A., Powner M.W., Nilsson M., Morris G.A., Sutherland J.D. // Chem. Eur. J. 2013. V. 19. P. 4586–4595.
Islam S., Bučar D.K., Powner M.W. // Nat. Chem. 2017. V. 9. P. 584–589.
Stairs S., Nikmal A., Bučar D.-K., Zheng S.-L., Szostak J.W., Powner M.W. // Nat. Communs. 2017. V. 8. 15 270.
Kim S.C., O’Flaherty D.K., Zhou L., Lelyveld V.S., Szostak J.W. // Proc. Natl. Acad. Sci. USA. 2018. V. 115. P. 13318–13323.
Becker S., Thoma I., Deutsch A., Gehrke T., Mayer P., Zipse H., Carell T. // Science. 2016. V. 352. P. 833–836.
Becker S., Schneider C., Okamura H., Crisp A., Amatov T., Dejmek M., Carell T. // Nat. Communs. 2018. V. 9. 163.
Roberts S.J., Szabla R., Todd Z.R., Stairs S., Bučar D.-K., Šponer J., Sasselov D.D., Powner M.W. // Nat. Communs. 2018. V. 9. 4073.
Noronha A.M., Wilds C.J., Lok C.-N., Viazovkina K., Arion D., Parniak M., Damha M.J. // Biochemistry. 2000. V. 39. P. 7050–7062.
Taylor A.I., Pinheiro V.B., Smola M.J., Morgunov A.S., Peak-Chew S., Cozens C., Weeks K.M., Herdewijn P., Holliger P. // Nature. 2015. V. 518. P. 427–430.
Li F., Sarkhel S., Wilds C.J., Wawrzak Z., Prakash T.P., Manoharan M., Egli M. // Biochemistry. 2006. V. 45. P. 4141–4152.
Pinheiro V.B., Taylor A.I., Cozens C., Abramov M., Renders M., Zhang S., Chaput J.C., Wengel J., Peak-Chew S.-Y., McLaughlin S.H., Herdewijn P., Holliger P. // Science. 2012. V. 336. P. 341–344.
Liu Z., Rossi J.-C., Pascal R. // Life. 2019. V. 9. 26.
Li L., Prywes N., Tam C.P., O’Flaherty D.K., Lelyveld V.S., Izgu E.C., Pal A., Szostak J.W. // J. Amer. Chem. Soc. 2017. V. 139. P. 1810−1813.
Fahrenbach A.C., Giurgiu C., Tam C.P., Li L., Hongo Y., Aono M., Szostak J.W. // J. Amer. Chem. Soc. 2017. V. 139. P. 8780–8783.
Mariani A., Russell D.A., Javelle T., Sutherland J.D. // J. Amer. Chem. Soc. 2018. V. 140. P. 8657–8661.
Yi R., Y. Hongo Y., Fahrenbach A.C. // Chem. Communs. 2018. V. 54. P. 511–514.
Acevedo O.L., Orgel L.E. // J. Mol. Biol. 1987. V. 197. P. 187–193.
Ferris J.P. // Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2006. V. 361. P. 1777–1786.
Joshi P.C., Aldersley M.F., Ferris J.P. // Biochem. Biophys. Res. Communs. 2011. V. 413. P. 594–598.
Jheeta S., Joshi P.C. // Life. 2014. V. 4. P. 318–330.
Saito H., Watanabe K., Suga H. // RNA. 2001. V. 7. P. 1867–1878.
Tamura K. // J. Mol. Evol. 2019. V. 87. P. 143–146.
Ando T., Takahashi S., Tamura K. // Nucl. Acids Res. 2018. V. 46. P. 11 144–11 152.
Gibard C., Bhowmik S., Karki M., Kim E.-K., Krishnamurthy R. // Nat. Chem. 2018. V. 10. P. 212–217.
Turk R.M., Chumachenko N.V., Yarus M. // Proc. Natl. Acad. Sci. USA. 2010. V. 107. P. 4585–4589.
Yarus M. // Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2011. V. 366. P. 2902–2909.
Vlassov A.V., Kazakov S.A., Johnston B.H., Landweber L.F. // J. Mol. Evol. 2005. V. 61. P. 264–273.
Wochner A., Attwater J., Coulson A., Holliger P. // Science. 2011. V. 332. P. 209–212.
Attwater J., Wochner A., Holliger P. // Nat. Chem. 2013. V. 5. P. 1011–1018.
Horning D.P., Joyce G.F. // Proc. Natl. Acad. Sci. USA. 2016. V. 113. P. 9786–9791.
Samanta B., Joyce G.F. // eLife. 2017. V. 6. e31 153.
Zhou L., Kim S.C., Ho K.H., O’Flaherty D.K., Giurgiu C., Wright T.H., Szostak J.W. // eLife. 2019. V. 8. e51 888.
Ferris J.P., Hill A.R., Jr., Liu R., Orgel L.E. // Nature. 1996. V. 381. P. 59–61.
Leman L., Orgel L., Ghadiri M.R. // Science. 2004. V. 306. P. 283–286.
Danger G., Boiteau L., Cottet H., Pascal R. // J. Amer. Chem. Soc. 2006. V. 128. P. 7412–7413.
Sibilska I.K., Feng T., Li L., Yin J. // Orig. Life Evol. Biosph. 2018. V. 48. P. 277–287.
Forsythe J.G., Yu S.-S., Mamajanov I., Grover M.A., Krishnamurthy R., Fernández F.M., Hud N.V. // Angew. Chem. Int. Ed. 2015. V. 54. P. 9871–9875.
Forsythe J.G., Petrov A.S., Millar W.C., Yu S.-S., Krishnamurthy R., Grover M.A., Hud N.V., Fernández F.M. // Proc. Natl. Acad. Sci. USA. 2017. V. 114. P. E7652–E7659.
Krishnamurthy R. // Chem. Eur. J. 2018. V. 24. P. 16 708–16 715.
Adamala K., Szostak J.W. // Science. 2013. V. 342. P. 1098–1100.
Toparlak O D., Mansy S.S. // Exptl. Biol. Med. 2019. V. 244. P. 304–313.
DeGuzman V., Vercoutere W., Shenasa H., Deamer D. // J. Mol. Evol. 2014. V. 78. P. 251–262.
Damer B., Deamer D. // Life. 2015. V. 5. P. 872–887.
Deamer D.W. // Assembling Life. How Can Life Begin on Earth and Other Habitable Planets? Oxford University Press, Oxford, UK, 2019.
Zhang S. // Interface Focus. 2017. V. 7. 20 170 028.
Vauthey S., Santoso S., Gong H.Y., Watson N., Zhang S.G. // Proc. Natl. Acad. Sci. USA. 2002. V. 99. P. 5355–5360.
Childers W.S., Ni R., Mehta A.K., Lynn D.G. // Curr. Opin. Chem. Biol. 2009. V. 13. P. 652–659.
Szostak J.W. // Angew. Chem. 2017. V. 129. P. 11 182–11 189.
Zahnle K., Nick A., Cockell C.S., Halliday A., Nisbet E., Selsis F., Sleep N.H. // Space Sci. Rev. 2007. V. 129. P. 35–78.
Mompeán C., Marun-Yaseli M.R., Espigares P., González-Toril E., Zorzano M.-P., Ruiz-Bermejo M. // Sci. Rep. 2019. V. 9. 1916.
Moretti J.E., Muller U.F. // Nucl. Acids Res. 2014. V. 42. P. 4767–4778.
Chandru K., Mamajanov I., Cleaves II, H.J., Jia T.Z. // Life. 2020. V. 10. 6.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия