

Биоорганическая химия, 2020, T. 46, № 5, стр. 466-485
Активные формы кислорода: участие в клеточных процессах и развитии патологии
Т. И. Шлапакова 1, Р. К. Костин 1, *, Е. Е. Тягунова 1
1 ФГАОУ ВО Первый Московский государственный медицинский университет имени И.М. Сеченова Министерства здравоохранения Российской Федерации (Сеченовский Университет)
119991 Москва, ул. Трубецкая, 8/2, Россия
* E-mail: rkostin2000@mail.ru
Поступила в редакцию 18.02.2020
После доработки 27.02.2020
Принята к публикации 29.02.2020
Аннотация
Активные формы кислорода (АФК) и азота (АФА) – не только побочные продукты химических реакций, но и участники различных клеточных процессов: защита от патогенных микроорганизмов (H2O2, HOCl, ONOO–, ${\text{O}}_{2}^{{\centerdot - }},$ OH•), оплодотворение (H2O2), деление клеток $\left( {{\text{O}}_{2}^{{\centerdot - }}} \right),$ апоптоз (H2O2), регенерация (H2O2), координация направления клеточного движения, регуляция тонуса сосудов (NO•) и т.д. Баланс между продукцией и устранением АФК и АФА приводит к внутриклеточному гомеостазу, в то же время их чрезмерное образование приводит к повреждению клеток и, скорее всего, изменению их метаболизма. АФК и АФА способны действовать как внутриклеточные мессенджеры, т.е. изменять внутриклеточное окислительно-восстановительное состояние и/или структуру и функцию белка путем модификации аминокислотных остатков (в основном цистеиновых), а редокс-состояние ряда белков может влиять на клеточный метаболизм. Перекись водорода является основной формой АФК, участвующей в окислительно-восстановительной передаче сигналов у эукариот. Нарушения в восстановительных системах способствуют старению и возникновению возрастных заболеваний. Старение в первую очередь связано с повышенным уровнем окислительного стресса, различными типами макромолекулярных изменений и накоплением повреждений дезоксирибонуклеиновой кислоты (ДНК). Поскольку большинство клеточных функций выполняется белками, старение может быть в какой-то степени следствием нарушения регуляции протеостаза или изменением функционирования протеома. Более того, не все клеточные белки могут быть ресинтезированы из-за возникающих в результате старения повреждений ДНК. Таким образом, активные формы кислорода и азота, постоянно генерируемые в организме, – важные участники регуляторных механизмов в клетке, но также и причина некоторых патологических состояний, включая рак. Известно, что АФК регулируют метаболизм сигнальных молекул, необходимых для осуществления клеточного цикла. Более того, АФК способны изменять активность железосодержащих белков. Связанное с неэффективной работой антиоксидантной защиты старение ассоциировано с окислительным стрессом, различными изменениями в клеточных структурах и макромолекулах, накоплением продуктов метаболизма, которые могут оказывать негативный эффект, повреждений дезоксирибонуклеиновой кислоты (ДНК), например, из-за совершения ошибок в ходе репликации ДНК-полимеразами, сбоев работы репарационных систем.
ВВЕДЕНИЕ
В норме в ходе клеточного метаболизма образуются активные формы кислорода (АФК) и активные формы азота (АФА), которые являются не только побочными продуктами химических реакций, но и участвуют в различных клеточных процессах: защита от патогенных микроорганизмов (H2O2, HOCl, ONOO–, ${\text{O}}_{2}^{{\centerdot - }},$ OH•), оплодотворение (H2O2), деление клеток $\left( {{\text{O}}_{2}^{{\centerdot - }}} \right),$ апоптоз (H2O2), регенерация (H2O2), координация направления клеточного движения (H2O2), регуляция тонуса сосудов (NO•) и т.д. Однако чрезмерно продуцируемые АФК и АФА – молекулы с высокой реакционной способностью, которые могут приводить к различным повреждениям клеток. Антиоксидантная защита обеспечивает инактивацию АФК и АФА, что способствует поддержанию гомеостаза. Недостаточная эффектность данной системы может приводить к окислительному стрессу, являющемуся одной из причин развития дегенеративных и онкологических заболеваний, а также клеточному старению. В данной работе мы рассмотрим участие АФК и АФА в различных клеточных процессах (в том числе редокс-сигнализации), а также механизмы антиоксидантной защиты.
МАТЕРИАЛЫ И МЕТОДЫ
Для подробного изучения клеточных антиоксидантных механизмов, влияния АФК на метаболизм и процессы старения были проанализировали опубликованные статьи из баз данных Elsevier, NCBI MedLine, Scopus, Scholar.Google, Embase, Web of Science, MedLine, The Cochrane Library, EMBASE, Global Health, CyberLeninka и RSCI. Для поиска англоязычных статей использовались следующие ключевые слова: “reactive oxygen species”, “reactive nitrogen species”, “antioxidant cell defense”, “redox signalling”, “ferroptosis”, “singlet molecular oxygen”, “hydroxyl radical”, “hypobromous acid”, “hypochlorous acid”, “superoxide”, “nitric oxide”, “peroxynitrite”, “hydrogen peroxide”, “hypothiocyanous acid oxidative stress”, “ROS pathways”, “senescence”, “aging”. Для поиска русскоязычных статей были использованы следующие ключевые слова: “перекисное окисление липидов”, “респираторный взрыв”, “АФК”, “СПОЛ”, “активные формы азота”, “активные формы кислорода”. Оценка приемлемости англоязычных и русскоязычных оригинальных источников осуществлялась в несколько этапов: просматривались заголовки, аннотации и полнотекстовые статьи. Кроме того, осуществлялся дополнительный поиск источников, указанных в отобранных статьях. Были исключены статьи, включающие оригинальные исследования на небольших группах пациентов (или подопытных животных), статьи, в которых были приведены предварительные результаты исследований или дублировались результаты предыдущих исследований.
УЧАСТИЕ АФК В КЛЕТОЧНОМ МЕТАБОЛИЗМЕ
Значительную роль в клеточном метаболизме играют активные формы кислорода (АФК) и активные формы азота (АФА). Активные формы азота представляют собой продукты метаболизма оксида азота. Они генерируются, когда оксид азота(II) (NO•), образующийся либо экзогенно, либо эндогенно, взаимодействует с такими активными формами кислорода, как супероксид $\left( {{\text{O}}_{2}^{{\centerdot - }}} \right)$ и перекись водорода (H2O2). Как АФА, так и АФК образуются как естественные продукты нормальной клеточной активности и участвуют в клеточной передаче сигналов [1]. Однако чрезмерное образование АФК и АФА может вызывать окислительный и нитрозирующий стрессы соответственно, что приводит к повреждению клеток и, как следствие этого, к их гибели [1–3]. В целом повреждение клетки свободными радикалами приводит к возникновению многих заболеваний, включая сердечно-сосудистые и нейродегенеративные заболевания, катаракту, рак молочной железы, легких, печени, толстой кишки, простаты, яичников и головного мозга [2, 3]. Было показано, что окислительный стресс, являясь причиной или следствием патологии, вовлечен в патогенез более, чем 100 заболеваний [4, 5].
Избыточный синтез АФК может быть результатом изменений во многих процессах, например, окислительном фосфорилировании, химических реакциях с участием ионов переходных металлов, оксидазной активности, фолдинге белков, катаболизме полиаминов и тимидина. Окислительное фосфорилирование и реакции с участием переходных металлов будут рассмотрены позже. Рассмотрим продукцию АФК в процессе фолдинга белков (1) и катаболизме полиаминов (2) и тимидина (3) [1].
1) Изменения в процессе фолдинга белков приводят к накоплению неправильно свернутых и развернутых белков в просвете эндоплазматического ретикулума (ЭПР), что негативно сказывается на ЭПР. Это нарушает клеточный гомеостаз и инициирует ответ развернутого белка (ОРБ) [6]. ОРБ способствует продукции АФК, а последние в свою очередь могут способствовать стрессорному воздействию на ЭПР [7].
2) Катаболизм полиаминов включает циклы взаимопревращения, при которых спермин разлагается до спермидина и спермидин до путресцина с образованием токсичных альдегидов и АФК. Например, сперминоксидаза катализирует превращение спермина в спермидин, что сопровождается выделением 3-аминопропаналя и H2O2 [8].
3) Тимидинфосфорилаза (ТФ) – скорость-лимитирующий фермент, участвующий в катаболизме тимидина. ТФ катализирует обратимое превращение тимидина в тимин и 2-дезокси-D-рибозо-1-фосфат (2dDR1P). ТФ активируется во многих опухолях и играет важную роль в ангиогенезе, противодействию апоптоза, инвазии и метастазировании, а также в ответе на химиотерапию [9]. Недавно Tabata et al. обнаружили, что активность TФ повышает уровни NADPH через пентозофосфатный путь, который активирует NADPH-оксидаза-зависимую продукцию АФК в раковых клетках [10]. Ранее было показано, что добавление тимидина к клеточной линии карциномы со сверхэкспрессией ТФ вызывает клеточный окислительный стресс [11]. Авторы предложили еще один потенциальный механизм продукции АФК, вызванного ТФ. Этот механизм основан на избыточной продукции 2dDR1P, образующемся во время фосфорилирования тимидина, которое может быть катализировано переходным металлом, что приводит к образованию АФК.
Активные формы кислорода и азота включают вещества как радикальной, так и нерадикальной природы: перекись водорода (H2O2), супероксид-анион $\left( {{\text{O}}_{2}^{{\centerdot - }}} \right),$ синглетный кислород (1O2), гидроксильный радикал (OH•), хлорноватистая кислота (HOCl), бромноватистая кислота (HOBr), гипотиоциановая кислота (HOSCN), оксид азота (NO•) и пероксинитрит (ONOO–) [1–4].
АФК образуются, когда наши клетки вырабатывают энергию из пищи, а также когда мы подвергаемся воздействию бактериальных и вирусных инфекций, интенсивных физических нагрузок, ксенобиотиков, сигаретного дыма, алкоголя, ионизирующего и ультрафиолетового излучений, пестицидов, озона и др. [3, 12].
АФК сконцентрированы практически во всех внутри- и внеклеточных структурах и жидкостях: в митохондриях, цитозоле, одиночных мембраносвязанных органеллах (пероксисомах, эндосомах и фагосомах), экзосомах, высвобождаемых плазматическими мембранами, и внеклеточных жидкостях, включая плазму. Наиболее важными источниками АФК являются митохондрии (рис. 1) ввиду происходящего переноса электронов по цепи переноса электронов [13, 14]. Супероксидный радикал $\left( {{\text{O}}_{2}^{{\centerdot - }}} \right)$ продуцируется в ряде участков митохондрии, включая комплекс I (сайты IQ и IF), комплекс III (сайт IIIQo), а также в процессе ферментативных реакций с глицерол-3-фосфатдегидрогеназой, Q-оксидоредуктазой, пируватдегидрогеназой и 2-оксоглутаратдегидрогеназой [15]. В процессе работы каталитического цикла цитохрома (ферменты данного цикла метаболизируют липиды, стероидные гормоны, ксенобиотики и т.д.) образуются супероксидный радикал и перекись водорода [16]. Кроме того, было показано, что некоторые другие белки млекопитающих, такие как NADH-цитохром b5 редуктаза [17], дигидрооротатдегидрогеназа [18], комплекс II (сукцинатдегидрогеназа) [19] и моноаминоксидазы (МАО) [20], генерируют АФК в митохондриях в процессе биохимических реакций.
Рис. 1.
Участие АФК экзогенного и эндогенного происхождения в различных клеточных процессах: защита, повреждение клетки, передача сигнала о повреждении для отдаленных тканей, адаптации и т.д.
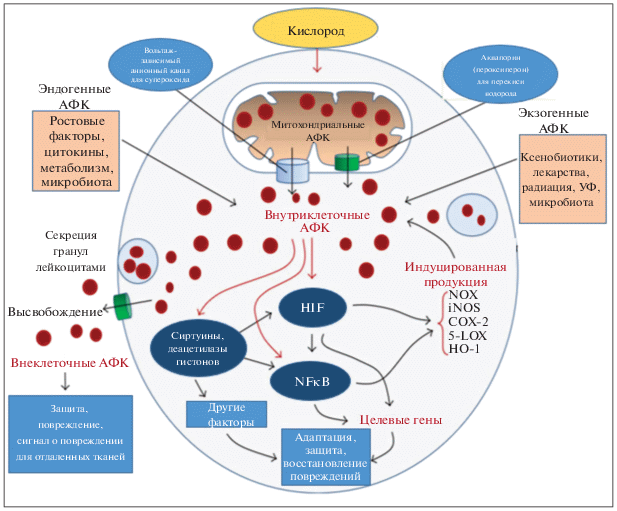
Рис. 2.
Механизм синтеза АФК фагоцитирующими клетками. Реакция 1 – gp91phox приобретает конформацию, способную передавать электрон, полученный от цитоплазматического NADPH, на молекулу кислорода О2. В результате этой реакции образуется короткоживущий супероксид-анион $\left( {{\text{O}}_{2}^{{\centerdot - }}} \right).$ Реакция 2 – супероксид-анион взаимодействует с H+, в результате чего образуется H2O2, обладающая выраженной антимикробной активностью. Эта реакция протекает в присутствии фермента супероксиддисмутазы. Реакция 3 – в присутствии миелопероксидазы формируются производные галогенов. При наличии H+ и Cl– реакция протекает с образованием хлорноватистой кислоты (HOCl), которая обладает выраженным антимикробным эффектом. Реакция 4 – НOCl способна окисляться H2O2 с образованием синглетного кислорода (1O2). Реакция 5 – НOCl может взаимодействовать с ${\text{O}}_{2}^{{\centerdot - }},$ в результате чего образуется гидроксильный радикал (HO•) – один из самых токсичных производных кислорода. Реакция 6 – спонтанная дисмутация в присутствии ионов железа (реакция Фентона). Реакция 7 – формирование пероксинитрита.
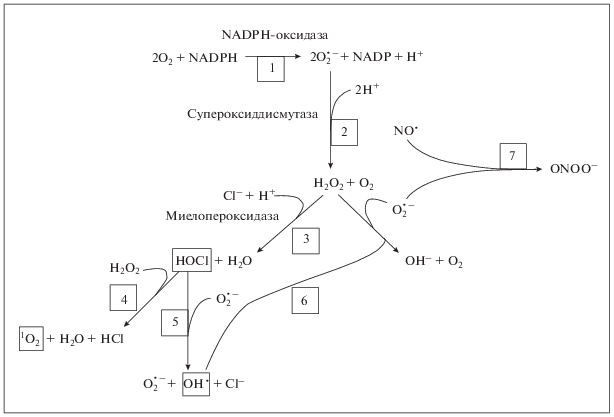
Рис. 3.
Окисление остатков цистеина активными формами кислорода и образование глутатионилированных белков. Цистеиновые остатки белка окисляются, при этом образуются сульфеновые группы. Последние могут образовывать дисульфидную связь с SH-группой другого цистеинового остатка. Сульфеновая группа и дисульфидная связь могут быть редуцированы с помощью различных антиоксидантов в клетках, таких как глутатион (GSH). Таким образом, белок может быть избирательно и обратимо окислен АФК, а такой измененный белок способен генерировать клеточные сигналы. Сульфеновая группа может быть в дальнейшем окислена с образованием сульфиновой и сульфонильной групп, которые не могут быть восстановлены в нормальной внутриклеточной системе.
Немитохондриальными источниками свободных радикалов являются:
1) реакция Фентона – реакция пероксида водорода с ионами железа (приведена цепочка превращений):
2) Фентон-подобные реакции (цепочка превращений):
Стоит отметить, что часть авторов понимают под Фентон-подобными реакциями взаимодействие между окислителем, отличным от перекиси водорода, с переходными металлами (кроме железа), такими как медь, хром, никель, кобальт и т.д. [22].
3) микросомальная система окисления, использующая цитохром P450,
4) пероксисомальное бета-окисление,
5) респираторный взрыв фагоцитирующих клеток [23].
Воспалительные индуцибельные ферменты (NOX (КФ 1.6.3.1)), индуцибельная синтаза оксида азота (iNOS) (КФ 1.14.13.39), индуцибельная циклооксигеназа (COX2) (КФ 1.14.99.1), 5-липоксигеназа (КФ 1.13.11.34) и индуцибельная гемоксигеназа-1 (HO-1) (КФ 1.14.99.3) могут вызывать дополнительный выброс АФК [13]. Образованные в процессе ферментативных реакций с участием миелопероксидазы, эозинофильной пероксидазы АФК высвобождаются во внеклеточное пространство путем их секреции активированными лейкоцитами или путем прохождения АФК через плазматическую мембрану по анионным каналам (супероксиды) или аквапоринам (гидропероксиды) [13, 14, 23]. Внеклеточные АФК важны для защиты, например, как АФК в случае высвобождения эозинофилами в борьбе с макропаразитами и вызывающими повреждение не только соседних здоровых тканей, но и отдаленных тканей и органов, сигнализируя о локальном повреждении и активируя механизмы адаптации и хроническое повреждение [13]. Умеренные количества митохондриального супероксида и перекиси водорода играют важную роль в ряде клеточных сигнальных процессов и могут активировать сигнальные пути, которые способствуют выживанию клеток и устойчивости к болезням вследствие гормезиса (стимулирующее действие умеренных доз стрессоров) [24].
Продукция O2–, H2O2, HOCl активированными фагоцитами является классическим примером запрограммированной метаболической генерации АФК для обеспечения гомеостаза организма в борьбе с антигенами [15, 25, 26]. Главным событием образования АФК фагоцитами является сборка ферментативного комплекса NADPH-оксидазы (NOX) (КФ 1.6.3.1), локализующегося преимущественно на цитоплазматической мембране и в некоторых органеллах [27]. Основой NOX являются две мембранные единицы: p91phox и p22phox (phox –phagocyte oxidase, что подчеркивает фагоцитарную роль фермента). Субъединица p91phox содержит участок связывания NADPH и простетическую группу FAD в C-концевой части. В процессе активации NOX к мембранным субъединицам присоединяются цитоплазматические субъединицы (p40phox, p47phox, p67phox), а также малый G-белок Rac1 [27]. Процесс сборки комплекса начинается с белка p47phox, который заранее должен быть фосфорилирован протеинкиназой С. Данный процесс запускают сигналы от рецепторов фагоцитов, отвечающих за распознавание патогенов. В результате сборка ферментативного комплекса приводит к тому, что gp91phox приобретает конформацию, которая способна передавать электрон, полученный от цитоплазматического NADPH, на молекулу кислорода О2. В результате этой реакции образуется супероксид-анион, обладающий низкой антибактериальной активностью [27]. В последующей реакции в присутствии супероксиддисмутазы супероксид-анион взаимодействует с H+ в присутствии супероксиддисмутазы, в результате чего образуется перекись водорода, которая обладает выраженной антимикробной активностью. H2O2 вызывает окисление SH-групп белков и способствует перекисному окислению ненасыщенных жирных кислот. В присутствии миелопероксидазы формируются производные галогенов (например, хлорноватистая кислота, обладающая выраженным антимикробным эффектом) [27]. Так при взаимодействии HOCl с аминокислотами происходит образование хлораминов, обладающих микробоцидным действием. НOCl может окисляться H2O2 с образованием синглетного кислорода (1O2), который окисляет полиненасыщенные жирные кислоты, в результате чего происходит разрушение поверхностной мембраны микроорганизмов [27]. Хлорноватистая кислота способна взаимодействовать с супероксид-анионом, что приводит к образованию гидроксильного радикала. OH• образуется также в процессе спонтанной дисмутации, которая протекает в присутствии ионов железа [27]. Гидроксильный радикал – один из самых токсичных производных кислорода: он разрушает нити ДНК и пептидные связи внутри белковых молекул, окисляет сульфгидрильные группы и т.д. ${\text{O}}_{2}^{{\centerdot - }}$ участвует в формировании пероксинитрита (OONO–), который окисляет SH-группы различных молекул. Кроме того, при взаимодействии супероксид-аниона с водой формируется молекула озона (О3), обладающего широким микробоцидным действием [27].
Необходимо рассмотреть характерные черты и биологические функции основных представителей АФК и АФА.
ПЕРЕКИСЬ ВОДОРОДА (H2O2)
Перекись водорода является основным видом АФК, образующимся в качестве побочного продукта во время разнообразных клеточных процессов и являющимся конечным продуктом многочисленных метаболических реакций. H2O2 способна передавать клеточные сигналы, но в избытке может наносить вред клеткам и тканям. H2O2 является сильным двухэлектронным окислителем [28]. Она является более сильным окислителем, чем хлорноватистая кислота или пероксинитрит, но в отличие от этих высокореактивных веществ H2O2 имеет высокий энергетический барьер активации, который необходимо преодолеть для проявления окислительной способности. Переходные металлы, такие, как железо(II) и медь(I), могут расщеплять связь O–O H2O2 с образованием гидроксильных радикалов или комплексных соединений с металлом [28].
Перекись водорода является продуктом таких многочисленных метаболических процессов, как спонтанная дисмутация супероксидных радикалов, катаболизм полиаминов, тимидина, каталитический цикл цитохрома, работой таких ферментов, как NOXs, моноаминоксидазы, лизилоксидазы, дигидрооротатдегидрогеназа, пероксисомальные ферменты (ацил-КоА-оксидазы, d-аминокислотная оксидаза, d-аспартат-оксидаза и др.), система микросомальной монооксигеназы, фолдинг белков (ответ развернутого белка), метаболизм полиненасыщенных жирных кислот [1].
Перекись водорода способна участвовать в таких окислительных процессах, как деградация гемовых белков, выделение железа, инактивация ферментов и окисление ДНК, липидов, селенопротеинов, тиоловых групп и кетокислот [29].
Было показано, что добавление экзогенной Н2О2 способствует активации внутриклеточных процессов, сопутствующих рецептор-зависимой стимуляции клеток. В эндотелиальных клетках низкие дозы Н2О2 активируют эндоцитоз, как показано на примере флуоресцентно меченного декстрана. Н2О2 также активирует сигнальные каскады тирозинкиназных рецепторов [30]. Внеклеточно добавленный Н2О2 подобно инсулину активировал PI3-киназу и ее мишень – киназу Аkt/РКВ – в инсулин-чувствительных преадипоцитах 3Т3-L1 [30].
Небольшие количества Н2О2 (1–10 µМ) не вызывают видимых повреждений клетки, но в то же время оказывают митогенный эффект: при добавлении к смешанной культуре лимфоцитов 10 µМ Н2О2 наблюдался значительный рост включения Н3-тимидина (маркера пролиферативной активности клеток) [30].
ГИДРОКСИЛЬНЫЙ РАДИКАЛ (OH•)
Источники гидроксильного радикала – реакции с переходными металлами (реакция Фентона и Фентон-подобные реакции), пероксисомы, реакция HOCl и OH–, метаболизм тимидина [1]. ОН• – высокоагрессивный вид радикалов, ответственный за окислительное повреждение большинства биомолекул [1]. Агрессивные гидроксильные радикалы, возникающие в результате Фентон-подобных реакций, могут также образовываться в результате радиолиза воды. Сообщалось, что ОН• является самым мощным окислительным радикалом, который может взаимодействовать в месте своего образования с большинством органических и неорганических молекул – ДНК, белков, липидов, аминокислот, сахаров и металлов [13]. Эти реакции характеризуются высокой скоростью из-за реактивности и короткого периода существования гидроксильных радикалов (период полураспада гидроксильных анионов составляет 10–9 секунд). Гидроксильный радикал – высокотоксичное вещество для клетки. Биологическая роль ОН• неизвестна [13].
СУПЕРОКСИД-АНИОН $\left( {{\text{O}}_{2}^{{\centerdot - }}} \right)$
Основными источниками супероксид-аниона являются митохондриальная цепь переноса электронов и мембраносвязанная NOX, а также супероксиддисмутазы, каталитический цикл цитохрома, митохондриальные ферменты (глицерол-3-фосфатдегидрогеназа, 2-оксоглутаратдегидрогеназа, NADH-цитохром b5 редуктаза и др.), ксантиноксидоредуктаза [1].
Разнообразные гормоны, включая факторы роста (тромбоцитарный фактор роста (PDGF), эпидермальный фактор роста (EGF), инсулин и IGF) и цитокины (ФНО-α и ангиотензин II) могут стимулировать выработку ${\text{O}}_{2}^{{\centerdot - }}$ NADPH-оксидазами неиммунных клеток [31]. ${\text{O}}_{2}^{{\centerdot - }},$ образованный NOX, затем быстро превращается в H2O2 с помощью SOD.
Митохондриальный ${\text{O}}_{2}^{{\centerdot - }}$ участвует в таких процессах, как
1) инактивация митохондриальной аконитазы (редокс-регулируемый фермент цикла Кребса) посредством разрушения железо-серного кластера данного фермента и
2) индукция митохондриального перекисного окисления липидов (данный процесс происходит после реакций Н2О2 с цитохромом с, что в свою очередь приводит к цитозольному высвобождению проапоптотических факторов [13]) [32].
После синтеза супероксид подвергается либо быстрой детоксикации с помощью митохондриальной MnSOD (Mn-зависимой супероксиддисмутазы (КФ 1.15.1.1)) (MnSOD также разрушает перекись водорода), либо транспорту через митохондриальную мембрану, при этом супероксид проходит через вольтаж-зависимый анионный канал (митохондриальный порин) [13].
СИНГЛЕТНЫЙ КИСЛОРОД (1O2)
Синглетный кислород (1O2) – это электронно-возбужденная форма кислорода [33, 34].
Различные нефотосенсибилизированные механизмы его образования встречаются в биологических системах, но важность эндогенного образования синглетного кислорода имеет противоречивую историю [34]. Совсем недавно было показано, что антитела или аминокислоты катализируют превращение синглетного кислорода (1O2) в озон (O3) и что эта реакция происходит во время уничтожения бактерий активированными нейтрофилами [33].
Под действием солнечного света происходит генерация окислителей электронным возбуждением. Фотовозбуждение эндогенных или экзогенных молекул сенсибилизатора (фотосенсибилизация) приводит к образованию реакционноспособных частиц, в частности синглетного молекулярного кислорода, электронно-возбужденных карбонилов и супероксидных анионных радикалов; это может вызвать молекулярное повреждение. Волнами, имеющими биологическое значение, являются ультрафиолетовое бета-излучение (290–320 нм) и ультрафиолетовое альфа-излучение (320–400 нм). Известно, что видимый свет и даже инфракрасное альфа-излучение вызывают фотобиологические реакции [33].
С точки зрения здоровья человека, наиболее подвержены воздействию ультрафиолета такие ткани, как кожа и глаза. Синглетный кислород опосредует общую делецию митохондрий (образуются мутации в ДНК митохондрий), связанную с фотостарением [33]. Одним из генерирующих синглетный кислород фотосенсибилизаторов является липофусцин, который образуется в пигментном эпителии сетчатки с возрастом или в связи с такими генетическими нарушениями, как болезнь Старгардта [33]. Основным продуктом окисления митохондриальной ДНК является 7,8-дигидро-8-оксогуанин (8-oxoG), а синглетный кислород способствует преимущественно образованию этого соединения в ДНК. Хотя реакция гидроксильного радикала с ДНК также приводит к образованию некоторого количества 8-oxoG, однако эта реакция малозначима. Кроме того, генерация синглетного кислорода в присутствии карбонилов, аминокислот и белков в митохондрии обеспечивает среду для генерации митохондриального озона, который может способствовать разрывам двухцепочечной митохондриальной ДНК [33].
ПЕРОКСИНИТРИТ (OONO–)
Пероксинитрит (ONOO–) является сильным биологическим окислителем, образуется в результате диффузионно-ограниченной реакции оксида азота (NO•) и супероксид-аниона $\left( {{\text{O}}_{2}^{{\centerdot - }}} \right)$ [35]. Его относительно большой биологический период полураспада и высокая реакционная способность позволяют пероксинитриту окислять ряд различных мишеней в клетке [36]. В физиологических условиях пероксинитрит может непосредственно реагировать с тиолами, или радикальные продукты разложения пероксинитрита могут косвенно окислять другие клеточные компоненты, например, липиды, белки и ДНК. Окисленные модификации, образовавшиеся под действием пероксинитрита, запускают гибель клеток различными механизмами в зависимости от концентрации окислителя [36]. Пероксинитрит стимулирует некроз, апоптоз, аутофагию, партанатоз (форма программируемой клеточной гибели, характеризующаяся гиперактивацией поли(АДФ-рибоза)полимеразы 1-белка, участвующего в клеточном ответе на повреждение ДНК) и некроптоз [36]. Помимо концентрации пероксинитрита вид клеточной гибели определяется также типом клеток. Низкая концентрация пероксинитрита активирует сигнальные пути гибели клеток, что приводит к апоптозу. Высокая концентрация пероксинитрита стимулирует некроз. Пероксинитрит может активировать различные виды гибели клеток в одном типе клеток, например, апоптоз и партанатоз в корковых нейронах [36].
ОКСИД АЗОТА(II) (NO•)
Оксид азота – молекула с неспаренным электроном, довольно слабый окислитель. NO• вырабатывается из L-аргинина тремя основными изоформами синтазы оксида азота (NOS): эндотелиальной NOS (eNOS), связанной с вазодилатацией и регуляцией сосудов; нейрональной NOS (nNOS), которая связана с внутриклеточной передачей сигналов; и индуцибельной NOS (iNOS), которая имеет различные функции в зависимости от метаболических потребностей [37, 38]. В то время как выработка оксида азота с помощью nNOS и eNOS жестко регулируется кальцием с помощью кальмодулин-зависимого механизма, iNOS активируется в ответ на различные сигналы эндотоксина или цитокинов, что может привести к быстрому образованию больших потоков оксида азота [38].
Взаимодействие NO с окислителями и восстановителями может привести к появлению таких АФА, как нитроксильный анион (NO–; образуется при восстановлении NO•), высшие оксиды азота (NO2, N2O4 и т.д.), S-нитрозотиолы (RSNO), и динитрозильные комплексы с Fe. Удаление одного электрона NO• приводит к образованию катиона нитрозония (NO+), который способен реагировать с нуклеофильными центрами, образуя нитрозосоединения. Нитрозилгалогениды выделяются, когда NO• реагирует с фтором, хлором или бромом [37].
Зависимая от оксида азота продукция цГМФ имеет широкий спектр мишеней, играет роль в регуляции ряда функций в нервной системе. Однако не все функции оксида азота опосредованы продукцией цГМФ [38]. Окислительные продукты оксида азота вскоре были обнаружены в макромолекулах и ряде белков. Было описано несколько механизмов нитрозилирования, включая окислительное S-нитрозирование, транс-нитрозилирование [38]. Нитрозилирование регулирует разнообразные процессы, происходит в функционально разнообразной группе белков в различных субклеточных структурах [38].
ХЛОРНОВАТИСТАЯ КИСЛОТА (HOCl)
Фермент миелопероксидаза превращает H2O2 и хлорид-ионы в HOCl, высокоактивная форма хлора, реагирующая с большинством биологически значимых соединений. HOCl (включая ее соль – гипохлорит натрия (NaOCl)) в качестве молекулярного (2-электронного) агента обладает сильной окислительной и хлорирующей способностью для обеспечения ярко выраженных бактерицидных и цитотоксических свойств [39]. Это делает HOCl одним из самых мощных окислителей in vivo [39].
БРОМНОВАТИСТАЯ КИСЛОТА (HOBr)
Эозинофильная пероксидаза (главный основный белок) и миелопероксидаза являются металлопротеидами, которые могут катализировать выработку HOBr [40, 41]. Реакция HOBr с гликозаминогликанами (гепарансульфатом, гепарином, хондроитинсульфатом и гиалуронаном) дает производные полимера N-бромпроизводные (бромамины, дибромамины, N-бромсульфонамиды и бромамиды). Разложение этих частиц, которое может происходить самопроизвольно и/или путем одноэлектронного восстановления низковалентными ионами переходных металлов (Cu+ и Fe2+), приводит к фрагментации и модификации полимера [40]. Реакция HOBr с белками приводит к генерации производных N-брома, а также к фрагментации полипептидного остова [40]. Реакция HOBr с внеклеточным матриксом, синтезируемым гладкомышечными клетками in vitro, индуцирует высвобождение углеводных и белковых компонентов. Разложение гликозаминогликанов и белков внеклеточного матрикса HOBr может способствовать повреждению ткани, связанному с такими воспалительными заболеваниями, как астма [40].
HOBr и HOCl окисляют тиолы, они также эффективно воздействуют на амины с образованием галоаминов в боковых цепях белка, свободных аминокислотах, нуклеиновых кислотах и аминосодержащих фосфолипидах или с образованием галоамидов на боковых цепях гликозаминогликанов [41]. Эти галоамины и галогенамиды сохраняют окислительную способность исходной гипогалогенной кислоты и, следовательно, могут генерировать последующие окислительные модификации [41]. HOBr и HOCl также эффективно реагируют с ароматическими кольцами в таких аминокислотах, как тирозин, триптофан и нуклеиновые кислоты. Их реакция с тирозином с образованием соответствующих 3-галотирозина и 3,5‑дигалотирозина количественно незначительна, но примечательна как специфический биомаркер окислительного повреждения, вызванного HOBr и HOCl [41].
ГИПОТИОЦИАНОВАЯ КИСЛОТА (HOSCN)
Гипотиоциановая кислота (HOSCN) вырабатывается из тиоцианата (SCN–) в биологических системах под действием ферментов пероксидазы [42, 43].
(1)
${{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}} + 2{\text{SC}}{{{\text{N}}}^{ - }} \to 2{{{\text{H}}}_{{\text{2}}}}{\text{O}} + {{\left( {{\text{SCN}}} \right)}_{2}}.$(2)
${{\left( {{\text{SCN}}} \right)}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{HOSCN}} + {{{\text{H}}}^{ + }} + {\text{SC}}{{{\text{N}}}^{ - }}.$(3)
${\text{HOSCN}} \leftrightarrow {\text{OSC}}{{{\text{N}}}^{ - }} + {{{\text{H}}}^{ + }}.\,\,\,[42].$HOSCN играет важную роль в защитных механизмах млекопитающих благодаря своим антибактериальным свойствам. HOSCN также может генерироваться in vivo посредством реакции SCN– с HOCl и HOBr. SCN– присутствует в миллимолярных уровнях в таких биологических жидкостях, как слюна, молоко и слеза. SCN– является основным субстратом для лактопероксидазы и близкородственной пероксидазы слюны, предпочтительным субстратом для миелопероксидазы и пероксидазы эозинофилов и также легко окисляется пероксидазой желудка и пероксидазой щитовидной железы [42].
Тиолы с низкой молекулярной массой, например, восстановленный глутатион GSH, являются важными мишенями для HOSCN. Это приводит к образованию нестабильных производных сульфенилтиоцианата (RS–SCN) [42]. При физиологическом рН эти вещества легко реагируют с другими молекулами тиола с образованием дисульфидов. Окисленный глутатион (GSSG) является единственным продуктом, наблюдаемым при обработке GSH с помощью HOSCN [42]. HOSCN реагирует преимущественно с тиоловыми группами белков, как было показано в исследованиях с изолированными белками и биологическими жидкостями, например, плазмой. Это приводит к генерации производных белка, содержащих группу RS–SCN, о чем свидетельствует обратимое включение в продукты реакции 14C из HOS14CN [42].
HOSCN часто называют относительно мягким окислителем, который безвреден для клеток млекопитающих. Сообщается, что присутствие SCN– и образование HOSCN пероксидазами является важным механизмом детоксикации, ответственным за удаление потенциально более вредных окислителей, таких как H2O2 или HOCl [42]. Однако HOSCN может вызывать лизис эритроцитов, действовать как вирулицидный агент, вызывать остановку роста или ингибировать деление клеток в ряде бактериальных клеток, ингибировать гликолиз и дыхание и снижение поглощения глюкозы [42]. Способность HOSCN вызывать гибель клеток через апоптоз и некроз, а также экспрессию тканевого фактора и молекул адгезии моноцитов показывает, что этот окислитель обладает значительным вредным воздействием.воздействие на клетки млекопитающих [42]. В частности, индукция ICAM-1 (Inter-Cellular Adhesion Molecule 1, также CD54), VCAM-1 (Vascular cell adhesion molecule 1, “васкулярная молекула клеточной адгезии 1”) и E-селектина (гликопротеин, находящийся на клеточной поверхности, относится к классу молекул клеточной адгезии; рецептор к некоторым углеводным лигандам лейкоцитов крови), вероятно, играет значительную роль в развитии атеросклеротического поражения и поддерживает недавние исследования, которые предоставляют in vivo доказательства участия SCN– – производных в патогенезе заболеваний коронарных артерий [42, 43].
Таким образом, существует большое разнообразие форм АФК и АФА, которые обладают уникальными характеристиками и метаболическими особенностями.
Клетки имеют антиоксидантные защитные механизмы, позволяющие инактивировать чрезмерно продуцируемые АФК и АФА. Баланс между продукцией и устранением АФК и АФА приводит к внутриклеточному гомеостазу, в то же время чрезмерное отклонение концентраций АФК и АФА в ту или иную сторону приводит к повреждению клетки, что в дальнейшем может приводить к различным негативным последствиям (в т.ч. их гибели, трансформации в опухолевую клетку, инсулинорезистентности и т.д.) [1, 3, 13, 14]. В умеренных концентрациях данные свободные радикалы являются посредниками реакций, с помощью которых уничтожаются поврежденные клетки и продукты их распада выводятся из организма [3]. В случае уменьшения концентрации АФК и АФА в организме есть вероятность столкнуться с изменением в таких необходимых защитных механизмах, как апоптоз, фагоцитоз и детоксикация [13]. Хорошо известно, что АФК и АФА в низких концентрациях являются сигнальными молекулами, модулирующими пролиферацию клеток и экспрессию генов посредством активации таких факторов транскрипции, как NF-каппа-B и индуцируемый гипоксией фактор-1α (HIF-1α) [23]. Повышение продукции АФК в результате действия на клетку различных провоспалительных молекул (ФНО- α, липополисахариды, тромбин) может опосредовать активацию NF-κB и последующую индукцию воспалительных генов, изменять активность протеасом, транскрипцию антиоксидантных генов, активацию воспалительных процессов и секрецию цитокинов (например, митоген-активируемые протеинкиназы (MAPKs) (КФ 2.7.11.24), которые активируют факторы транскрипции AP-1, и IκB киназы (IKK), которые затем осуществляют активацию NF-каппа-B и интерлейкина-1-бета (IL-1β)) [1, 3, 13, 23]. Провоспалительные цитокины, ФНО-α, IL-1β и IFN-γ, могут дополнительно усиливать окислительный стресс у человека, вызывая выработку АФК [23]. Кроме этого, АФК играют важную роль в процессах передачи сигналов клеток сосудов, включая активацию эндотелиальной синтазы оксида азота (eNOS) (КФ 1.14.13.39) (H2O2 индуцирует экспрессию гена eNOS в эндотелиальных клетках посредством Ca2+/кальмодулин-зависимой протеинкиназы II/янус киназа 2 (нерецепторная тирозинканаза)-зависимого пути) и стимуляцию роста и миграцию клеток посредством модуляции внутриклеточного кальция, активации NF-каппа-B и p38 митоген-активируемой протеинкиназы (p38MAPK) ((КФ 2.7.11.24) и протеинкиназы B (PKB) (КФ 2.7.11.1) [5, 10, 11].
АФК И ФЕРРОПТОЗ
АФК задействованы в многочисленных клеточных процессах, это подтверждается еще одной их важной ролью: активация апоптоз-независимой клеточной смерти – ферроптоза [44]. Ферроптоз является регулируемой формой гибели клеток, вызванной потерей активности фермента репарации липидов глутатионпероксидазы 4 (GPX4) и последующим накоплением активных форм кислорода на основе липидов (АФК), в частности гидропероксидов липидов (липидов, подвергшихся окислению перекисью водорода). Эта форма железозависимой гибели клеток генетически, биохимически и морфологически отличается от других форм гибели клеток (апоптоза, нерегулируемого некроза и некроптоза) [44].
Активация митохондриальных вольтаж-зависимых анионных каналов и MAPKs, стрессорное воздействие на эндоплазматический ретикулум и ингибирование цистин – глутаматного антипортера вовлечены в индукцию ферроптоза [45, 46]. Этот процесс характеризуется накоплением продуктов перекисного окисления липидов (ППОЛ) и АФК, образовавшихся в результате метаболизма железа (реакция Фентона). ППОЛ оказывают токсическое воздействие через два основных механизма. Поскольку липиды отвечают за поддержание целостности клеточных мембран, ППОЛ изменяют состав, структуру и динамику липидных мембран. Будучи высокореактивными соединениями, ППОЛ также могут способствовать большему образованию АФК или превращаться в реакционноспособные соединения, способные сшивать ДНК и белки [46]. Ферроптоз может быть фармакологически ингибирован хелаторами железа (например, дефероксамином и мезилатом десферриоксамина) и ингибиторами перекисного окисления липидов (например, ферростатином, липроксстатином) [46]. Глутатионпероксидаза 4, белок теплового шока бета-1 и эритроидный ядерный фактор (Nrf2) функционируют как подавляющие ферроптоз факторы, ограничивая выработку АФК и снижая клеточное поглощение железа. Напротив, NADPH-оксидаза и p53 (особенно мутантный p53 с дефектом ацетилирования) [45] действуют в качестве активаторов ферроптоза, стимулируя продукцию АФК и ингибируя экспрессию SLC7A11 (специфическая легко-цепочечная субъединица цистин/глутаматного антипортера) соответственно [46].
АФК КАК ВНУТРИКЛЕТОЧНЫЕ МЕССЕНДЖЕРЫ
АФК способны действовать как внутриклеточные мессенджеры, т.е. изменять внутриклеточное окислительно-восстановительное состояние и/или структуру и функцию белка путем модификации аминокислотных остатков, а редокс-состояние ряда белков может влиять на клеточный метаболизм [47–49]. Среди АФК перекись водорода представляет собой небольшой, незаряженную, свободно диффундирующую молекулу, которая может быть синтезирована и быстро разрушена в ответ на внешние раздражители. Эти свойства молекулы соответствуют всем важным критериям внутриклеточного мессенджера. Многочисленные исследования подтверждают тот факт, что перекись водорода – это повсеместный внутриклеточный мессенджер [47–50].
Другой вид АФК, гидроксильный радикал (OH•), является наиболее реакционноспособным ввиду его наименьшего периода полураспада в тканях (10–9 с) по сравнению с другими АФК [1]. Он окисляет практически любой клеточный компонент, с которым сталкивается, и является токсичным для клетки соединением из-за высокой вероятности окислительного повреждения. Ввиду этой реакционной способности и отсутствия специфичности считается, что гидроксильный радикал не играет сигнальной роли в клетках [49].
Перекись водорода – это наиболее стабильная форма АФК, с предполагаемым периодом полураспада в клетках примерно 1 мс. H2O2 способна диффундировать через мембраны. Клетки, по-видимому, регулируют транспорт H2O2 путем изменения липидного состава мембран, тем самым поддерживая клеточную концентрацию H2O2 [51]. Мембранный транспорт перекиси водорода дополнительно облегчается аквапориновыми каналами [52, 53].
Таким образом, данные о молекуле дают основание предположить, что перекись водорода является основной формой АФК, участвующей в окислительно-восстановительной передаче сигналов у эукариот. Рассмотрим отличия клеточных мишеней супероксида и перекиси водорода. В клетках бактерий одной из мишеней супероксида являются железо-серные кластеры в белках, как это видно при супероксид-опосредованной инактивации митохондриального фермента аконитазы (КФ 4.2.1.3) [54]. Известно, что различные железо-серные белки участвуют в репликации, транскрипции и репарации ДНК [55]. Еще один представитель семейства АФК, супероксид-анион, несет отрицательный заряд, что ограничивает его способность к диффузии через мембраны [49]. “Однако существуют доказательства того, что супероксид может транспортироваться из митохондрий в цитоплазму с помощью вольтаж-зависимого анионного канала” [49]. Супероксид обладает высокой реакционной способностью в отношении железо-серных кластеров в белках и реагирует с ними со скоростью, ограниченной только диффузией [49]. Данные реакции могут способствовать высвобождению свободного железа и вызывать структурные изменения, что в результате приводит к изменению активности белка. В растворах очищенных белков супероксид реагирует с остатками цистеина с образованием тиильного радикала [49]. Последующая реакция тиильного радикала с кислородом приводит к образованию супероксида, который затем превращается в пероксид водорода. In vivo скорость спонтанной или катализируемой ферментами реакции дисмутации супероксида в пероксид водорода очень высокая, что делает участие супероксида в окислении тиолов белков маловероятным способом окислительно-восстановительной сигнализации [49].
Супероксид может реагировать с оксидом азота с образованием пероксинитрита с различными последствиями. Основной мишенью окислительно-восстановительной передачи сигнала перекисью водорода являются остатки цистеина в белках [49].
Одним из правил в путях передачи сигнала является то, что образуемые модификации регуляторных молекул должны быть обратимыми; то есть каждый компонент клеточной сигнализации имеет два возможных состояния: включенное и выключенное. Двухэлектронное окисление остатков цистеина перекисью водорода приводит к образованию сульфеновой кислоты. Это промежуточное соединение может образовывать меж- или внутримолекулярные дисульфидные связи или образовывать ковалентную связь с глутатионом. Тирозинфосфатазы являются хорошо известными мишенями для перекиси водорода. Эти белки обычно отключают пути передачи сигналов тирозинкиназы и, следовательно, их инактивация может приводить к передаче сигналов [36, 49].
Второе правило в путях передачи сигнала – это специфичность. Центральные вопросы в области окислительно-восстановительного баланса в отношении этого правила:
1) какие белки нацелены на АФК для инициирования сигнального пути;
2) как достигается специфичность.
Стоит подчеркнуть одно ключевое различие между окислительно-восстановительной передачей сигналов и традиционными путями передачи сигнала. Последние начинаются со связывания лиганда, например, факторов роста, цитокинов или стероидных гормонов с мембранными или внутриклеточными рецепторами. Гироксилирование крупных молекулярных структур приводит к индуцированному соответствию [49]. Напротив, окислительно-восстановительная передача сигналов инициируется через ковалентное взаимодействие, перекись водорода реагирует с тиольной группой определенного остатка цистеина [49]. В клетках содержится большое количество цистеин-содержащих белков, которые являются носителями большого количества потенциальных рецепторов, и это ставит вопрос о том, как можно достичь специфичности [49]. Ограничения, налагаемые критериями передачи сигналов, дают ответ на то, как достигается специфичность редокс-сигнализации: ключевые взаимодействия ограничены быстрыми реакциями, нацеленными на специфический цистеин в определенном белке [49]. Вероятность окисления уникального остатка цистеина в определенном белке зависит от концентрации этого остатка/белка в клетке, аминокислот, окружающих цистеин, расположения белка относительно источника образования перекиси водорода [49].
Пероксиредоксины (КФ 1.11.1.15) представляют собой семейство белков, которые проявляют особенно высокие константы скорости реакции с перекисью водорода, порядка 105–107 М– 1 с– 1 [56]. Они присутствуют в клетках в относительно высокой концентрации для белков: приблизительно 20 µМ [57]. Модельные исследования показывают, что уникальная белковая среда вокруг каталитического остатка цистеина в пероксиредоксине стабилизирует переходное состояние реакции, что делает эти белки особенно хорошо приспособленными к реакции с перекисью водорода [49]. Тиолы в белках частично окружены водной, но все же их основным окружением являются белковые структуры. При этом белковые аминокислотные остатки обеспечивают взаимодействия зарядов, диполей и водородных связей, которые окружают тиолы [49].
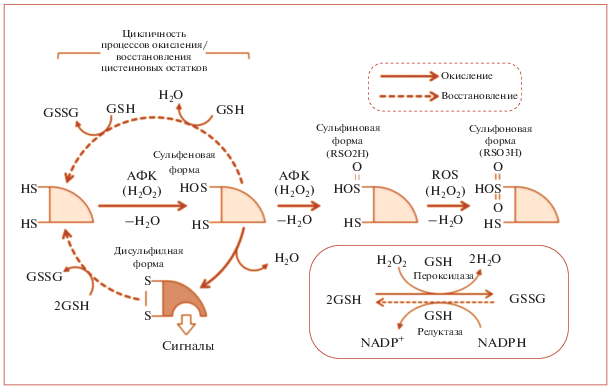
Высокая реакционная способность, относительно большое количество и распределение членов семейства пероксиредоксинов по разным клеточным компартментам соответствуют ожидаемым критериям для белков, которые участвуют в клеточных сигнальных путях. Расположение потенциальных мишеней относительно источника генерации перекиси водорода является еще одним компонентом специфичности в редокс-сигнализации [49]. Это иллюстрируется протеин-тирозин-фосфатазой (PTP1B) (КФ 3.1.3.48) и окислительно-восстановительной передачей сигналов после связывания лиганда с рецепторной тирозинкиназой. PTP1B должен конкурировать с пероксиредоксинами в качестве мишени для перекиси водорода [49]. Константы скорости реакции перекиси водорода с Prx2 (пероксиредоксином) и PTP1B составляют 2 × 107 и 20 М–1 с–1 соответственно [49]. Согласно “модели шлюза” (рис. 4), передача сигналов достигается посредством локальной инактивации пероксиредоксинов. Связывание лиганда с рецепторами тирозинкиназы приводит к фосфорилированию и инактивации Prx1. Переокисление таргетированного остатка цистеина в Prx2 превращает тиоловую группу в сульфиновую кислоту и инактивирует этот фермент. В результате могут накапливаться достаточно высокие уровни перекиси водорода для таргетирования белков сигнального пути (таких, как PTP1B) [49].
Рис. 4.
В так называемой модели шлюза преодолены кинетические ограничения окислительно-восстановительной сигнализации, а специфичность достигается благодаря непосредственной близости сигнальной мишени и источника генерации перекиси водорода [58]. Модель шлюза показана здесь с протеин-тирозин-фосфатазой 1B (PTP1B) в качестве сигнальной мишени. Пероксиредоксины (Prx) присутствуют в клетках в относительно высоких концентрациях и очень эффективно разрушают перекись водорода по сравнению со скоростью реакции PTP1B и перекиси водорода. Однако для окисления PTP1B может накапливаться достаточное количество перекиси водорода, тем не менее, после инактивации Prx1 фосфорилированием, вызванным активацией рецепторной тирозинкиназы факторами роста (ФР), остаток цистеина, принадлежащий Prx2, необратимо окисляется до сульфиновой кислоты (SO2H) [49].
АФК передают сигналы во многих метаболических путях, таких как:
1) пути, определяющие пролиферацию и срок жизни клеток через MAP-киназы, PI3-киназу (КФ 2.7.1.137), PTEN (phosphatase and tensin homolog deleted on chromosome 10, фосфатаза с двойной субстратной специфичностью) (КФ 3.1.3.67) и протеин-тирозин-фосфатазы (КФ 3.1.3.48) [59–61];
2) гомеостаз АФК и регуляция антиоксидантных генов с помощью Ref-1 (англ. redox factor-1), Nrf-2, тиоредоксина и др. [62, 63];
3) старение, процесс опосредован p66Shc (данный белок участвует в регуляции клеточного уровня АФК) [64];
4) реакция повреждения ДНК – через киназу ATM (англ. аtaxia telangiectasia mutated, серин/треониновая протеинкиназа, которая рекрутируется и активируется двунитевыми разрывами ДНК) (КФ 2.7.11.1); это может способствовать ингибированию mTORC1, что приведет к подавлению синтеза белка и активации аутофагии [65, 66];
5) гомеостаз железа – с помощью железорегуляторных белков (IRP) и чувствительных к железу элементов (IRE) и т.д. [67, 68];
АНТИОКСИДАНТНАЯ ЗАЩИТА КЛЕТОК. СТАРЕНИЕ
Антиоксидантная защита человека сложна и выполняет задачу установления физиологически-важного уровня АФК в клетке с возможностью функционирования клеточной передачи сигналов, в то же время минимизирует уровни АФК, чтобы не допустить окислительного повреждения.
Клеточное окислительно-восстановительное равновесие тщательно поддерживается эндогенной антиоксидантной защитной системой, которая включает эндогенные антиоксидантные ферменты и участники этих реакций, такие как супероксиддисмутаза (SOD) (КФ 1.15.1.1), каталаза (CAT) (КФ 1.11.1.6), глутатионпероксидаза (GPx) (КФ 1.11.1.9), глутатион (GSH), глутатионредуктаза (GR) (КФ 1.8.1.7), частично фермент глутатион-S-трансфераза (GST) (КФ 2.5.1.18), белки и неферментативные низкомолекулярные акцепторы (мочевая кислота, коэнзим Q и липоевая кислота). Данными свойствами обладают также экзогенные антиоксиданты: бутилированный гидроксианизол (BHA), бутилированный гидрокситолуол (BHT), пропилгаллат (PG) и трет-бутилгидрохинон (TBHQ) [3, 13, 23].
Антиоксидантную защиту в клетке можно разделить на два вида в соответствии с механизмом действия.
Первая группа – антиоксиданты, которые снижают возможность образования новых свободных радикалов или предотвращают развитие цепных реакций. Антиоксиданты имеют эндогенное и экзогенное происхождение и составляют гетерогенную группу, которая включает супероксиддисмутазу (SOD), каталазу (CAT) и глутатионпероксидазу (GPX), глутатион, альбумин, белки, которые связывают металлы (ферритин и церулоплазмин), ионы металлов (Se, Cu и Zn) [9, 29], витамины С и Е, каротиноиды и флавоноиды [9, 30]. Вторая группа – ферменты, которые восстанавливают повреждение, вызванное свободными радикалами. Это липазы, протеазы, ферменты репарации ДНК, трансферазы и метионин-сульфоксидредуктазы (КФ 1.8.4.11) [72–74].
Интересно отметить, что в физиологических условиях баланс между прооксидантными и антиоксидантными веществами поддерживается с перевесом в пользу прооксидантных продуктов, что способствует умеренному окислительному стрессу, так как данное состояние необходимо для оптимального функционирования иммунной системы и процессов передачи сигналов в клетке [23]. Однако ввиду непрерывного образования АФК и поддержания его на физиологическом уровне постоянно происходят небольшие повреждения внутри клетки. Именно поэтому существует потребность во второй группе эндогенной антиоксидантной системы защиты, которая удаляет или восстанавливает поврежденные биомолекулы до того, как произошло их накопление, что привело бы к изменению клеточного метаболизма и необратимому повреждению [75]. Поврежденные таким образом нуклеиновые кислоты восстанавливаются специфическими ферментами, окисленные белки удаляются протеолитическими системами, а окисленные липиды восстанавливаются фосфолипазами, пероксидазами и ацилтрансферазами [76].
Установлено, что нарушения в некоторых или во всех восстановительных системах в большей степени способствуют старению и возникновению возрастных заболеваний, чем умеренные колебания уровня веществ с антиоксидантными свойствами и образование АФК [2, 14]. В стареющих клетках значительно снижается эффективность работы многих систем репарации, накапливаются повреждения клеток, например, происходит накопление липофусцина в цитоплазме [2]. Возрастные окислительные изменения наиболее заметны в непролиферирующих клетках, таких как нейроны и кардиомиоциты, так как у них не происходит “разбавления” поврежденных структур посредством деления клеток [2, 77, 78].
Сложный процесс биологического старения является результатом действия генетических и экологических факторов, а также времени. Ключевыми изменениями во время старения являются воспаление, иммунное старение и клеточное старение. Старение – это необратимая форма длительной остановки клеточного цикла, вызванная чрезмерным внутриклеточным или внеклеточным стрессом, или повреждением [79]. Цель этой остановки клеточных циклов – ограничение пролиферации поврежденных клеток, устранение накопленных вредных факторов и предотвращение трансформации злокачественных клеток. Поскольку биологический возраст не обязательно должен соответствовать хронологическому возрасту, важно найти конкретные признаки и биомаркеры, которые могли бы объективно определять темп старения человека [79]. Эти биомаркеры могут быть ценными показателями физиологического возраста. Биомаркеры должны соответствовать нескольким критериям. Например, они должны предсказывать скорость старения, контролировать основной процесс, лежащий в основе процесса старения, иметь возможность многократно тестироваться, не нанося вреда человеку [79]. Кроме того, биомаркеры должны быть индикаторами биологических процессов, патогенных процессов или фармакологических реакций на терапевтическое вмешательство. Считается, что длина теломер является недостаточно объективным биомаркером (с плохой прогностической точностью), и в настоящее время нет надежного биомаркера, отвечающего всем необходимым критериям [79].
С возрастом увеличивается продукция АФК, при этом эффективность работы некоторых эндогенных защитных механизмов снижается [2, 80–82]. Например, взаимодействие митохондрий с ядром и изменения митохондриального гомеостаза приводят к возрастным изменениям. Неэффективный контроль АФК над митохондриальными суперкомплексами вызывает изменение сигнала АФК, направляя реакцию клеток на стресс в сторону возрастозависимого повреждения [83, 84]. Прогрессирующее снижение АФК-поглотителей приводит к сдвигу статуса стареющих клеток в сторону прооксидантного статуса [85]. Именно поэтому существует потребность во второй группе эндогенной антиоксидантной системы защиты, которая удаляет или восстанавливает поврежденные биомолекулы до того, как произошло их накопление, что привело бы к изменению клеточного метаболизма и необратимому повреждению [85, 86]. Этот дисбаланс приводит к прогрессирующему повреждению клеточных структур, предположительно приводящему к фенотипу старения. Данное предположение поддерживается двумя направлениями исследований [87–89].
Первое направлено на измерение концентрации продуктов/маркеров окислительного стресса в стареющих клетках, тканях или организмах. Было показано, что карбонилированные белки (маркеры тяжелого и хронического оксидативного стресса) находятся в повышенных концентрациях в последней трети жизни. Эти белки были обнаружены в культуре тканей дермальных фибробластов человека, хрусталике глаза и мозге человека, полученных при вскрытии, печени крысы и цельных мухах [90–92].
Другое направление исследований основано на искуственном индуцировании окислительного стресса в клетках с помощью оксидативных веществ. В модели, названной стресс-индуцированным преждевременным старением, клетки были смешаны с субтоксическими концентрациями оксиданта Н2О2 или генераторами оксидантов (паракват, УФ, железо или медь) [93–95]. Таким образом, индуцировалась реакция хронического стресса, что приводило к фенотипическому старению. При этом проявляются общие особенности старения: сверхэкспрессия p21 и p16INK4a, увеличение активности SA-β-Gal, увеличение клеточного объема и приобретение клеткой круглой формы [96, 97]. Кроме того, в эксперименте, в котором клетки были подвержены гипероксии (как известно, она индуцирует хронический окислительный стресс), было показано сходство паттерна экспрессии генов у клеток с индуцированным оксидативным стрессом по сравнению со стареющими фибробластами [98, 99]. Существуют и другие способы воспроизведения отдельных признаков клеточного старения, например, накопление внутриклеточного белка путем искусственного воздействия липофусцином на клетки [2, 100].
Однако, еще раз подчеркнем, старение – это многофакторный естественный процесс, зависящий от времени, с характерным прогрессирующим снижением большинства физиологических функций. Старение связано с повышенным уровнем окислительного стресса, различными типами макромолекулярных изменений, накоплением продуктов метаболизма, которые могут оказывать негативный эффект, повреждений дезоксирибонуклеиновой кислоты (ДНК), например, из-за совершения ошибок в ходе репликации ДНК-полимеразами, сбоев работы репарационных систем [101]. Поскольку большинство клеточных функций выполняется белками, старение может быть в какой-то степени следствием нарушения регуляции протеостаза или изменением функционирования протеома [2]. Более того, не все клеточные белки могут быть ресинтезированы из-за возникающих в результате старения повреждений ДНК [2, 101, 102].
В случае преждевременного возникновения окислительного стресса происходит накопление внутриклеточных повреждений, обычно наблюдаемых в пожилом возрасте – это так называемое преждевременное старение. Преждевременное старение чаще затрагивает определенные ткани/органы, а не организм в целом. Например, хроническая обструктивная болезнь легких (ХОБЛ), вызванная курением, вероятно, является результатом ускоренного старения легких, вызванного сигаретным дымом [103, 104]. Ожирение и диабет могут способствовать возникновению сердечно-сосудистых заболеваний и заболеваний почек, потенциально это происходит через индукцию преждевременного старения клеток в других тканях [105]. Было показано, что высокий уровень глюкозы, связанный с диабетом, способствует старению эпителиальных клеток in vitro. Высокий уровень глюкозы в крови может привести к гликированию белков, что нарушает их нормальную функцию. Кроме того, конечные продукты гликирования (белки или липиды, которые подверглись гликированию углеводами) могут взаимодействовать с мембранными рецепторами клеток, которые изменяют внутриклеточную передачу сигналов и способствуют выработке провоспалительных цитокинов и АФК [106, 107]. Было проведено ограниченное количество исследований, которые предполагают, что конечные продукты гликирования могут выступать в качестве пускового механизма для стимулирования старения клеток [108, 109].
Факторы, способствующие ускорению старения, включают генетические, хронические, а также такие связанные с образом жизни заболевания, как ожирение, сердечно-сосудистые заболевания и диабет 2 типа [2, 110].
ВЫВОДЫ
1) Активные формы кислорода и азота, постоянно генерируемые в организме, необходимы для осуществления регуляторных механизмов в клетке и защиты от микроорганизмов, но также являются и причиной некоторых патологических состояний, включая рак;
2) Активные АФК непрерывно продуцируются во время нормального клеточного метаболизма;
3) Антиоксидантная защита человека сложна и выполняет задачу установления физиологически-важного уровня АФК в клетке с возможностью функционирования клеточной передачи сигналов, в то же время минимизирует уровни АФК, чтобы не допустить окислительного повреждения;
4) Старение ассоциировано с окислительным стрессом, различными изменениями в клеточных структурах и макромолекулах, а также повреждением ДНК;
5) Несмотря на новые открытия на клеточном и молекулярном уровне, понимание процесса старения все еще ограничено.
Рис. 5.
Предполагаемые механизмы АФК-опосредованной активации путей MAPK. АФК генерируются в процессе реакций с различными NOX, которые активируются факторами роста, цитокинами и разнообразными стрессорами и быстро удаляются внутриклеточными антиоксидантами. АФК (как только выработка АФК превышает способность антиоксидантов) могут индуцировать окислительную модификацию сигнальных белков МАРК, включая RTKs и MAP3K (см. путь 1), тем самым приводя к активации МАРК. АФК могут активировать пути MAPK посредством ингибирования и/или деградации MKP (см. путь 2). MKPs – MAPK фосфатазы, они дефосфорилируют MAPKs, тем самым инактивируя их. MAPK фосфатазы обеспечивают отрицательную обратную связь, тем самым модулируя продолжительность, величину и пространственно-временной профиль активности MAPK в ответ на физиологические и патологические стимулы [72].
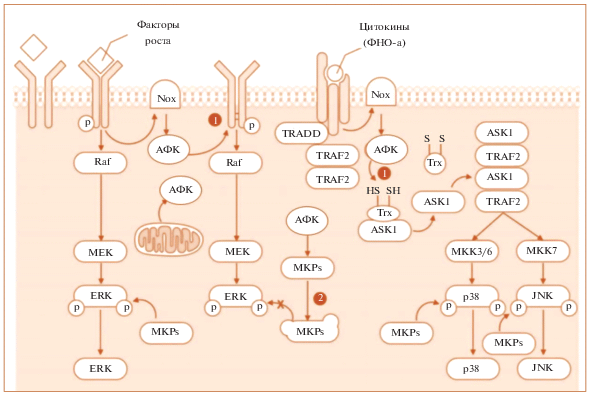
Рис. 6.
Сравнение структур молодой и старой клеток: в старых клетках выражены процессы окисления, накопление липофусцина, повреждение органоидов.
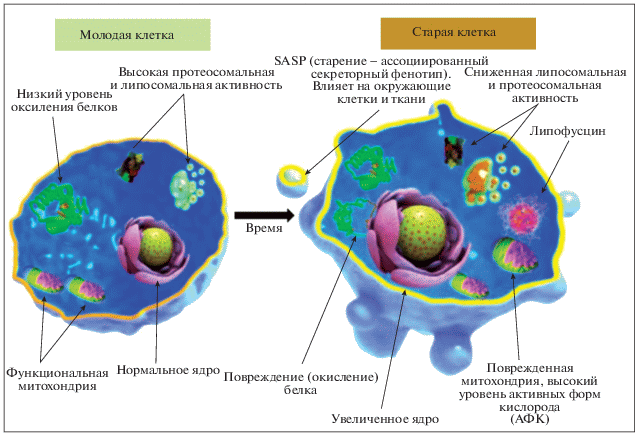
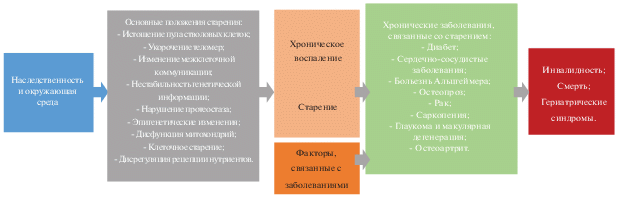
Рис. 7.
Схема концепции геронтологии. Представлен подход к изучению патогенеза возрастных заболеваний. Факторы окружающей среды и генетические факторы оказывают влияние на ряд ключевых клеточных процессов и путей, которые недавно были определены как отличительные признаки старения. Многие из этих путей способствуют возникновению хронического воспаления и старению [111].
Список литературы
Snezhkina A., Kudryavtseva A., Kardymon O., Savvateeva M.V., Melnikova N.V., Krasnov G.S. // Oxid. Med. Cell. Longev. 2019. V. 2019. P. 1–17. https://doi.org/10.1155/2019/6175804
Höhn A., Weber D., Jung T., Ott C., Hugo M., Kochlik B., Kehm R., König J., Grune T., Castro J.P. // Redox. Biol. 2017. V. 11. P. 482–501. https://doi.org/10.1016/j.redox.2016.12.001
Poljsak B. // Oxid. Med. Cell. Longev. 2011. V. 2011. P. 1–15. https://doi.org/10.1155/2011/194586
Halliwell B., Gutteridge J.M.C., Cross C.E. // J. Lab. Clin. Med. 1992. V. 119. P. 598–620.
Gutteridge J.M.C. // Free Radic. Res. Commun. 1993. V. 19. P. 141–158. https://doi.org/10.3109/10715769309111598
Scriven P., Brown N.J., Pockley A.G., Wyld L. // J. Mol. Med. 2007. V. 85. P. 331–341. https://doi.org/10.1007/s00109-006-0150-5
Santos C.X.C., Tanaka L.Y., Wosniak J., Laurindo F.R.M. // Antioxid. Redox. Sign. V. 11. P. 2409–2427. https://doi.org/10.1089/ars.2009.2625
Pegg A.E. // IUBMB Life. 2009. V. 61. P. 880–894. https://doi.org/10.1002/iub.230
Elamin Y.Y., Rafee S., Osman N., O’Byrne K.J., Gately K. // Cancer Microenviron. 2016. V. 9. P. 33–43. https://doi.org/10.1007/s12307-015-0173-y
Tabata S., Yamamoto M., Goto H., Hirayama A., Ohishi M., Kuramoto T., Mitsuhashi A., Ikeda R., Haraguchi M., Kawahara K., Shinsato Y., Minami K., Saijo A., Hanibuchi M., Nishioka Y., Sone S., Esumi H., Tomita M., Soga T., Furukawa T., Akiyama S. // Sci. Rep. 2018. V. 8. P. 6760–6767. https://doi.org/10.1038/s41598-018-25189-y
Brown N.S., Jones A., Fujiyama C., Harris A.L., Bicknell R. // Cancer Res. 2000. V. 60. P. 6298–6302.
Halliwell B. // Trends Pharmacol. Sci. 2011. V. 32. P. 125–130. https://doi.org/10.1016/j.tips.2010.12.002
Tafani M., Sansone L., Limana F., Arcangeli T., De Santis E., Polese M., Fini M., Russo M.A. // Oxid. Med. Cell. Longev. 2016. V. 2016. P. 1–18. https://doi.org/10.1155/2016/3907147
Sosnowska B., Mazidi M., Penson P., Gluba-Brzózka A., Rysz J., Banach M. // Atherosclerosis. 2017. V. 265. P. 275–282. https://doi.org/10.1016/j.atherosclerosis.2017.08.027
Brand M.D. // Exp. Gerontol. 2010. V. 45. P. 466–472. https://doi.org/10.1016/j.exger.2010.01.003
Yasui H., Hayashi S., Sakurai H. // Drug Metab. Pharmacokinet. 2005. V. 20. P. 1–13. https://doi.org/10.2133/dmpk.20.1
Whatley S.A., Curti D., Gupta F.D., Ferrier I.N., Jones S.J., Taylor C., Marchbanks R.M. // Mol. Psychiatry. 1998. V. 3. P. 227–237. https://doi.org/10.1038/sj.mp.4000375
Hey-Mogensen M., Goncalves R.L.S., Orr A.L., Brand M.D. // Free Radic. Biol. Med. 2014. V. 72. P. 149–155. https://doi.org/10.1016/j.freeradbiomed.2014.04.007
Zhang L., Yu L., Yu C.A. // J. Biol. Chem. 1998. V. 273. P. 33 972–33 976. https://doi.org/10.1074/jbc.273.51.33972
Kaludercic N., Mialet-Perez J., Paolocci N., Parini A., Di Lisa F. // J. Mol. Cell. Cardiol. 2014. V. 73. P. 34–42. https://doi.org/10.1016/j.yjmcc.2013.12.032
Wang S. // Dyes and Pigments. 2008. V. 76. P. 714–720. https://doi.org/10.1016/j.dyepig.2007.01.012
Narváez R., Bandala O., Sol E., Hernández L., Manuel J. // Int. J. Environ. Sci. Technol. 2018. V. 16. P. 1515–1526. https://doi.org/10.1007/s13762-018-1764-1
Poljsak B., Šuput D., Milisav I. // Oxid. Med. Cell. Longev. 2013. V. 2013. P. 1–11. https://doi.org/10.1155/2013/956792
Hunt P.R., Son T.G., Wilson M.A., Yu Q.S., Wood W.H., Zhang Y., Becker K.G., Greig N.H., Mattson M.P., Camandola S., Wolkow C.A. // PLoS One. 2011. V. 6. P. e21 922. https://doi.org/10.1371/journal.pone.0021922
Deng W.-M. // Adv. Exp. Med. Biol. 2019. V. 1167. P. 8–250. https://doi.org/10.1371/journal.pone.0021922
Mut-Salud N., Álvarez P., Garrido J., Carrasco E., Aránega A., Rodríguez-Serrano F. // Oxid. Med. Cell. Longev. 2016. V. 2016. P. 1–19. https://doi.org/10.1155/2016/6719534
Савченко А.А., Кудрявцев И.В., Борисов А.Г. // Инфекция и иммунитет. 2017. Т. 7. С. 327–340. [Savchenko A.A., Kudryavtsev I.V., Borisov A.G. // J. Infect. 2017. V. 7. P. 327–340.] https://doi.org/10.15789/2220-7619-2017-4-327-340
Winterbourn C.C. // Antioxid. Redox. Sign. 2018. V. 29. P. 541–551. https://doi.org/10.1089/ars.2017.7425
Son Y., Kim S., Chung H.-T., Pae H.-O. // Methods Enzymol. 2013. V. 528. P. 27–48. https://doi.org/10.1016/b978-0-12-405881-1.00002-1
Ткачук В.А., Тюрин-Кузьмин П.А., Белоусов В.В., Воротников А.В. // Биол. мембраны. 2012. Т. 29. С. 21–37. [Tkachuk V.A., Tyurin-Kuzmin P.A., Belousov V.V., Vorotnikov A.V. // Biol. Membrany. 2012. V. 29. P. 21–37.]
Veal E.A., Day A.M., Morgan B.A. // Mol. Cell. 2007. V. 26. P. 1–14. https://doi.org/10.1016/j.molcel.2007.03.016
Radi R. // Proc. Natl. Acad. Sci. 2018. V. 115. P. 5839–5848. https://doi.org/10.1073/pnas.1804932115
Onyango A.N. // Oxid. Med. Cell. Longev. 2016. V. 2016. P. 1–22. https://doi.org/10.1155/2016/2398573
Stanley C.P., Maghzal G.J., Ayer A., Talib J., Giltrap A.M., Shengule S., Wolhuter K., Wang Y., Chadha P., Suarna C., Prysyazhna O., Scotcher J., Dunn L.L., Prado F.M., Nguyen N., Odiba J.O., Baell J.B., Stasch J.P., Yamamoto Y., Mascio P.D., Eaton P., Payne R.J., Stocker R. // Nature. 2019. V. 566. P. 548–552. https://doi.org/10.1038/s41586-019-0947-3
Radi R. // J. Biol. Chem. 2013. V. 288. P. 26 464–26 472. https://doi.org/10.1074/jbc.r113.472936
Ramdial K., Franco M.C., Estevez A.G. // Brain Res. Bull. 2017. V. 133. P. 4–11. https://doi.org/10.1016/j.brainresbull.2017.05.008
Alhasawi A., Legendre F., Jagadeesan S., Appanna V., Appanna V. // Microb. Divers. Genom. Era. 2019. V. 2017. P. 153–169. https://doi.org/10.1016/b978-0-12-814849-5.00010-1
Adams L., Franco M.C., Estevez A.G. // Exp. Biol. Med. 2015. V. 240. P. 711–717. https://doi.org/10.1177/1535370215581314
Schröter J., Schiller J. // Oxid. Med. Cell. Longev. 2016. V. 2016. P. 1–26. https://doi.org/10.1155/2016/8386362
Rees M.D., McNiven T.N., Davies M.J. // J. Biochem. 2007. V. 401. P. 587–596. https://doi.org/10.1155/2016/8386362
Colon S., Page-McCaw P., Bhave G. // Antioxid. Redox Sign. 2017. V. 27. P. 839–854. https://doi.org/10.1089/ars.2017.7245
Hawkins C.L. // Free Radic. Res. 2009. V. 43. P. 1147–1158. https://doi.org/10.3109/10715760903214462
Ismael F.O., Barrett T.J., Sheipouri D., Brown B.E., Davies M.J., Hawkins C.L. // PLoS One. 2016. V. 11. P. e0168 844. https://doi.org/10.1371/journal.pone.0168844
Yang W.S., Stockwell B.R. // Trends Cell Biol. 2016. V. 26. P. 165–176. https://doi.org/10.1016/j.tcb.2015.10.014
Jiang L., Kon N., Li T., Wang S.J., Su T., Hibshoosh H., Baer R., Gu W. // Nature. 2015. V. 520. P. 57–62. https://doi.org/10.1038/nature14344
Xie Y., Hou W., Song X., Yu Y., Huang J., Sun X., Kang R., Tang D. // Cell Death Differ. 2016. V. 23. P. 369–379. https://doi.org/10.1038/cdd.2015.158
Schieber M., Chandel N.S. // Curr. Biol. 2014. V. 24. P. R453–R462. https://doi.org/10.1016/j.cub.2014.03.034
Saha S., Lee S., Won J., Choi H.Y., Kim K., Yang G.-M., Dayem A.A., Cho S.-G. // Int. J. Mol. Sci. 2017. V. 18. P. 1544–1574. https://doi.org/10.3390/ijms18071544
Briehl M. // Redox Biol. 2015. V. 5. P. 124–139. https://doi.org/10.1016/j.redox.2015.04.002
Nikolenko V.N., Gridin L.A., Oganesyan M.V., Rizaeva N.A., Podolskiy Y.S., Kudryashova V.A., Kochurova E.V., Kostin R.K., Tyagunova E.E., Mikhaleva L.M., Avila-Rodriguez M., Somasundaram S.G., Kirkland C.E., Aliev G. // Curr. Top. Med. Chem. 2019. V. 19. P. 2991–2998. https://doi.org/10.2174/1568026619666191127122452
Bienert G.P., Schjoerring J.K., Jahn T.P. // Biochim. Biophys. Acta Biomembr. 2006. V. 1758 P. 994–1003. https://doi.org/10.1016/j.bbamem.2006.02.015
Sies H. // Redox Biol. 2017. V. 11. P. 613–619. https://doi.org/10.1016/j.redox.2016.12.035
Hara-Chikuma M., Satooka H., Watanabe S., Honda T., Miyachi Y., Watanabe T., Verkman A.S. // Nat. Commun. 2015. V. 6. P. 7454. https://doi.org/10.1038/ncomms8454
Gardner P.R., Raineri I., Epstein L.B., White C.W. // J. Biol. Chem. 1995. V. 270. P. 13 399–13 405. https://doi.org/10.1074/jbc.270.22.13399
White M.F., Dillingham M.S. // Curr. Opin. Struct. Biol. 2012. V. 22. P. 94–100. https://doi.org/10.1016/j.sbi.2011.11.004
Winterbourn C.C. // Nat. Chem. Biol. 2008. V. 4. P. 278–286. https://doi.org/10.1038/nchembio.85
Ferrer-Sueta G., Manta B., Botti H., Radi R., Trujillo M., Denicola A. // Chem. Res. Toxicol. 2011. V. 24. P. 434–450. https://doi.org/10.1021/tx100413v
Dickinson B.C., Chang C.J. // Nat. Chem. Biol. 2011. V. 7. P. 504–511. https://doi.org/10.1038/nchembio.607
Marinho H.S., Real C., Cyrne L., Soares H., Antunes F. // Redox Biol. 2014. V. 2. P. 535–562. https://doi.org/10.1016/j.redox.2014.02.006
McCord J.M., Fridovich I. // Antioxid. Redox Sign. 2014. 20. P. 1548–1549. https://doi.org/10.1089/ars.2013.5547
Sarsour E.H., Kalen A.L., Goswami P.C. // Antioxid. Redox Sign. 2014. 20. P. 1618–1627. https://doi.org/10.1089/ars.2013.5303
Moldogazieva N.T., Mokhosoev I.M., Feldman N.B., Lutsenko S.V. // Free Radic. Res. 2018. V. 52. P. 507–543. https://doi.org/10.1080/10715762.2018.1457217
Zhu L., Lu Y., Zhang J., Hu Q. // Adv. Exp. Med. Biol. 2017. V. 967. P. 385–398. https://doi.org/10.1007/978-3-319-63245-2_25
Ferrer-Sueta G., Manta B., Botti H., Radi R., Trujillo M., Denicola A. // Chem. Res. Toxicol. 2011. V. 24. P. 434–450. https://doi.org/10.1021/tx100413v
Prasad S., Gupta S., Pandey M., Tyagi A.K., Deb L. // Oxid. Med. Cell. Longev. 2016. V. 2016. P. 1. https://doi.org/10.1155/2016/5010423
Balani S., Nguyen L.V., Eavesa C.J. // Nat. Commun. 2017. V. 8. P. 1–10. https://doi.org/10.1038/ncomms15422
Ray P.D., Huang B.-W., Tsuji Y. // Cell Signal. 2012. V. 24. P. 981–990. https://doi.org/10.1016/j.cellsig.2012.01.008
Geybels M.S., van den Brandt P.A., van Schooten F.J., Verhage B.A.J. // Cancer Epidem. Biomar. 2015. V. 24. P. 178–186. https://doi.org/10.1158/1055-9965.epi-14-0968
Jaramillo M.C., Zhang D.D. // Gene Dev. 2013. V. 27. P. 2179–2191. https://doi.org/10.1101/gad.225680.113
Carocho M., Ferreira I.C.F.R. // Food Chem. Toxicol. 2013. V. 51. P. 15–25. https://doi.org/10.1016/j.fct.2012.09.021
Jungwoon L., Yee S.C., Haiyoung J., Inpyo C. // Oxid. Med. Cell. Longev. 2018. V. 2018. P. 1–13. https://doi.org/10.1155/2018/4081890
Boutros T., Chevet E., Metrakos P. // Pharmacol. Rev. 2008. V. 60. P. 261–310. https://doi.org/10.1124/pr.107.00106
Zhong S., Jeong J.-H., Chen Z., Chen Z., Luo J.-L. // Transl. Oncol. 2020. V. 13. P. 57–69. https://doi.org/10.1016/j.tranon.2019.10.001
Doppler H., Storz P. // Front. Oncol. 2017. V. 7. P. 1–41. https://doi.org/10.3389/fonc.2017.00041
Carrier A. // Antioxid. Redox Signal. 2017. V. 26. P. 429–431. https://doi.org/10.1089/ars.2016.6929
Kallaur A.P., Reiche E.M.V., Oliveira S.R., Simao A.N.C., Pereira W.L.D.J., Alfieri D.F., Flauzino T., Proenca C.D., Lozovoy M.A.B., Kaimen-Maciel D.R., Maes M. // Mol. Neurobiol. 2017. V. 54. P. 31–44. https://doi.org/10.1007/s12035-015-9648-6
Lindqvist D., Dhabhar F.S., James S.J., Hough C.M., Jain F.A., Bersani F.S., Reus V.I., Verhoeven J.E., Epel E.S., Mahan L., Rosser R., Wolkowitz O.M., Mellon S.H. // Psychoneuroendocrinology. 2017. V. 76. P. 197–205. https://doi.org/10.1016/j.psyneuen.2016.11.031
Krakhmal N.V., Zavyalova M.V., Denisov E.V., Vtorushin S.V., Perelmuter V.M. // Acta Naturae. 2015. V. 7. P. 17–28. https://doi.org/10.32607/20758251-2015-7-2-17-28
Dodig S., Čepelak I., Pavić I. // Biochem. Med. (Zagreb). 2019. V. 29. P. 483–497. https://doi.org/10.11613/bm.2019.030501
Rinnerthaler M., Bischof J., Streubel M.K., Trost A., Richter K. // Biomol. 2015. V. 5. P. 545–589. https://doi.org/10.3390/biom5020545
Espinosa-Diez C., Miguel V., Mennerich D., Kietzmann T., Sánchez-Pérez P., Cadenas S., Lamas S. // Redox Biol. 2015. V. 6. P. 183–197. https://doi.org/10.1016/j.redox.2015.07.008
Sies H, Berndt C., Jones D.P. // Annu. Rev. Biochem. 2017. V. 86. P. 715–748. https://doi.org/10.1146/annurev-biochem-061516-045037
Yang L., Zheng L., Tian Y., Zhang Z.Q., Dong W.L., Wang X.F., Zhang X.Y., Cao C. // Exp. Cell. Res. 2015. V. 332. P. 47–59. https://doi.org/10.1016/j.yexcr.2014.12.017
Harisa G.I. // Saudi Pharm. J. 2015. V. 23. P. 48–54. https://doi.org/10.1016/j.jsps.2014.04.006
Hall S.R., Blundon H.L., Ladda M.A., Robertson A.W., Martinez-Farina C.F., Jakeman D.L., Goralski K.B. // Pharmacol. Res. Perspect. 2015. V. 3. P. e00110. https://doi.org/10.1002/prp2.110
Wilson A.J., Kerns J.K., Callahan J.F., Moody C.J. // J. Med. Chem. 2013. V. 56. P. 7463–7476. https://doi.org/10.1021/jm400224q
Corcoran A., Cotter T.G. // FEBS J. 2013. V. 280. P. 1944–1965. https://doi.org/10.1111/febs.12224
Truong T.H., Carroll K.S. // Crit. Rev. Biochem. Mol. Biol. 2013. V. 48. P. 332–356. https://doi.org/10.3109/10409238.2013.790873
Sabharwa S.S., Schumacker P.T. // Nat. Rev. Cancer. 2014. V. 14. P. 709–721. https://doi.org/10.1038/nrc3803
Porter K.M., Kang B.-Y., Adesina S.E., Murphy T.C., Hart C.M., Sutliff R.L. // PLoS One. 2014. V. 9. P. e98 532. https://doi.org/10.1371/journal.pone.0098532
Zhao W., Ma G., Chen X. // Vascul. Pharmacol. 2014. V. 63. P. 162–172. https://doi.org/10.1016/j.vph.2014.06.008
Spencer N.Y., Engelhardt J.F. // Biochem. 2014. V. 53. P. 1551–1564. https://doi.org/10.1021/bi401719r
Dang W. // Drug Discov. Today Technol. 2014. V. 12. P. e9–e17. https://doi.org/10.1016/j.ddtec.2012.08.003
Woo H.A., Yim S.H., Shin D.H., Kang D., Yu D.Y., Rhee S.G. // Cell. 2010. V. 140. P. 517–528. https://doi.org/10.1016/j.cell.2010.01.009
Koppenol W.H., Bounds P.L., Dang C.V. // Nat. Rev. Cancer. 2011. V. 11. P. 325–337. https://doi.org/10.1038/nrc3038
Reuter S., Gupta S.C., Chaturvedi M.M., Aggarwal B.B. // Free Radic. Biol. Med. 2010. V. 49. P. 1603–1616. https://doi.org/10.1016/j.freeradbiomed.2010.09.006
Rudolf E., Rudolf K. // Apoptosis. 2015. V. 20. P. 1651–1665. https://doi.org/10.1007/s10495-015-1182-5
Zeng L., Yang Y., Hu Y., Sun Y., Du Z., Xie Z., Zhou T., Kong. W. // PLoS One. 2014. V. 9. P. e88 019. https://doi.org/10.1371/journal.pone.0088019
Castro J.P., Grune T., Speckmann B. // Biol. Chem. 2016. V. 397. P. 709–724. https://doi.org/10.1515/hsz-2015-0305
Pernodet N., Dong K., Pelle E. // Int. J. Cosmet. Sci. 2016. V. 67. P. 13–20.
König J., Besoke F., Stuetz W., Malarski A., Jahreis G., Grune T., Höhn A. // BioFactors. 2016. V. 42. P. 307–315. https://doi.org/10.1002/biof.1274
Kudryavtseva A.V., Krasnov G.S., Dmitriev A.A., Alekseev B.Y., Kardymon O.L., Sadritdinova A.F., Snezhkina A.V. // Oncotarget. 2016. V. 7. P. 7–25. https://doi.org/10.18632/oncotarget.9821
Mercado N., Ito K., Barnes P.J. // Thorax. 2015. V. 70. P. 482–489. https://doi.org/10.1136/thoraxjnl-2014-206084
Hepple R.T. // Free Radic. Biol. Med. 2016. V. 98. P. 177–186. https://doi.org/10.1016/j.freeradbiomed.2016.03.017
Burton D.G.A., Faragher R.G.A. // Biogerontology. 2018. V. 19. P. 447–459. https://doi.org/10.1007/s10522-018-9763-7
Singh V.P., Bali A., Singh N., Jaggi A.S. // Korean J. Physiol. Pharmacol. 2014. V. 18. P. 1–14. https://doi.org/10.4196/kjpp.2014.18.1.1
Reeg S., Jung T., Castro J.P., Davies K., Henze A., Grune T. // Free Radic. Biol. Med. 2016. V. 10. P. 153–166. https://doi.org/10.1016/j.freeradbiomed.2016.08.002
Toda N., Okamura T. // J. Clin. Pharmacol. 2013. V. 53. P. 1228–1239. https://doi.org/10.1002/jcph.179
Flohe L. // Free Radic. Res. 2016. V. 50. P. 126–142. https://doi.org/10.3109/10715762.2015.1046858
Sies H. // Redox biol. 2015. V. 4. P. 180–183. https://doi.org/10.1016/j.redox.2015.01.002
Campisi J., Kapahi P., Lithgow G.J., Melov S., Newman J.C., Verdin E. // Nature. 2019. V. 571. P. 183–192. https://doi.org/10.1038/s41586-019-1365-2
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия