

Биоорганическая химия, 2020, T. 46, № 6, стр. 676-685
Разработка пилотной технологии получения рекомбинантной легкой цепи энтеропептидазы человека в растворимой и иммобилизованной формах
Д. А. Макаров 1, *, А. А. Зинченко 1, В. Н. Степаненко 1, Д. С. Калинин 1, 2, Т. Д. Мелихова 1, Е. А. Нокель 1, М. Э. Гаспарян 1, И. В. Мягких 1, Д. А. Долгих 1, 2
1 ФГБУН Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова Российской академии наук
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
2 ФГБОУ ВО Московский государственный университет имени М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: youngchemist@mail.ru
Поступила в редакцию 27.02.2020
После доработки 17.03.2020
Принята к публикации 22.03.2020
Аннотация
Энтеропептидаза, протеолитический фермент EC 3.4.21.9, представляет собой гликопротеин, состоящий из двух тяжелых и легких полипептидных цепей. Легкая цепь фермента представляет собой сериновую протеиназу, рекомбинантные варианты которой применяются в биотехнологии для расщепления пептидных связей в гибридных белках после специфического сайта узнавания DDDDK- или DDDDR-. Мы разработали пилотный способ получения рекомбинантной легкой цепи энтеропептидазы человека, содержащей каскад из двух аффинных меток. Сконструированный экспрессионный плазмидный вектор рL-HEP-H6-CBD, кодирующий гибридный белок, содержащий последовательность легкой цепи энтеропептидазы человека, металлсвязывающий и хитин-связывающий домены был транформирован в штамм Escherichia coli BL21(DE3) и после экспрессии гибридного белка HEP-H6-CBD была разработана относительно простая схема ренатурации и очистки фермента. После иммобилизации очищенного препарата на хитиновый носитель фермент сохранял активность специфического расщепления субстратов в течении как минимум 6 циклов.
ВВЕДЕНИЕ
Простота выделения, очистки и идентификации многих рекомбинантных белков часто достигается путем создания гибридных конструкций, включающих дополнительные аффинные пептидные метки [1]. Удаление аффинной метки может быть осуществлено ферментативным расщеплением гибридного белка, однако в промышленности использование подобного подхода часто ограничивается высокой стоимостью ферментных препаратов. В связи с этим, создание удобных в работе гибридных конструкций рекомбинантных ферментов с приемлемой активностью, пригодных для использования в промышленных процессах получения белковых препаратов различного назначения, а также разработка недорогих технологичных процессов выделения и очистки таких ферментных препаратов является актуальной задачей. В число наиболее широко используемых в биотехнологии рекомбинантных протеолитических ферментов входит легкая цепь энтеропептидазы человека [2].
Энтеропептидаза (EC 3.4.21.9) является высокоспецифичной протеиназой и играет ключевую роль в пищеварении позвоночных [3]. В начале каскада активации ферментов пищеварения энтеропептидаза с высокой эффективностью катализирует гидролиз трипсиногена строго по остатку лизина в специфическом сайте узнавания -DDDDK–I-, высвобождая трипсин, который далее активирует целый ряд панкреатических зимогенов [4, 5].
Нативная энтеропептидаза представляет собой дисульфид-связанный гетеродимер, состоящий из “тяжелой” и “легкой” цепей с молекулярной массой ~120 и ~26 кДа соответственно [3]. Легкая цепь энтеропептидазы, каталитический домен фермента, является сериновой эндопептидазой трипсинового типа [6] и содержит четыре дисульфидных мостика, которые важны для функционирования и стабильности молекулы фермента [7].
Описано несколько способов получения энтеропептидазы из природных источников [8, 9]. Однако в биотехнологических целях чаще используют рекомбинантную легкую цепь энтеропептидазы [10, 11].
В исследовании Ла-Валли показана возможность клонирования и функциональной экспрессии легкой цепи бычьей энтеропептидазы в клетках млекопитающих COS-1 [12]. Легкая цепь энтеропептидазы была успешно экспрессирована в разнообразных биологических системах, таких как Aspergillus niger [13] и Saccharomyces cerevisiae [14]. Также были опубликованы многочисленные работы по экспрессии легкой цепи энтеропептидазы в Pichia pastoris [15]. Кроме того, легкая цепь энтеропептидазы может быть получена даже в клетках насекомых [16].
Наиболее предпочтительной системой для экспрессии легкой цепи энтеропептидазы является бактерия Е. coli, поскольку получаемый белок продуцируется в большом количестве за короткий срок. Однако исследователи сталкивались с тем, что энтеропептидаза продуцировалась в нерастворимой форме. До сегодняшнего дня было опубликовано несколько исследований, посвященных повышению растворимости и изучению фолдинга рекомбинантной легкой цепи энтеропептидазы [17–20].
В настоящей работе описана оригинальная, эффективная, легко масштабируемая технология получения рекомбинантного гибридного белка t-L-HEP содержащего хитин-связывающий домен на С-конце, что позволило иммобилизовать фермент на хитиновый сорбент и использовать многократно для расщепления слитных белков медицинского назначения.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Разработка пилотной технологии получения рекомбинантного белка L-HEP
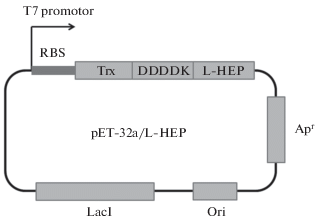
В лаборатории инженерии белка ИБХ РАН под руководством академика М.П. Кирпичникова ранее была разработана методика получения рекомбинантной легкой цепи энтеропептидазы человека (регистрационный номер GenBank U09860) [11, 17] и создан штамм-продуцент Е. coli BL21(DE3)/pET-32a/L-HEP гибридного белка Trx-DDDDK-L-HEP, содержащего аминокислотную последовательность тиоредоксина (Trx), специфического сайта узнавания энтеропептидазы (DDDDK-) и легкой цепи энтеропептидазы человека (L-HEP) (рис. 1).
Рис. 1.
Физическая карта плазмиды pET-32a/L-HEP. ori – ориджин репликации, AmpR – ген резистентности к ампициллину, lacI – ген репрессора лактозного оперона, T7 promoter – промотор бактериофага T7, RBS – сайт связывания с рибосомой, Trx-DDDDK-L-HEP – ген, кодирующий гибридный белок, состоящий из тиоредоксина, сайта узнавания легкой цепи энтеропептидазы человека и непосредственно легкой цепи энтеропептидазы человека.
Особенность данной конструкции заключалась в том, что ГБ после ренатурации автокаталитически расщеплялся с высвобождением свободной L-HEP и белка-партнера.
Лабораторная методика позволяла получить 10 мг высокоочищенной активной L-HEP с 1 л клеточной культуры после двух циклов ренатурации [11, 17]. Для наработки рекомбинантной L-HEP в количествах, достаточных для производственных целей, требовалось масштабировать и оптимизировать процесс получения фермента. Наличие в лабораторной методике нескольких стадий диализа и использование дорогостоящего аффинного сорбента (STI-агароза) осложняло масштабирование процесса. Поэтому была проведена оптимизация технологии получения L-HEP.
Переход от культивирования штамма-продуцента в качалочных колбах на промышленный ферментер объемом 200 л увеличил средний выход влажной биомассы до 40 г с 1 л культуральной среды. Экспрессия гибридного белка при этом достигала 30% от общего белка клетки.
Тельца включения, полученные после разрушения биомассы, отмывали буферными растворами, содержащими мочевину и/или тритон Х-100, как описано в Экспериментальной части. Отмытые ТВ растворяли в денатурирующих условиях, используя буферный раствор 50 мМ Tрис, 6 M мочевина, pH 10. Ренатурацию осуществляли методом разбавления растворенных ТВ до постоянной концентрации гибидного белка 0.7 мг/мл в соответствующих буферных растворах.
Оптимизация стадии ренатурации включала исследование влияния ряда факторов на эффективность процесса. В процессе исследований варьировали состав и pH буферных растворов, температуру проведения процесса, время реакции, а также изучали влияние на ход ренатурации различных химических реагентов (NaCl, ЭДТА, аргинина, глицерина, цистеина, сорбитола и др.).
Наиболее удачными для ренатурации ГБ оказались буферные растворы, в состав которых входили 20% глицерин или 1 мМ цистеин. Остальные исследованные реагенты не оказывали положительного эффекта на течение процесса (данные не приведены). В правильно подобранных условиях ренатурации наблюдалось автокаталитическое отщепление энтеропептидазы с высвобождением L-HEP.
Активность фермента в ренатуратах оценивали электрофоретически по степени расщепления мГБ-1 и/или мГБ-2 (рис. 2, 3, 4). По результатам исследований было установлено, что наиболее эффективная ренатурация, и, как следствие, более глубокое расщепление модельных гибридных белков образующейся энтеропептидазой, наблюдалась в буферном растворе, содержащем 20% глицерин.
Рис. 2.
Электрофореграмма расщепления модельного гибридного белка 2 с помощью L-HEP: 1 – стандарт молекулярных масс, 2 – ренатурат легкой цепи энтеропептидазы человека с добавкой 20% глицерина, 3–4 – расщепление мГБ-2, 25°C в течение 8 и 16 ч соответственно, в массовом соотношении 1 : 50, 5 – ренатурат легкой цепи энтеропептидазы человека с добавкой 1 мМ цистеина, 6–7 – расщепление мГБ-2, 25°C в течение 8 и 16 ч соответственно, в массовом соотношении 1 : 50.
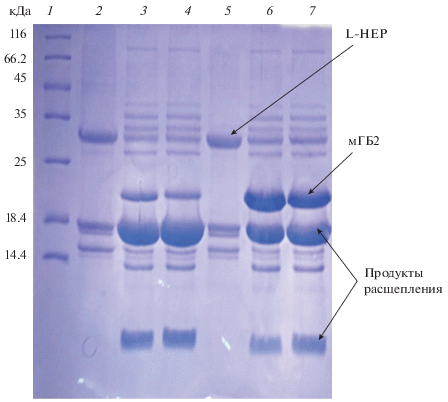
Рис. 3.
Электрофореграмма кинетики расщепления модельного гибридного белка 2 ферментом L-HEP. Массовое соотношение 1 : 50: 1 – мГБ-2, 2 – расщепление энтеропептидазой 8 ч, 3 – расщепление энтеропептидазой 16 ч, 4 – расщепление энтеропептидазой 24 ч.
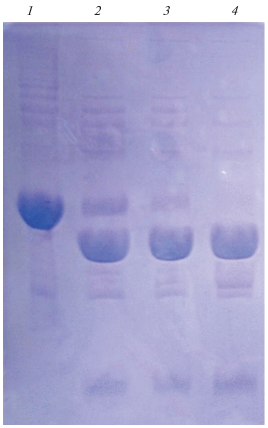
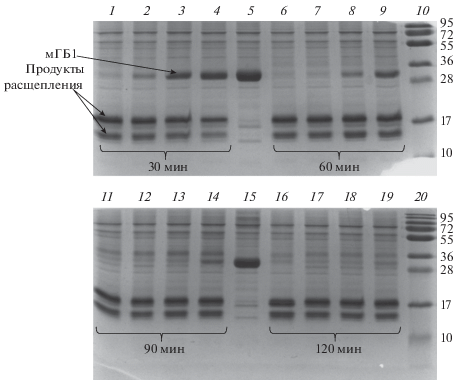
Рис. 4.
Электрофореграмма кинетики расщепления модельного гибридного белка 1 ферментом L-HEP. Расщепление мГБ-1 с L-HEP в массовом соотношении фермент : субстрат 1 : 50; 1 : 100; 1 : 200; 1 : 400 (массовое соотношение с учетом активной L-HEP 1 : 2500; 1 : 5000; 1 : 10 000; 1 : 20 000), соответственно, за 30 мин (дорожки 1–4), за 60 мин (дорожки 6–9), за 90 мин (дорожки 11–14) и за 120 мин (дорожки 16–19). Дорожки 5 и 15 представляют белок мГБ-1 до расщепления. Дорожки 10 и 20 – маркеры молекулярных масс.
Для оценки количества активного фермента в ренатуранте, содержащем 20% глицерин, препарат очистили на сорбенте STI-Sepharose (сефароза, конъюгированная с ингибитором трипсина сои). Аффинная хроматография использовалась только для контроля процесса ренатурации и не применялась в технологическом процессе. Аликвоту ренатурата пропускали через аффинный сорбент и определяли количество связавшейся L-HEP по отношению к общему количеству фермента. В результате оказалось, что на сорбенте связалось около 2% фермента от общей массы L-HEP, что составило 0.02 мг активного фермента в 1 мг L-HEP. Таким образом, в ходе разработанной технологии ренатурации был достигнут такой же выход активного фермента, как и в методике лабораторной технологии [11], но с меньшими трудозатратами и сокращением времени производства.
Подбор условий расщепления модельных гибридных белков и определение активности полученной легкой цепи энтеропептидазы человека привел к следующим результатам: расщепление мГБ-1 прошло более чем на 85% в течение 30 минут при температуре 25°С и массовом соотношении фермент субстрат 1 : 100 (массовое соотношение с учетом активной L-HEP составило 1 : 5000), или в течение 60 мин при массовом соотношении фермент–субстрат 1 : 200 (массовое соотношение с учетом активной L-HEP составило 1 : 10 000). Расщепление мГБ-2 прошло более чем на 85% в течение 24 часов при температуре 25°С и массовом соотношении фермент/субстрат 1 : 50 (массовое соотношение с учетом активной L-HEP составило 1 : 2500). На основе полученных результатов была разработана опытно-промышленная технология получения легкой цепи энтеропептидазы человека и наработана партия ферментного препарата. Выход активного фермента составил 100 мг с 1 л культуральной жидкости. Полученный препарат был успешно использован при получении инновационного анальгетика Пуротоксин-6 и Интерферона альфа-2b.
Разработка технологии получения рекомбинантного гибридного белка t-L-HEP
При использовании ферментных препаратов для получения рекомбинантных белков различного назначения часто возникают проблемы, связанные с необходимостью удаления фермента из реакционной смеси, например, для остановки ферментативной реакции, очистки целевого продукта от следовых количеств фермента и т.д. Для этого в структуру целевого белка вводят аффинные метки, позволяющие существенно облегчить такие процедуры [20, 21]. Для решения подобных задач мы создали новую генно-инженерную конструкцию гибридной L-HEP, содержащую в своей структуре пептидные аффинные метки, позволяющие количественно удалять фермент из любой смеси и на любой стадии получения, выделения и очистки белков.
Для этого был сконструирован экспрессионный плазмидный вектор рL-HEP-H6-CBD, кодирующий гибридный белок, содержащий аминокислотную последовательность легкой цепи энтеропептидазы человека, в N-концевой части которой расположен специфический сайт узнавания L-HEP, DDDDK-, а в С-концевой – полигистидиновая последовательность (H6 и хитин-связывающий домен (CBD) (рис. 5).
Рис. 5.
Физическая карта плазмиды pL-HEP-H6-CBD. ori – ориджин репликации, Apr – ген резистентности к ампициллину, lacI – ген репрессора лактозного оперона, T7 promoter – промотор бактериофага T7, RBS – сайт связывания с рибосомой, N-seq – DDDDК-L-HEP-H6-CBD – нуклеотидная последовательность, кодирующая гибридный белок, включающий аминокислотные последовательности специфического сайта узнавания энтеропептидазы, легкой цепи энтеропептидазы человека, гексагистидинового домена и хитин-связывающего домена.
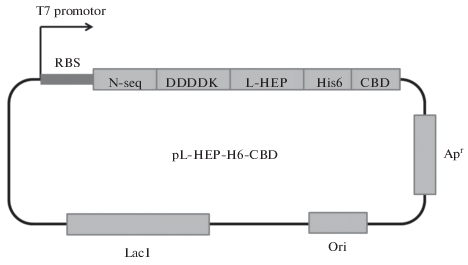
Экспрессионный вектор был трансформирован в E. coli BL21(DE3). В первичной структуре L-HEP содержатся остатки цистеина (которые образуют четыре дисульфидные связи), что значительно затрудняет правильный фолдинг экспрессированного белка [20]. В полученном штамме-продуценте E. coli BL21(DE3)/рL-HEP-H6-CBD был зафиксирован высокий уровень экспрессии ГБ N-seq – DDDDK – L-HEP – H6 – CBD – 40 г влажной биомассы с 1 л культуры. После индукции большая часть ГБ (~95%) формировала тельца включения. Отмывка, экстракция и ренатурация новой гибридной конструкции проводилась по схеме, отработанной для получения L-HEP.
Оптимизация стадии очистки t-L-HEP
Для очистки t-L-HEP использовали металл-хелатную аффинную хроматографию. Обессоливание элюата после аффинной хроматографии проводили с помощью гель-фильтрации. Хроматографическая чистота полученной t-L-HEP составила более 80% (рис. 6).
Рис. 6.
Электрофореграмма белковых фракций при очистке t-L-HEP c помощью металл-хелатной аффинной хроматографии: 1 – маркеры молекулярных масс, 2 – исходное нанесение, 3 – элюат примесных белков, 4 – элюат t-L-HEP.
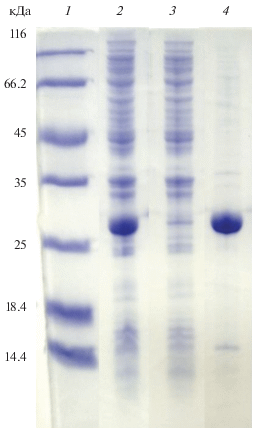
Таким образом, была создана пилотная технология получения ферментного препарата t-L-HEP. Эффективность ренатурации сохранилась и осталась соответствующей лабораторным результатам, полученным для L-HEP [17]. Однако оптимизация процесса и масштабирование лабораторной методики, позволили получить значительные количества ферментного препарата. Невысокий выход на стадии ренатурации компенсируется простотой и хорошей воспроизводимостью технологии выделения и очистки t-L-HEP. Выход активного гибридного фермента t-L-HEP (L-HEP – H6 – CBD) составил 65.2 мг с 1 л культуральной жидкости.
Иммобилизация t-L-HEP на хитиновой смоле
Иммобилизация ферментов является одним из возможных вариантов решения проблемы снижения материальных издержек при производстве рекомбинантных белков различного назначения. Такая возможность была предусмотрена нами в структуре гибридного белка t-L-HEP, содержащей аминокислотную последовательность хитин-связывающего домена – CBD. Иммобилизацию t-L-HEP осуществляли на хитиновой смоле (рис. 7), условия имммобилизации приведены в Экспериментальной части. Емкость сорбента по белку составила 1 мг/мл.
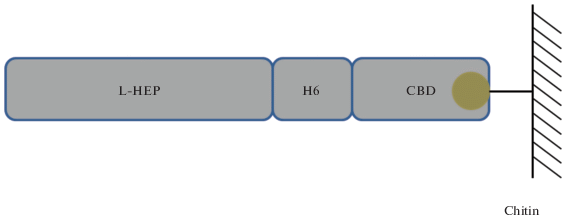
Активность i-t-L-HEP определяли по расщеплению мГБ-1. Через 0.5 мл сорбента с иммобилизованным ферментом (i-t-L-HEP) пропускали 10 мл раствора мГБ-1 (3 мг/мл). В ходе эксперимента время контакта субстрата с i-t-L-HEP контролировали скоростью потока через сорбент (от 0.5 до 4 мл/мин). В результате установлено, что 12 секунд контакта мГБ-1 с i-t-L-HEP (0.5 мг в объеме 0.5 мл) достаточно для количественного расщепления мГБ-1, данное время соответствует скорости потока 2.5 мл/мин. Была определена стабильность иммобилизованного фермента. Показано вплоть до 3 цикла отсутствие снижения ферментативной активности i-t-L-HEP, при этом активность иммобилизованного фермента после шестого цикла снизилась не более чем на 10%, что позволит использовать фермент многократно (рис. 8). При скоростях потока больше 2.5 мл/мин происходила деформация сорбента и десорбция иммобилизованного фермента. Это свидетельствует о невысокой операционной стабильности препарата иммобилизованного фермента, что может ограничить возможности его использования.
Рис. 8.
Электрофореграмма расщепления модельного гибридного белка 1 (25°C, время контакта 12 с) иммобилизованным ферментом i-t-L-HEP; 1 – третий цикл, 2 – шестой цикл, 3 – отрицательный контроль.
Таким образом, в результате проведенных исследований создана новая структура гибридного белка t-L-HEP (L-HEP – H6 – CBD) способного расщеплять белки по специфическому сайту энтеропептидазы, разработан эффективный биотехнологический способ получения двух вариантов рекомбинантной легкой цепи энтеропептидазы человека, L-HEP и t-L-HEP, содержащей каскад из двух аффинных меток, осуществлена иммобилизация ферментного препарата t-L-HEP. Показано сохранение активности иммобилизованного фермента в течение 6 циклов. Обнаружены особенности функционирования i-t-L-HEP, связанные с механическими свойствами носителя. В дальнейшем планируется оптимизация условий использования иммобилизованных ферментов на хитиновых носителях.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
В исследовании использовали следующие реактивы: натрия хлорид (Panreac, Испания), едкий натр (Panreac, Испания), дрожжевой экстракт (Sigma-Aldrich, США), этанол (Panreac, Испания), ледяную уксусную кислоту (Panreac, Испания) мочевину (Sigma-Aldrich, США); акриламид (Panreac, Испания), Трис (трис(гидроксиметил)аминометан) (Panreac, Испания) акриламид/бисакриламид 29/1 (Sigma Aldrich, США), персульфат аммония (Merck), бромфеноловый синий (BioRad, США); Кумасси R 250 (BioRad, США), хлороводородную кислоту (Panreac, Испания), дитиотреитол (DTT) (Sigma-Aldrich, США), додецилсульфат натрия SDS (Sigma-Aldrich, США), этилендиаминтетрауксусная кислота (ЭДТА) (Sigma-Aldrich, США), ампициллин (Sigma-Aldrich, США), бактотриптон (Sigma-Aldrich, США), N,N,N',N'-тетраметилэтилендиамин (TEMED), молекулярный маркер Unstained Protein Molecular Weight Marker #26610 (“Thermo Fisher Scientific”, США). Среда LB [23].
В исследовании использовался штамм E. coli. BL21(DE3) генотип: E. coli str.BF– ompTgaldcmlonhsdSB(rB–mB–) λ (DE3 [lacI lacUV5-T7 p07 ind1 sam 7 nin 5]) [malB+] K-12 (λS).
В работе использовали хитиновый сорбент производства NEB (Англия). Емкость сорбента – 2 мг мальтоза-связывающего белка/мл сорбента, размер частиц 50–60 микрон.
Все генно-инженерные конструкции выполнены по аутсорсингу в компании ЗАО Евроген (Россия).
Модельные гибридные белки, мГБ-1 и мГБ-2, получены на Опытном биотехнологическом производстве ИБХ РАН с использованием технологии рекомбинантной ДНК и экспрессией в E. coli.
Создание штамма продуцента E. coli BL21(DE3)/р-L-HEP-H6-CBD (pET-32a/L-HEP)
Плазмидами рL-HEP-H6-CBD или pET-32a/ L-HEP трансформировали компетентные клетки (E. coli штамм BL21(DE3)). Трансформанты высевали на агаризованную среду с ампицилином и инкубировали в течение 20 ч при 37°С. Выросшие колонии с помощью петли в стерильных условиях переносили в питательную среду LB с ампицилином объемом 4 мл и культивировали в течение 16 часов при 37°С. Полученной ночной культурой засевали 6 мл питательной среды LB, содержащей ампициллин в концентрации 100 мкг/мл, и инкубировали на инкубаторе-шейкере INNOVA 40R (New Brunswick, США), при 37°С 200 об./мин. до значений оптической плотности культуры 0.9 опт. ед. Оптическую плотность измеряли на спектрофотометре ПЭ-5300ВИ (Экохим, Россия) при длине волны 600 нм, используя в качестве контроля питательную среду. Затем производили индукцию биосинтеза целевого ГБ добавлением 1000 кратного раствора изопропил-β-D-тиогалактопиранозида (ИПТГ) до конечной концентрации 0.22 ммоль/л, после чего культуру инкубировали в течение 4 часов при 37°С. По окончании культивирования осадок отделяли путем центрифугирования на центрифуге 260D (DENVILL, США) 14 000 g в течение 10 мин при комнатной температуре. Клеточный лизат анализировали на наличие целевого гибридного белка с помощью электрофореза по Лэммли [23]. По результатам анализа был выбран клон с наибольшим уровнем экспрессии целевого гена. Суспензию клеток замораживали при –72°С с добавлением глицерина до конечной концентрации 15%.
Выращивание инокулята штамма E. coli BL21(DE3)/рL-HEP-H6-CBD (pET-32a/L-HEP) для засева ферментера
Два литра среды LB, содержащей ампициллин в концентрации 100 мкг/мл, засевали размороженной суспензией клеток E. coli BL21(DE3)/рL-HEP-H6-CBD (или pET-32a/L-HEP) в количестве 0.1% и культивировали в качалочных колбах в течение ночи (14 ч) при 32°С (170 об./мин). Оптическая плотность посевной культуры составила (4.9) опт. ед. Общий объем инокулята составил 2 л.
Культивирование штамма-продуцента E. coli BL21(DE3)/рL-HEP-H6-CBD (pET-32a/L-HEP) в ферментере
Ферментацию штамма-продуцента E. coli BL21(DE3)/рL-HEP-H6-CBD (или pET-32a/L-HEP) проводили в 200 л ферментере (MBR, Швейцария), содержащем стерильную питательную среду LB. Культивирование штамма-продуцента проводили при температуре 37°С, рН 7.0. Продолжительность культивирования 12.0 ч. Через 7–7.5 часов от начала культивирования (при достижении оптической плотности культуральной жидкости, равной 28–30 оптическим единицам) проводили индукцию биосинтеза гибридного белка в клетках культуры посредством введения в ферментер водного раствора ИПТГ до конечной концентрации 0.22 ммоль/л. Через 4–4.5 часа после введения в аппарат индуктора биосинтеза гибридного белка процесс останавливали. Во время культивирования осуществляли наблюдение за процессом, фиксируя значения показателей: время роста, температура культивирования, рО2, рН, оптическая плотность при длине волны 600 нм. Сбор биомассы клеток осуществляли на проточной центрифуге Z81 G (SEPA, Германия) при температуре 20°С. Было получено 8 кг влажной биомассы.
Разрушение биомассы
Полученную биомассу в количестве 500 г суспендировали в буферном растворе (50 мМ Tрис/HCl, 10 мМ ЭДТА, рН 8) в соотношении 1 : 10 и гомогенизировали на лабораторном гомогенизаторе APV–1000 (Германия) при температуре 5°С. Степень разрушения клеток определяли микроскопированием. Разрушенную клеточную суспензию центрифугировали на центрифуге 34 Z383K Hermle Labor Technik (Германия) при 11500 об./мин, 40 мин, 5°С. Полученные тельца включения (ТВ) взвешивали. Было получено 40 г биомассы с одного литра культуральной жидкости.
Отмывка телец включения, экстракция и ренатурация
ТВ отмывали буферным раствором, содержащим тритон Х-100 (50 мМ Tрис/HCl, 0.1% Triton Х-100, рН 8) или мочевину (50 мМ Tрис/HCl, 2 М мочевина, pH 8) в соотношении 1 : 10, после чего суспензию центрифугировали (5000 g, 20 мин, 4°С). Если использовали буферный раствор, в состав которого входил тритон Х-100, то ТВ далее трижды промывали буфером 50 мМ Tрис/HCl, pH 8. Если использовали буфер, содержащий мочевину, то ТВ промывали буфером 50 мМ Tрис/HCl, pH 8 один раз.
ТВ экстрагировали буферным раствором 50 мМ Tрис, 6 M мочевина, pH 10 в соотношении 1 : 10. Солюбилизированый белок разбавляли буфером для ренатурации (50 мМ Tрис, 20% глицерин, рН 10) до конечной концентрации белка 0.7 мг/мл и инкубировали при 25°С в течение 18 ч.
Аффинная хроматография гибридного белка
В работе использовали хроматографическую систему Akta Basic 100 (GE, США), и колонку XK 26/40 (GE, США), заполненную сорбентом Ni Sepharose® 6 Fast Flow (GE, США) объемом 43 мл, который перед нанесением раствора белка был уравновешен сначала тремя объемами 100 мМ раствора NiSO4, а затем тремя объемами буферного раствора (50 мМ Трис/HCl, 0.5 M NaCl, pH 8). Перед нанесением на колонну pH ренатурационной смеси доводили до 8 добавлением раствора соляной кислоты, а также добавляли хлористый натрий до конечной концентрации 0.5 М. После нанесения белкового раствора сорбент промывали двумя объемами буфера (50 мМ Трис/HCl, 0.5 M NaCl, pH 8) и элюировали сорбировавшийся белок двумя объемами буфера, содержащего имидазол (0.250 M имидозол, 25 мM Трис/HCl, 0.5 M NaCl, pH 8). Скорость потока составляла 5 мл/мин.
Гель-фильтрация
В работе использовали хроматографическую систему Akta Basic 100 (GE, США) с колонкой XK 16/100 (GE, США), заполненной сорбентом Sephadex G-25 (GE, США), объемом 170 мл. Сорбент перед нанесением раствора белка уравновешивали тремя объемами буферного раствора (50 мМ Tрис/HCl, pH 8), 20 мл элюата после аффинной хроматографии наносили со скоростью 1 мл/мин, элюцию белка проводили тем же буфером.
Иммобилизация t-L-HEP на хитиновом сорбенте
1 мл хитинового сорбента (NEB, Англия) уравновешивали 3 объемами буферного раствора 50 мM Трис/HCl, 100 мM NaCl, pH 8, а затем наносили на него 10 мл раствора t-L-HEP с концентрацией 1 мг/мл со скоростью 1 мл/мин. После нанесения раствора белка сорбент промывали тремя объемами исходного буферного раствора.
Расщепление модельных ГБ
Активность рекомбинантной L-HEP, t-L-HEP и ее иммобилизованного варианта, i-t-L-HEP, определяли с помощью электрофоретического метода по степени расщепления модельных гибридных белков, Trx-интерферон альфа-2b (модельный гибридный белок 1, мГБ-1) или Trx-пуротоксин-6 (модельный гибридный белок 2, мГБ-2), содержащих специфический сайт узнавания энтеропептидазы. В 10 мл белкового раствора, содержащего модельный гибридный белок с концентрацией 3 мг/мл, вносили L-HEP или t-L-HEP в соотношениях фермент/субстрат 1 : 50, 1 : 100, 1 : 200 и 1 : 400. Общие условия реакции: 50 мМ Трис/HCl, рН 8, 25°С, 1–24 ч. При использовании мГБ-1 добавляли ДТТ до конечной концентрации 0.5 мМ.
Через 0.5 мл i-t-L-HEP пропускали 10 мл раствора мГБ-1 в 50 мМ Трис/HCl, рН 8, 0.5 мМ ДТТ при 25°С, время контакта субстрата с ферментом от 12 до 60 с. Процедуру проводили 6 раз. В процессе отбирали пробы для электрофоретического анализа.
Аналитические методы контроля
Электрофоретический анализ белковых растворов осуществляли, используя систему PowerPac HC (BioRad, США), в 15% ПААГ по Лэммли [23]. Гели окрашивали раствором Coumassi R-250 (2.5 мг/мл). Концентрацию белка в растворах измеряли с помощью спектрофотометра Ultrospec – 1000 (Amersham Pharmacia Biotech, Great Britain) по методу Бредфорда [24].
Список литературы
Kaur J., Kumar A., Kaur J. // Int. J. Biol. Macromol. 2018. V. 106. P. 803–822. https://doi.org/10.1016/j.ijbiomac.2017.08.080
Waugh D.S. // Protein Expr. Purif. 2011. V. 80. P. 283–293. https://doi.org/10.1016/j.pep.2011.08.005
Barrett A., Rawlings N.D., Woessner J. // Elsevier, London, 2004. 2nd ed. P. 1513–1517.
Zheng X.L., Kitamoto Y., Sadler J.E. // Front. Biosci. (Elite Ed). 2009. V. 1. P. 242–249.
Kitamoto Y., Veile R.A., Donis-Keller H., Sadler J.E. // Biochemistry. 1995. V. 34. P. 4562–4568. https://doi.org/10.1021/bi00014a008
Lu D., Yuan X., Zheng X., Sadler J.E. // J. Biol. Chem. 1997. V. 272. P. 31 293–31 300. https://doi.org/10.1074/jbc.272.50.31293
Lu D., Fütterer K., Korolev S., Zheng X., Tan K., Waksman G., Sadler J.E. // J. Mol. Biol. 1999. V. 292. P. 361–373. https://doi.org/10.1006/jmbi.1999.3089
Light A., Janska H. // Trends Biochem. Sci. 1989. V. 14. P. 110–112. https://doi.org/10.1016/0968-0004(89)90133-3
Mann N.S., Mann S.K. // Exp. Biol. Med. 1994. V. 206. P. 114–118. https://doi.org/10.3181/00379727-206-43728
Light A., Janska H. // J. Protein Chem. 1991. V. 10. P. 475–480. https://doi.org/10.1007/bf01025475
Gasparian M.E., Ostapchenko V.G., Dolgikh D.A., Kirpichnikov M.P. // Biochem. 2006. V. 71. P. 113–119. https://doi.org/10.1134/S0006297906020015
LaVallie E.R., Rehemtulla A., Racie L.A., DiBlasio E.A., Ferenz C., Grant K.L., Light A., McCoy J.M. // J. Biol. Chem. 1993. V. 268. P. 23 311–23 317.
Svetina M., Krasevec N., Gaberc-Porekar V., Komel R. // J. Biotechnol. 2000. V. 76. P. 245–251. https://doi.org/10.1016/S0168-1656(99)00191-1
Choi S.I., Song H.W., Moon J.W., Seong B.L. // Biotechnol. Bioeng. 2001. V. 75. P. 718–724. https://doi.org/10.1002/bit.10082
Melicherová K., Krahulec J., Šafránek M., Lišková V., Hopková D., Széliová D., Turňa J. // Appl. Microbiol. Biotechnol. 2017. V. 101. P. 1927–1934. https://doi.org/10.1007/s00253-016-7960-3
Azhar M., Somashekhar R. // Prep. Biochem. Biotechnol. 2015. V. 45. P. 268–278. https://doi.org/10.1080/10826068.2014.907185
Gasparian M.E., Ostapchenko V.G., Schulga A.A., Dolgikh D.A., Kirpichnikov M.P. // Protein Expr. Purif. 2003. V. 31. P. 133–139. https://doi.org/10.1016/S1046-5928(03)00159-1
Chun H., Joo K., Lee J., Shin H.-C. // Biotechnol. Lett. 2011. V. 33. P. 1227–1232. https://doi.org/10.1007/s10529-011-0562-3
Simeonov P., Berger-Hoffmann R., Hoffmann R., Strater N., Zuchner T. // Protein Eng. Des. Sel. 2011. V. 24. P. 261–268. https://doi.org/10.1093/protein/gzq104
Skala W., Goettig P., Brandstetter H. // J. Biotechnol. 2013. V. 168. P. 421–425. https://doi.org/10.1016/j.jbiotec.2013.10.022
Tan H., Wang J., (Kent) Zhao Z. // Protein Expr. Purif. 2007. V. 56. P. 40–47. https://doi.org/10.1016/j.pep.2007.07.006
Bertani G. // J. Bacteriol. 1951vol. 62. P. 293–300.
Laemmli U.K. // Nature. 1970. V. 227. P. 680–685.
Bradford M.M. // Anal. Biochem. 1976. V. 72. P. 248–254.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия