

Биоорганическая химия, 2020, T. 46, № 6, стр. 686-692
Cинтез (3R,10R)- и (3S,10S)-диастереомеров 3,10-диметилспермина
М. А. Хомутов 1, М. Т. Хивонен 2, А. И. Салихов 1, А. О. Чижов 3, И. М. Рыжов 4, С. Н. Кочетков 1, Й. Вепсалайнен 2, Т. А. Кейнанен 2, А. Р. Хомутов 1, *
1 Институт молекулярной биологии им. В.А. Энгельгардта РАН
119991 Москва, ул. Вавилова, 32, Россия
2 Департамент Фармации, Биоцентр Куопио, Университет Восточной Финляндии
FI-70210 Куопио, Финляндия
3 Институт органической химии им. Н.Д. Зелинского РАН
119991 Москва, Россия
4 ФГБУН Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
* E-mail: alexkhom@list.ru
Поступила в редакцию 18.04.2020
После доработки 26.04.2020
Принята к публикации 30.04.2020
Аннотация
Предложен простой и удобный десятистадийный способ синтеза неизвестных ранее диастереомеров 1,12-диамино-3,10-диметил-4,9-диазадодекана (3,10-Me2Spm), исходя из коммерчески доступных энантиомеров 2-(Boc-амино)-1-пропанола. Целевые соединения были получены в граммовых количествах с высокой энантиомерной чистотой. Поскольку биохимические свойства диастереомеров 3,10-Me2Spm определяются конфигурацией хиральных центров, эти соединения представляют интерес не только для исследования ферментов метаболизма спермина и изучения его клеточных функций, но также и для изучения особенностей заболеваний, обусловленных нарушениями метаболизма спермина.
ВВЕДЕНИЕ
Биогенные полиамины спермин (Spm), спермидин (Spd) и их предшественник путресцин (Put) представляют собой низкомолекулярные поликатионы и присутствуют в клетках всех типов в μM–mM концентрациях, что, соответственно, и определяет множественность их клеточных функций, большинство из которых жизненно необходимы [1, 2]. Нарушение внутриклеточного содержания полиаминов сопутствует различным заболеваниям, таким как атеросклероз, инсульт, некоторые виды панкреатита, синдром Шнайдера-Робинсона, болезни Альцгеймера и Паркинсона, а также сопровождает воспалительные процессы и заболевания, связанные со снижением иммунного ответа [3–8]. Содержание полиаминов в опухолевых клетках повышено, а соединения, способные понижать их уровень, обладают противоопухолевой активностью [9] и используются также в период ремиссии [10].
Синдром Шнайдера-Робинсона – редкое заболевание, возникающее вследствие мутаций в гене сперминсинтазы человека, приводящих к значительному снижению количества Spm и увеличению содержания Spd во многих тканях, включая и мозг. Заболевание характеризуется умственной отсталостью, остеопорозом, гипотонией, нарушениями речи и судорогами [11]. Обработка Spm клеточных линий, полученных от пациентов с этим диагнозом, нормализует соотношение Spd/Spm, но для терапии (in vivo) подобный подход не пригоден [12] из-за легкости превращения Spm в Spd (рис. 1). Кроме того, Spm легко подвергается окислительному расщеплению как с участием сперминоксидазы (SMOX), так и с участием N1-ацетилполиаминооксидазы (РАОХ) (рис. 1), вследствие чего образуются токсичные акролеин и перекись водорода [1, 9].
Рис. 1.
Катаболизм и взаимопревращения полиаминов. PАОX – ацетилполиаминоксидаза; SSAT – спермидин/спермин-N1-ацетилтрансфераза; SMOX – сперминоксидаза; SpdSy – спермидинсинтаза; SpmSy – сперминсинтаза.
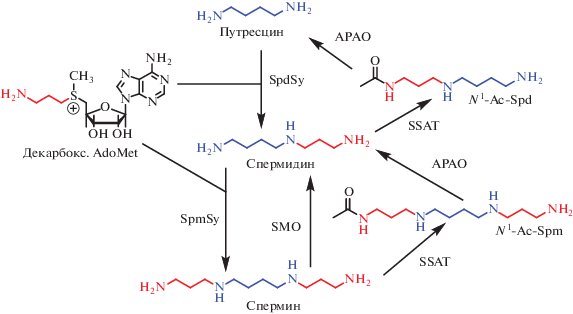
Однако для лечения синдрома Шнайдера-Робинсона могут быть полезны функционально-активные, метаболически устойчивые миметики Spm. (1R,12R)-1,12-Me2Spm (рис. 2) является аналогом Spm c требуемым набором свойств и не проявляет субстратных свойств ни в отношении спермидин/ спермин-N1-ацетилтрансферазы (SSAT), ни в отношении SMOX (рис. 1), тогда как (1S,12S)-1,12-Me2Spm оказался в два раза лучшим субстратом SMOX по сравнению с природным Spm [13]. Недавно было показано, что (1R,12R)-1,12-Me2Spm (рис. 2) – первое и единственное соединение, которое существенно снижает уровень Spd и возвращает соотношение Spd/Spm к нормальному уровню в различных органах модельных трансгенных мышей, используемых для изучения синдрома Шнайдера-Робинсона [14]. Однако использование (1R,12R)-1,12-Me2Spm в дозах выше 50 мг/кг невозможно из-за его токсичности, обусловленной индукцией SSAT [14].
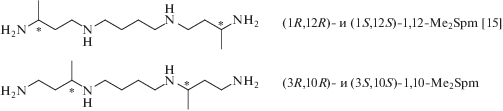
Клеточные эффекты рацемических бис-метилированных аналогов Spm определяются положением метильной группы. Так, рацемический 3,10-Me2Spm, в отличие от 1,12-Me2Spm, не индуцирует SSAT в клетках DU145 [16]. Соответственно, один из диастереомеров 3,10-Me2Spm может оказаться удобным инструментом для изучения синдрома Шнайдера-Робинсона in vivo в модельных системах. В настоящей статье мы описываем простой и удобный способ синтеза неизвестных ранее диастереомеров (3R,10R)- и (3S,10S)-3,10-Me2Spm (рис. 2), которые обладают очевидным биохимическим и, возможно, терапевтическим потенциалом.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Ключевой стадией синтеза аналогов и производных полиаминов служит создание C−N-связей в “скелете” полиаминов, и для этих целей используются разнообразные методы [17]. Ранее мы описали удобный способ синтеза, позволяющий получить рацемический 3,10-Me2Spm, исходя из 4-амино-2-бутанола, в качестве предшественника трех-углеродного фрагмента этого аналога [16]. Однако изомеры 4-амино-2-бутанола коммерчески недоступны.
В настоящей работе в качестве исходных веществ для получения диастереомеров 3,10-Me2Spm нами были использованы (2R)- и (2S)-изомеры 2-(Boc-амино)-1-пропанола ((Iа) и (Iб) соответственно), которые сначала превращали в Вос-защищенные аминонитрилы согласно описанному в работе [18], а последующее мягкое восстановление с использование LiAlH4 при –5°C без рацемизации приводило к N-Boc-диаминам (IIа) и (IIб) [15]. Карбобензоксилирование свободной аминогруппы давало (3R)- и (3S)-N1-Cbz-N3-Boc-диамины (III), а последующее удаление Boc-защитной группы позволило синтезировать (3R)- и (3S)-N1-Cbz-диамины (IV) c практически количественным выходом. Дальнейшие превращения включали в себя получение нозильных производных (Vа) и (Vб) и их алкилирование 1,4-дийодбутаном в DMF при 40°C в течение 48 ч в присутствии K2CO3 (схема 1 ). Для удаления не вошедших в реакцию изомеров (Vа) и (Vб), к реакционной смеси прибавляли BnBr (15 мол. %) и инкубировали еще 14 ч при 20°C. Затем Ns-защитные группы удаляли “one-pot” действием PhSH/K2CO3 в DMF, а соединения (VIa) и (VIб) выделяли колоночной хроматографией на силикагеле. Последующее удаление Cbz-групп каталитическим гидрированием над Pd-чернью и кристаллизация из MeOH-EtOH позволили получить целевые тетрагидрохлориды (3R,10R)- и (3S,10S)-3,10-Me2Spm’s (VIIa) и (VIIб) с суммарными выходами 18 и 16% (10-ти стадийный синтез), считая на исходные (2R)- и (2S)-N-Boc-аланинолы соответственно.
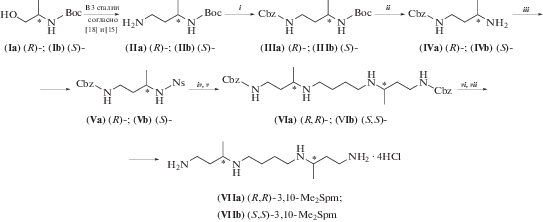
Схема 1 . Синтез (3R,10R)- и (3S,10S)-диастереомеров 3,10-Me2Spm. i – CbzCl/H2O/NaHCO3/THF; ii – HCl/MeOH; iii – NsCl/CH2Cl2/Et3N; iv – J(CH2)4J/DMF/K2CO3/45°C; v – PhSH/DMF/K2CO3; vi – H2/Pd/AcOH/MeOH; vii – вод. HCl.
Предложены и реализованы удобные схемы синтезов, позволяющие получать граммовые количества диастереомеров 3,10-Me2Spm с высокими суммарными выходами и оптической чистотой. Полученные диастереомеры 3,10-Me2Spm могут быть полезными инструментами в изучении ферментов метаболизма полиаминов, и, весьма вероятно, будут обладать различной активностью в экспериментах с культурами клеток и in vivo в случае моделей синдрома Шнайдера-Робинсона.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
При выполнении работы были использованы следующие реактивы: бензиловый эфир хлоругольной кислоты (CbzCl), тиофенол, хлорангидрид 2-нитробензолсульфокислоты (NsCl), триэтиламин (Et3N), – все фирмы Aldrich (США). Синтезы (3R)- и (3S)-N3-(трет-бутилоксикарбонил)-1,3-диаминобутанов были осуществлены, исходя из соответствующих (2R)- и (2S)-2-(Boc-амино)-1-пропанолов, (Aldrich (США), кат. № 469 505 и 469 513, соответственно; оба вещества обладали >98% оптической чистотой) согласно описанному ранее [15].
Колоночную хроматографию выполняли на силикагеле Kieselgel (40–63 μm, Merck, Germany), системы для элюции указаны в тексте. ТСХ проводили на пластинках Kieselgel 60 F254 plates (Merck, Germany) в системах: CH2Cl2–MeOH, 100 : 1 (A), диоксан–25% NH4OH, 95 : 5 (Б); EtOAc–гексан, 2 : 3 (В); и n-BuOH–AcOH–Py–H2O, 4 : 2 : 1 : 2 (Г). Вещества на хроматограммах визуализировали по УФ-поглощению, Boc-производные при помощи бромфенолового синего, а соединения со свободной аминогруппой – используя цветную реакцию с нингидрином.
Спектры 1Н и 13С ЯМР регистрировали на спектрометре Bruker AM-300 в CDCl3 и D2O, внутренний стандарт – Me4Si (CDCl3) и натриевая соль 3-триметилсилилпропан-сульфокислоты (D2O). Химические сдвиги приведены в миллионных долях, а КССВ – в герцах. Температуру плавления определяли в открытом капилляре на приборе Mel-Temp 1202D фирмы Electrotermals. Величину удельного вращения определяли на приборе 341 Polarimeter фирмы Perkin-Elmer, растворители и концентрации растворов указаны в тексте. Элементный анализ выполняли на CHN-анализаторе Carlo Erba 1106. Масс-спектры высокого разрешения (HR-MS) регистрировали на приборе Bruker maXis методом электрораспылительной ионизации (ESI) [19]. Измерения выполняли на положительных ионах (напряжение на капилляре – 4500 В) в диапазоне m/z 50–3000 с использованием внешней калибровки (ESI Tuning Mix, Agilent). Шприцевой ввод тетрагидрохлоридов (VIIa)/(VIIб) осуществляли в виде их водных растворов, для остальных веществ в качестве растворителя использовали ацетонитрил; скорость потока – 5 мкл/мин; газ-распылитель – азот; температура интерфейса – 180°C.
(3R)-N1-(Бензилоксикарбонил)-N 3-(трет-бутилоксикарбонил)-1,3-диаминобутан (IIIa). К охлажденному до 8°С раствору 3.7 г (19.7 ммоль) (3R)-N3-(трет-бутилоксикарбонил)-1,3-диаминобутана (IIa) и 2.53 г (20.5 ммоль) Et3N в 60 мл смеси С6Н6/Et2O (1 : 2) при перемешивании прибавляли за 40 мин раствор 3.5 г (20.5 ммоль) CbzCl в 15 мл C6H6, перемешивали 1 ч при 8°С и 4 ч при 20°С. Осадок отфильтровывали, фильтрат промывали последовательно H2O (3 × 15 мл), 10% лимонной кислотой (4 × 15 мл), H2O (10 мл), 5 М NaCl (2 × 15 мл) и высушивали над MgSO4. Растворитель отгоняли в вакууме, остаток очищали колоночной хроматографией на SiO2 (90 г), элюируя смесью CH2Cl2–МеOH, 98 : 2. Фракции, содержащие соединение (IIIa), упаривали в вакууме досуха, что привело к густому маслу, затвердевшему при высушивании в вакууме (25°С/0.5 мм Hg). После перекристаллизации из гексана получали 5.2 г (82%) соединения (IIIa), т.пл. 82.5–83°С, Rf 0.39 (A), $[\alpha ]_{{\text{D}}}^{{20}}$ –50.4° (c 2.0, CH2Cl2). 1H-ЯМР (CDCl3) δ: 7.39–7.27 (5H, м, Ph), 5.55 (1H, уш.с., NHCbz), 5.09 (2H, с, CH2Ph), 4.31 (1H, уш.с., NHBoc), 3.8–3.69 (1H, м, CH), 3.55–3.35 (1H, м, NHCH2), 3.08–2.91 (1H, м, NHCH2), 1.77–1.63 (1H, м, СН2CH), 1.52–1.32 (10H, м, С(СН3)3 + СН2CH), 1.14 (3H, д, J 6.5, CH3). 13C-ЯМР (CDCl3) δ: 156.59, 156.07, 136.93, 128.58, 128.12, 128.04, 79.50, 66.55, 43.93, 37.94, 37.15, 28.45, 21.60. Найдено, %: С 63.47; Н 8.21; N 8.58. C17H26N2O4. Вычислено, %: С 63.33; Н 8.13; N 8.69.
(3S)-N1-(Бензилоксикарбонил)-N3-(трет-бутилоксикарбонил)-1,3-диаминобутан (IIIб). Получали аналогично соединению (IIIа), исходя из 2.76 г (14.7 ммоль) соединения (IIб) и 2.6 г (15.2 ммоль) Cbz-Cl, что приводило к соединению (IIIб) (4.07 г, 86%), т. пл. 82.5–83°С, Rf 0.39 (A), $[\alpha ]_{{\text{D}}}^{{20}}$ +48.9° (c 2.0, CH2Cl2). 1H-ЯМР (CDCl3) δ: 7.40–7.28 (5H, м, Ph), 5.55 (1H, уш.с., NHCbz), 5.05 (2H, с, CH2Ph), 4.35 (1H, уш.с., NHBoc), 3.82–3.65 (1H, м, CH), 3.51–3.33 (1H, м, NHCH2), 3.10–2.88 (1H, м, NHCH2), 1.78–1.61 (1H, м, СН2CH), 1.50–1.34 (10H, м, С(СН3)3 + СН2CH), 1.14 (3H, д, J 6.5, CH3). 13C-ЯМР (CDCl3) δ: 156.61, 156.11, 136.95, 128.58, 128.17, 128.09, 79.54, 66.61, 43.96, 37.97, 37.19, 28.50, 21.69. Найдено, %: С 63.41; Н 8.18; N 8.66. C17H26N2O4. Вычислено, %: С 63.33; Н 8.13; N 8.69.
(3R)-N1-(Бензилоксикарбонил)-N3-(2-нитрофенилсульфонил)-1,3-диаминобутан (Va). К охлажденному до 0°С раствору 4.76 г (14.8 ммоль) соединения (IIIa) в 15 мл абс. EtOH прибавляли 6 мл 10 М HCl/EtOH и через 3 ч при 20°С реакционную смесь упаривали в вакууме досуха. Остаток соупаривали с EtOH (2 × 20 мл), растворяли в EtOH (15 мл), выливали в абс. Et2O (150 мл) и оставляли на ночь при –20°С. Выделившееся масло отделяли, к остатку прибавляли 2 М NaOH (15 мл) и экстрагировали CH2Cl2 (4 × 15 мл). Объединенные CH2Cl2-вытяжки последовательно промывали H2O (2 × 15 мл), 5 М NaCl (2 × 15 мл) и высушивали над K2CO3. Растворитель отгоняли в вакууме, остаток высушивали в вакууме над P2O5 и получали соединение (IVa) (2.7 г, 82%) в виде густого масла, Rf 0.35 (Б). 1Н-ЯМР (CDCl3) δ: 7.39–7.30 (5H, м, Ph), 5.46 (1Н, уш.с, NH), 5.10 (2Н, с, CH2Ph), 3.46–3.30 (1H, м, NHCH2), 3.29–3.16 (1H, м, NHCH2), 3.04–2.90 (1H, м, CH), 1.66–1.52 (1Н, м, NHCH2CH2), 1.50–1.30 (3Н, м, NHCH2CH2 + NH2), 1.10 (3H, д, J 6.4, CH3). 13С-ЯМР (CDCl3) δ: 156.64, 136.91, 128.64, 128.23, 128.19, 66.71, 45.62, 39.23, 29.84, 24.97.
К охлажденному до 4°C раствору 2.64 г (11.9 ммоль) соединения (IVa) и 2.13 мл (15.4 ммоль) Et3N в 25 мл абс. CH2Cl2 прибавляли при перемешивании в течение 10 мин раствор 2.68 г (12.1 ммоль) NsCl в 25 мл абс. CH2Cl2, перемешивали 1 ч при 4°C и еще 3 ч при 20°C. Осадок отфильтровывали, фильтрат разбавляли 25 мл CH2Cl2 и последовательно промывали H2O (3 × 15 мл), 10% лимонной кислотой (4 × 20 мл), H2O (15 мл), 5 М NaCl (2 × × 15 мл) и высушивали над MgSO4. Растворитель отгоняли в вакууме, остаток очищали колоночной хроматографией на SiO2 (160 г), элюируя смесью СН2Сl2 → СН2Сl2/MeOH = 100 : 0.5. Фракции, содержащие соединение (Va), упаривали в вакууме досуха и получали 4.1 г (85%, считая на (IVa)) соединения (Va), в виде полузакристаллизовавшегося масла, Rf 0.21 (В), $[\alpha ]_{{\text{D}}}^{{20}}$ –150.8° (c 3.0, СН2Сl2). 1H-ЯМР (CDCl3) δ: 8.17–8.09 (1H, м, Ns), 7.89–7.80 (1H, м, Ns), 7.76–7.67 (2H, м, Ns), 7.41–7.29 (5H, м, Ph), 5.25–5.14 (2H, м, NH-Cbz + NH-Ns), 5.10 (2H, с, CH2Ph), 3.66–3.50 (1H, м, CH), 3.47–3.33 (1H, м, NHCH2), 3.29–3.15 (1H, м, NHCH2), 1.87–1.69 (1H, м, NHCH2CH2), 1.61–1.48 (1H, м, NHCH2CH2), 1.04 (3H, д, J 6.6, CH3). 13C-ЯМР (CDCl3) δ: 156.59, 147.98, 136.75, 133.73, 133.07, 130.83, 128.66, 128.23 (2C), 125.57, 66.78, 48.80, 37.65, 37.58, 21.71. HRESIMS: m/z вычислено C18H22N3O6S+, [M + H]+: 408.1224. Найдено: 408.1221.
(3S)-N1-(Бензилоксикарбонил)-N3-(2-нитрофенилсульфонил)-1,3-диаминобутан (Vб). Получали аналогично соединению (Vа), исходя из 2.96 г (9.2 ммоль) соединения (IIIб) через промежуточное соединение (IVб), Rf 0.35 (Б), 1Н-ЯМР (CDCl3) δ: 7.38–7.28 (5H, м, Ph), 5.48 (1Н, уш.с., NH), 5.09 (2Н, с, CH2Ph), 3.43–3.30 (1H, м, NHCH2), 3.28–3.17 (1H, м, NHCH2), 3.02–2.92 (1H, м, CH), 1.64–1.52 (1Н, м, NHCH2CH2), 1.48–1.37 (1Н, м, NHCH2CH2), 1.22 (2H, уш.с., NH2), 1.09 (3H, д, J 6.4, CH3). 13С-ЯМР (CDCl3) δ: 156.66, 136.83, 128.63, 128.21, 128.18, 66.55, 45.49, 39.11, 29.82, 24.78, что проводило к соединению (Vб) (2.51 г, 67%, считая на IIIб) в виде полузакристаллизовавшегося масла, Rf 0.22 (В), $[\alpha ]_{{\text{D}}}^{{20}}$ +149.6° (c 3.0, СН2Сl2). 1H-ЯМР (CDCl3) δ: 8.17–8.10 (1H, м, Ns), 7.88–7.81 (1H, м, Ns), 7.75–7.68 (2H, м, Ns), 7.41–7.29 (5H, м, Ph), 5.20–5.08 (м, 4H, NH-Ns + + NH-Cbz + CH2Ph), 3.65–3.52 (1H, м, CH), 3.46–3.35 (1H, м, NHCH2), 3.29–3.17 (1H, м, NHCH2), 1.87–1.71 (1Н, м, NHCH2CH2), 1.64–1.49 (1H, м, NHCH2CH2), 1.04 (3H, д, J 6.6, CH3). 13C-ЯМР (CDCl3) δ: 156.60, 148.03, 136.78, 133.72, 133.07, 130.82, 128.67, 128.25 (2C), 125.59, 66.81, 48.82, 37.72, 37.22, 21.73. HRESIMS: m/z вычислено для C18H22N3O6S+, [M + H]+: 408.1224. Найдено: 408.1220.
(3R,10R)-N1,N12-Бис-(бензилоксикарбонил)-1,12-диамино-3,10-диметил-4,9-диазадодекан (VIa). Суспензию 3.6 г (8.86 ммоль) соединения (Va), 3.8 г (27.2 ммоль) К2СО3 и 1.18 г (3.81 ммоль) 1,4-дийодбутана в абс. DMF (15 мл) перемешивали в течение 24 ч при 45°С, прибавляли 0.5 мл (4.2 ммоль) BnBr и перемешивали еще 12 ч при 20°С. Затем к реакционной смеси прибавляли 2.0 г (14 ммоль) K2CO3 и 2.0 мл (19.2 ммоль) PhSH в DMF (5 мл) и перемешивали 12 ч при 20°C, осадок отделяли центрифугированием, промывали DMF (2 × 15 мл) и объединенные органические вытяжки упаривали в вакууме. Остаток растворяли в CH2Cl2 (25 мл), последовательно промывали Н2О (2 × 5 мл), 5М NaCl (2 × 5 мл) и высушивали над K2CO3. Растворитель отгоняли в вакууме, остаток очищали колоночной хроматографией на SiO2 (90 г), элюируя смесью диоксан — 25% NH4OH (95 : 5). Фракции, содержащие соединение (VIa), упаривали в вакууме досуха и остаток высушивали над P2O5, что приводило к соединению (VIa) (3.18 г, 72%) в виде бесцветного масла. Rf 0.24 (Б), $[\alpha ]_{{\text{D}}}^{{20}}$ –16.9° (c 2.0, СН2Сl2). 1H-ЯМР (CDCl3) 7.41–7.28 (10H, м, 2*Ph), 5.98 (2H, уш.с, 2*NHCbz), 5.09 (4H, с, 2*CH2Ph), 3.41–3.15 (4H, м, 2*CbzNHCH2), 2.78–2.57 (4H, м, 2*CH + CHCH2), 2.55–2.42 (2H, м, CHCH2), 1.65–1.39 (10H, м, 2*NHCH2CH2, 1.05 (6H, д, J 6.4, 2*CH3). 13C-ЯМР (CDCl3) δ: 156.58, 137.07, 128.58, 128.08 (2C), 66.50, 52.29, 47.01, 39.11, 35.85, 28.41, 20.54. HRESIMS: m/z вычислено для ${{{\text{C}}}_{{{\text{28}}}}}{{{\text{H}}}_{{{\text{43}}}}}{{{\text{N}}}_{{\text{4}}}}{\text{O}}_{4}^{ + },$ [M + H]+: 499.3279. Найдено: 499.3274.
(3S,10S)-N1,N12-Бис-(бензилоксикарбонил)-1,12-диамино-3,10-диметил-4,9-диазадодекан (VIб). Получали аналогично соединению (VIa), исходя из 2.4 г (5.83 ммоль) соединения (Vб) и 0.84 г (2.7 ммоль) 1,4-дийодбутана в абс. DMF, что после очистки при помощи колоночной хроматографии на силикагеле приводило к соединению (VIб) (2.03 г, 70%) в виде бесцветного масла. Rf 0.24 (Б), $[\alpha ]_{{\text{D}}}^{{20}}$ +16.3° (c 2.0, СН2Сl2). 1H-ЯМР (CDCl3) 7.40–7.27 (10H, м, 2*Ph), 5.98 (2H, уш.с, 2*NHCbz), 5.09 (4H, с, 2*CH2Ph), 3.40–3.15 (4H, м, 2*CbzNHCH2), 2.78–2.56 (4H, м, 2*CH + CHCH2), 2.55–2.41 (2H, м, CHCH2), 1.67–1.38 (10H, м, NHCH2CH2), 1.05 (6H, д, J 6.4, 2*CH3). 13C-ЯМР (CDCl3) δ: 156.58, 137.08, 128.59, 128.09 (2C), 66.51, 52.31, 47.02, 39.13, 35.85, 28.42, 20.55. HRESIMS: m/z вычислено для ${{{\text{C}}}_{{{\text{28}}}}}{{{\text{H}}}_{{{\text{43}}}}}{{{\text{N}}}_{{\text{4}}}}{\text{O}}_{4}^{ + },$ [M + H]+: 499.3279. Найдено: 499.3275.
Тетрагидрохлорид (3R,10R)-1,12-диамино-3,10-диметил-4,9-диазадодекана (VIIa). К раствору 2.86 г (5.74 ммоль) соединения (VIa) в 35 мл смеси AcOH : MeOH (1 : 1) прибавляли 1 мл суспензии Pd-черни в MeOH и гидрировали при атмосферном давлении до прекращения выделения CO2. Pd-чернь отфильтровывали, промывали MeOH, объединенные фильтраты упаривали в вакууме досуха. Остаток соупаривали с EtOH (2 × 15 мл), растворяли в 20 мл EtOH, прибавляли 7 мл 5 M HCl и полученный раствор упаривали в вакууме досуха. Остаток соупаривали с EtOH (3 × 15 мл), высушивали в вакууме над Р2О5 и перекристаллизовывали из смеси MeOH-EtOH, что после высушивания в вакууме над P2O5/КОН приводило к целевому соединению (VIIa) (1.6 г, 74%) в виде бесцветных кристаллов: т.пл. 242–243°C, Rf 0.16 (Г), $[\alpha ]_{{\text{D}}}^{{20}}$ +5.9° (c 2.0, H2O). 1Н-ЯМР (D2O) δ: 3.50–3.37 (2H, м, 2*СН), 3.23–3.03 (8H, м 2*NH2СН2 + + 2*NHСН2), 2.26–2.12 (2H, м, 2*CHCH2), 2.02–1.86 (2H, м, 2*CHCH2), 1.85–1.72 (4H, м, 2*NHСН2СН2), 1.36 (6H, д, J 6.6 Hz, 2*СН3). 13С-ЯМР (D2O) δ: 52.74, 44.87, 36.56, 30.90, 23.71, 15.79. HRESIMS: m/z вычислено для ${{{\text{C}}}_{{{\text{12}}}}}{{{\text{H}}}_{{{\text{31}}}}}{\text{N}}_{4}^{ + },$ [M + H]+: 231.2543. Найдено: 231.2545.
Тетрагидрохлорид (3S,10S)-1,12-диамино-3,10-диметил-4,9-диазадодекана (VIIб). Получали аналогично соединению (VIIа), исходя из 1.69 г (3.4 ммоль) соединения (VIб), что приводило к соединению (VIIб) (0.90 г, 70%) в виде бесцветных кристаллов: т.пл. 240–242°C, Rf 0.16 (Г), $[\alpha ]_{{\text{D}}}^{{20}}$ –6.3° (c 2.0, H2O). 1Н-ЯМР (D2O) δ: 3.51–3.36 (2H, м, 2*СН), 3.23–3.03 (8H, м 2*NH2СН2 + 2*NHСН2), 2.26–2.12 (2H, м, CHCH2), 2.02–1.87 (2H, м, 2*CHCH2), 1.85–1.72 (4H, м, 2*NHСН2СН2), 1.36 (6H, д, J 6.6 Hz, 2*СН3). 13С-ЯМР (D2O) δ: 52.72, 44.88, 36.55, 30.88, 23.71, 15.75. HRESIMS: m/z вычислено для ${{{\text{C}}}_{{{\text{12}}}}}{{{\text{H}}}_{{{\text{31}}}}}{\text{N}}_{4}^{ + },$ [M + H]+: 231.2543. Найдено: 231.2542.
Список литературы
Miller-Fleming L., Olin-Sandoval V., Campbell K., Ralser M. // J. Mol. Biol. 2015. V. 427. P. 3389–3406. https://doi.org/10.1016/j.jmb.2015.06.020
Pegg A.E. // J. Biol. Chem. 2016. V. 291. P. 14 904–14 912. https://doi.org/10.1074/jbc.R116.731661
Ramani D., De Bandt J.P., Cynober L. // Clin. Nut. 2014. V. 33. P. 14–22. https://doi.org/10.1016/j.clnu.2013.09.019
Inoue1 K., Tsutsui H., Akatsu H., Hashizume Y., Matsukawa N., Yamamoto T., Toyo’oka T. // Sci. Rep. 2013. V. 3. P. 2364. https://doi.org/10.1038/srep02364
Alhonen L., Parkkinen J.J., Keinanen T., Sinervirta R., Herzig K.H., Janne J. // Proc. Natl. Acad. Sci. USA. 2000. V. 97. P. 8290–8295. https://doi.org/10.1073/pnas.140122097
Eisenberg T., Abdellatif M., Schoeder S., Primessnig U., Stekovic S., Pendl T., Harger A., Schipke J., Zimmermann A., Schmidt A., Tong M., Ruckenstuhl Ch., Dammbrueck Ch., Gross A.S., Herbst V., Magnes Ch., Trausinger G., Narath S., Meinitzer A., Hu Z., Kirsch A., Eller K., Carmona-Gutierrez D., Büttner S., Pietrocola F., Knittelfelder O., Schrepfer E., Rockenfeller P., Simonini C., Rahn A., Horsch M., Moreth K., Beckers J., Fuchs H., Gailus-Durner V., Neff F., Janik D., Rathkolb B., Rozman J., Hrabe de Angelis M., Moustafa T., Haemmerle G., Mayr M., Willeit P., von Frieling-Salewsky M., Pieske B., Scorrano L., Pieber T., Pechlaner R., Willeit J., Sigrist S.J., Linke W.A., Mühlfeld Ch., Sadoshima J., Dengjel J., Kiechl S., Kroemer G., Sedej S., Madeo F. // Nature Med. 2016. V. 22. P. 1428–1438. https://doi.org/10.1038/nm.4222
Lewandowski N.M., Ju S., Verbitsky M., Ross B., Geddie M.L., Rockenstein E., Adame A., Muhammad A., Vonsattel J.P., Ringe D., Cote L., Lindquist S., Masliah E., Petsko G.A., Marder K., Clark L.N., Small S.A. // Proc. Natl. Acad. Sci. USA. 2010. V. 107. P. 16 970–16 975. https://doi.org/10.1073/pnas.1011751107
Igarashi K., Kashiwagi K. // Mol. Nutr. Food Res. 2011. V. 55. P. 1332–1341. https://doi.org/10.1002/mnfr.201100068
Casero R.A., Murray Stewart T., Pegg A.E. // Nature Rev. Cancer. 2018. V. 18. P. 681–695. https://doi.org/10.1038/s41568-018-0050-3
Gerner E.W., Bruckheimer E., Cohen A. // J. Biol. Chem. 2018. V. 293. P. 18 770–18 778. https://doi.org/10.1074/jbc.TM118.003343
Cason A.L., Ikeguchi Y., Skinner C., Wood T.C., Lubs H.A., Martinez F., Simensen R.J., Stevenson R.E., Pegg A.E., Schwartz C.E. // Eur. J. Hum. Genet. 2003. V. 11. P. 937–944. https://doi.org/10.1038/sj.ejhg.5201072
Murray-Stewart T., Dunworth M., Foley J.R., Schwartz C.E., Casero R.A. // Med. Sci. (Basel). 2018. V. 6. P. E112. https://doi.org/10.3390/medsci6040112
Hyvonen M.T., Keinanen T.A., Cerrada-Gimenez M., Sinervirta R., Grigorenko N., Khomutov A.R., Vepsalainen J., Alhonen L., Janne J. // J. Biol. Chem. 2007. V. 282. P. 34700–34706. https://doi.org/10.1074/jbc.M704282200
Murray Stewart T., Khomutov M., Foley J.R., Guo X., Holbert C.E., Dunston T.T., Schwartz C.E., Gabrielson K., Khomutov A., Casero R.A. // J. Biol. Chem. 2020. V. 295. P. 3247–3256. https://doi.org/10.1074/jbc.RA119.011572
Grigorenko N.A., Khomutov A.R., Keinänen T.A., Järvinen A., Alhonen L., Jänne J., Vepsäläinen J. // Tetrahedron. 2007. V. 63. P. 2257–2262. https://doi.org/10.1016/j.tet.2006.12.065
Khomutov M., Hyvönen M.T., Simonian A., Formanovsky A.A., Mikhura I.V., Chizhov A.O., Kochetkov S.N., Alhonen L., Vepsäläinen J., Keinänen T.A., Khomutov A.R. // J. Med. Chem. 2019. V. 62. P. 11 335–11 347. https://doi.org/10.1021/acs.jmedchem.9b01666
Хомутов M.A., Михура И.В., Кочетков С.Н., Хомутов A.Р. // Биоорган. химия. 2019. Т. 45. С. 588–614. [Khomutov M.A., Mikhura I.V., Kochetkov S.N., Khomutov A.R. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 463–487.] https://doi.org/10.1134/S013234231906023X
Lebreton L., Jost E., Carboni B., Annat J., Vaultier M., Dutartre P., Renaut P. // J. Med. Chem. 1999. V. 42. P. 4749–4763. https://doi.org/10.1021/jm991043x
Tsedilin A.M., Fakhrutdinov A.N., Eremin D.B., Zalesskiy S.S., Chizhov A.O., Kolotyrkina N. G., Ananikov V. P. // Mend. Commun. 2015. V. 25. P. 454–456. https://doi.org/10.1016/j.mencom.2015.11.019
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия