

Биоорганическая химия, 2020, T. 46, № 6, стр. 792-796
Особенности межфазного каталитического гликозилирования 2-меркаптобензимидазола
Т. А. Чупахина 1, *, В. О. Курьянов 1
1 Крымский федеральный университет им. В.И. Вернадского
295007 Симферополь, просп. Вернадского, 4, Россия
* E-mail: tachup@rambler.ru
Поступила в редакцию 27.03.2020
После доработки 03.04.2020
Принята к публикации 15.04.2020
Аннотация
Исследована реакция 2-ацетамидо-3,4,6-три-O-ацетил-2-дезокси-α-D-глюкопиранозилхлорида с 2-меркаптобензимидазолом в условиях межфазного катализа и выявлены особенности протекания межфазного процесса. Установлено, что основными продуктами глюкозаминилирования являются соответствующие N- и S-β-D-глюкозаминиды. Показано, что глюкозаминилирование 2-меркаптобензимидазола в межфазной каталитической системе “безводный хлористый метилен–гидрид натрия” протекает региоспецифично. Строение синтезированных соединений охарактеризовано с помощью 1H-ЯМР-спектроскопии и масс-спектрометрии.
ВВЕДЕНИЕ
Синтез и исследование биологической активности веществ охватывает различные области современной органической химии. Большое влияние на биологическую активность оказывает присутствие в структуре соединений гетероциклического фрагмента. Так, бензимидазольный фрагмент присутствует в структуре многих препаратов с различной биологической активностью. Среди широко применяемых лекарственных средств следует упомянуть дибазол (спазмолитик), пимозид и дроперидол (нейролептики), астемизол (антигистаминный препарат), омепразол (противоязвенный препарат) и др. [1, 2]. В настоящее время для лечения заболеваний, вызываемых патогенными грибами, используется ряд различных по происхождению и механизму действия лекарственных препаратов. Наиболее многочисленную группу синтетических противогрибковых средств представляют производные азотсодержащих циклических соединений. Не стали исключением и соединения ряда бензимидазола [3]. В последнее время отмечена тенденция роста грибковых заболеваний и устойчивости возбудителей грибковых инфекций к имеющимся лекарственным препаратам. В связи с этим серьезное внимание уделяется поиску новых противогрибковых препаратов. Введение углеводных фрагментов в молекулы биологически активных гетероциклических соединений может послужить одним из примеров решения подобной проблемы и оказаться весьма перспективным. Ранее, в рамках изучения спектра биологической активности синтетических гликозидов, была показана определенная фунгицидная и фунгистатическая активность S- и N-β-глюкозаминидов меркапто-1,2,4-триазолов и 2-меркаптобензимидазолов методом диффузии и серийных разведений в жидкой питательной среде на тест-культурах дрожжевых грибов Candida tenuis и плесневых грибов Aspergillus niger [4]. Таким образом, избирательная модификация 2-меркаптобензимидазола, протекающая в различных условиях межфазной каталитической реакции с перацетатом α-D-глюкозаминилхлорида, представляет интерес с целью получения набора замещенных меркаптобензимидазолов с потенциальной биологической активностью.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Ранее нами неоднократно в реакциях гликозилирования исследовались в качестве гликозил-акцепторов близкие структурные аналоги – тиол-ионные и амидо-имидольные таутомеры. Было показано, что в случае тиол-тионных таутомеров, например, в качестве продуктов гликозилирования образуется либо смесь региоизомеров, либо только один из региоизомеров. Использование гликозил-донора, несущего соучаствующие группы у C2, обеспечивало строгую стереоселективность процесса [5, 6].
В научной литературе Э. Ашри с соавторами описал гликозилирование бензимидазол-2-тиона в различных условиях. Проведение процесса в среде безводного DMFA/ацетон в присутствии K2CO3 или Et3N, в водной щелочи/ацетоне позволило авторам получить S-гликозид с выходами 51–71% [7].
Успешное применение катализируемой краун-эфирами МФ реакции α-D-глюкозаминилхлорида (I) с гетероароматическими соединениями [8] побудило нас к исследованию региоселективного процесса глюкозаминилирования 2-меркаптобензимидазола в условиях МФК.
Нами были последовательно изучены условия и состав продуктов МФ реакции α-D-глюкозаминилхлорида (I) с 2-меркаптобензимидазолом и однозначно доказано строение образующихся региоизомерных глюкозаминидов.
Реакцию α-хлорида (I) с 2-меркаптобензимидазолом проводили по методике, описанной нами ранее [9], используя безводные K2CO3 и ацетонитрил, при температуре 20–22°С и мольном соотношении (I)–(II)–K2CO3–15C5 равном 1 : 1 : 4.5 : 0.2. Обнаружено (ТСХ), что в условиях эксперимента конверсия гликозил-донора (I) в N-β- и S-β-глюкозаминиды (III) и (IV) происходила за 1.5 ч (рис. 1). Целевые соединения получены после очистки с помощью колоночной хроматографии с выходами 62 и 20% соответственно (табл. 1). В отсутствие межфазного катализатора процесс шел медленнее, и полная конверсия глюкозаминилхлорида (I) в продукты реакции завершалась за 5 ч. Выходы целевых гликозидов (III) и (IV) фактически остались прежними и составили 60 и 20% соответственно. Таким образом, использование 15C5 позволило сократить время реакции гликозилирования.
Рис. 1.
Строение исходного субстрата (I), 2-меркаптобензимидазола и конечных гликозидов (III), (IV).
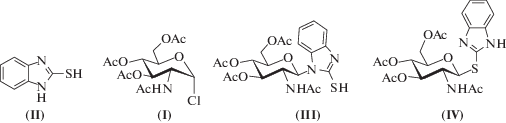
Таблица 1.
Условия реакции глюкозаминилирования 2-меркаптобензимидазола, время 100%-ной конверсии субстрата (I) (по данным ТСХ) и выходы гликозидов (III), (IV)*
| Гликозид** | Основание, моль | Катализатор, моль | Время реакции, ч | Выход (III)/(IV), % |
|---|---|---|---|---|
| (III)A, (IV)A | K2CO3, 4.5 | 15C5, 0.2 | 1.5 | 62/20 |
| (III)B, (IV)B | K2CO3, 4.5 | – | 5 | 60/20 |
| (III)C, (IV)C | K2CO3, 4.5 | Peg, 0.1 | 3 | 64/19 |
| (III)D | NaH, 1 | 15C5, 0.2 | 2.5 | 65 |
Известно, что олигоэтиленгликоли или полиэтиленгликоли, ввиду близости их химической природы, могут выступать как межфазные катализаторы во многих органических реакциях наряду с краун-эфирами [10]. Данное предположение нами также подтверждалось в изучении реакции глюкозаминилирования фенолов [11].
Поэтому следующим шагом в изучении обсуждаемой МФ реакции явилась замена катализатора межфазного переноса 15C5 на Peg (Мr 1500). Реакцию проводили при эквимолярном соотношении гликозил-донора (I) и гликозил-акцептора (II) в среде безводного ацетонитрила при использовании 4.5-кратного избытка твердого карбоната калия и 10 моль% Peg [11]. Конверсия глюкозаминилхлорида (I) в продукты глюкозаминилирования протекала за 3 ч. По данным ТСХ в реакционной среде были идентифицированы, как и в случае с 15C5, два основных продукта реакции. Выходы N-β- и S-β-гликозидов (III) и (IV) оказались сравнимыми с выходами, полученными в первом случае, и составили 64 и 19% соответственно.
Так как условия реакции различных гликозил-доноров с гетероциклами могут существенно влиять на регионаправленность превращений [12], нами проведено глюкозаминилирование 2-меркаптобенимидазола α-D-глюкозаминилхлоридом (I) в среде безводного хлористого метилена в присутствии гидрида натрия и 15C5. Первоначально в стехиометрических условиях в течение часа выдерживали 2-меркаптобензимидазол и гидрид натрия (60% суспензия в парафиновом масле) в среде безводного хлористого метилена, после этого в реакционную смесь вносили 20 моль% 15C5 и эквимолярное количество гликозил-донора (I). Было обнаружено, что превращение α-хлорида (I) протекало с образованием единственного продукта реакции – N-β-гликозида (III). Процесс завершался за 2.5 ч с выходом продукта реакции 65% после очистки с помощью колоночной хроматографии.
Интенсивные пики молекулярных ионов [M + H]+ с m/z 480 в масс-спектрах N-β- и S-β-гликозидов (III) и (IV) подтверждают введение углеводного остатка по одному из реакционных центров 2-меркаптобензимидазола и свидетельствуют о том, что гликозиды между собой являются изомерами. Для установления структуры и природы гликозидной связи в соединениях (III) и (IV), являющихся S- и N-глюкозаминидами, была привлечена 1H-ЯМР-спектроскопия. 1,2-транc-Конфигурация гликозидной связи подтверждается наличием в их 1H-ЯМР- спектрах дублетов аномерных протонов с химическими сдвигами 6.36 и 5.67 м.д. и константами спин-спинового взаимодействия 9.6 и 10.8 Гц. Отметим, что в 1H-ЯМР-спектре N-гликозида (III) наблюдается смещение в слабое поле сигнала аномерного протона по сравнению с дублетом H-1 его региоизомера – S-глюкозаминида (IV). Аналогичное смещение идентифицировано и для оставшихся сигналов скелетных протонов гликозидного остаткав N-гликозида (III) по сравнению с изомерным (IV). Химические сдвиги сигналов протонов гликозидного и гетероциклического остатков соединений (III) и (IV) соответствовали значениям, полученным нами ранее [5, 8], и литературным данным [7, 12–14].
Таким образом, установлено, что МФ реакции α-D-глюкозаминилхлорида (I) с 2-меркаптобензимидазолом протекает региоспецифично в зависимости от условий реакции. Применение в качестве МФ катализатора 15C5 и альтернативная замена его на Peg приводит к получению смеси региоизомеров (III) и (IV) в системе “безводный ацетонитрил–карбонат калия”, причем процесс образования N-гликозида является преобладающим. Образование N-гликозида (III) как единственного продукта реакции установлено в системе гидрид натрия – 15C5.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Температуры плавления определяли на приборе ПТП-1, оптическое вращение (20–22°С) – на поляриметре Polamat-A (длина волны λ, 546 нм). 1H-ЯМР-спектры (δ, м.д.; КССВ, Гц) получены для растворов в DMSO-d6 на приборе Varian Mercury-400 (400 МГц), внутренний стандарт – Me4Si. ТСХ проводили на пластинках Sorbfil АФВ-УФ (Сорбполимер, Россия). Зоны веществ обнаруживали обработкой 2% раствором серной кислоты в пропан-2-оле с последующим нагреванием при 200–300°С. Использовали систему растворителей бензол–пропан-2-ол, 10 : 1 (система А), хлороформ–пропан-2-ол 10 : 1 (система В). Для разделения веществ колоночной хроматографией применяли Kieselgel 60 (0.063–0.200 мм, Merck). ESI+-MS регистрировали на масс-спектрометре Thermo Scientific MS/MS TSQ Quantum Access MAX.
α-D-Глюкозаминилхлорид (I) синтезировали по методике [15]. Ацетонитрил, дихлорметан и K2CO3 подготавливали, как описано нами ранее [9].
Общая методика гликозилирования в системе “твердый карбонат калия–ацетонитрил”. К раствору 1.10 ммоль α-хлорида (I) в 12 мл CH3CN добавляли 1.10 ммоль гликозил-акцептора (II) и 4.95 ммоль безводного тонкоизмельченного K2CO3, 0.22 ммоль или 0.11 ммоль соответствующего катализатора (15C5 [8], способ A, полиэтиленгликоль [10], способ C) и перемешивали при комнатной температуре до полного исчезновения гликозил-донора (I) (ТСХ, система А). Аналогично, в безводном CH3CN, проводили гликозилирование 2-меркаптобензимидазола (II) в отсутствие катализатора (способ B). Твердую фазу отделяли фильтрованием, осадок промывали на фильтре ацетонитрилом (3 × 4 мл), растворитель удаляли при пониженном давлении. Выделяли целевые гликозиды (III) и (IV) колоночной хроматографией (градиентное элюирование хлороформ → хлороформ-изопропиловый спирт 100 : 1 → 10 : 1).
Гликозилирование по способу D. Смесь 0.82 ммоль 2-меркаптобензимидазола (II), 0.82 ммоль гидрида натрия в 9 мл безводного дихлорметана выдерживали 1 час при комнатной температуре. Затем в раствор гликозил-акцептора (II) вносили 0.16 ммоль 15C5, 0.82 ммоль α-хлорида (I) и реакционную смесь перемешивали до полной конверсии гликозил-донора (I) (ТСХ, система В). Твердую фазу отделяли фильтрованием, осадок промывали на фильтре дихлорметаном (3 × 4 мл). Маточный раствор нейтрализовали катионитом КУ-2 (H+), смолу отфильтровали, растворитель удаляли при пониженном давлении. Выделяли целевой гликозид (III), как описано выше.
1-(2-Ацетамидо-3,4,6-три-O-ацетил-2-дезокси-β-D-глюкопиранозил)-2-меркаптобензимидазол (III): получали из 0.4 г (1.1 ммоль) α-хлорида (I) и 165 мг (1.1 ммоль) 2-меркаптобензимидазола (II); т. пл. 252–254°С, [α]546 –106° (с 1.0; CHСl3). 1H ЯМР (DMSO-d6): 1.61 (c, 3H, NAc), 1.94 (с, 3H, OAc), 2.05 (с, 9H, OAc), 4.16 (м, 3H, H-5, H-6a,b), 4.67 (ддд, 1H, H-2, J2,3 9.6 Гц), 5.27 (дд, 1H, H-4, J4,5 9.6 Гц), 5.45 (дд, 1H, H-3, J3,4 9.6 Гц), 6.36 (д, 1H, H-1, J1,2 9.6 Гц), 7.20 (м, 4H, СНаром), 7.92 (д, 1H, NH, J2,NH 8.8 Гц), 12.88 (с, 1H, SH). MS, m/z: 480 (479 + 1) (M+ + H+).
2-(2-Ацетамидо-3,4,6-три-O-ацетил-2-дезокси-β-D-глюкопиранозилтио)бензимидазол (IV): получали из 0.4 г (1.1 ммоль) α-хлорида (I) и 165 мг (1.1 ммоль) 2-меркаптобензимидазола (II); аморф., [α]546 –38° (с 1.0; CHСl3). 1H-ЯМР (DMSO-d6): 1.78 (c, 3H, NAc), 1.85, 1.93, 1.97 (3c, 9H, 3OAc), 3.94 (ддд, 1H, H-5, J5,6a, J5,6b 1.6, 4.8 Гц), 3.95, 4.16 (2 дд, 2H, Н-6a,b, Jгем 12.6 Гц), 4.05 (ддд, 1H, H-2, J2,3 9.6 Гц), 4.90 (дд, 1H, H-4, J4,5 9.2 Гц), 5.20 (дд, 1H, H-3, J3,4 9.6 Гц), 5.67 (д, 1H, H-1, J1,2 10.8 Гц), 8.25 (д, 1H, NH, J2,NH 9.6 Гц), 7.15, 7.41, 7.54 (2м, д, 4H, CHаром), 12.53 (с, 1H, NHhet). MS, m/z: 480 (479 + 1) (M+ + H+).
Список литературы
Машковский М.Д. // Лекарственные средства: в 2-х т. / Ред. Литвина Н.А., Машковский С.А. М.: Новая волна, 2002.
Andersson T. // Clin. Pharmacokinet. 1996. V. 31. P. 9–28. https://doi.org/10.2165/00003088-199631010-00002
Bouzard D. // Antibiotics and Antiviral Compounds. Chemical Synthesis and Modification / Eds. Krohn R., Kirst H.A., Maag H. Weinheim: VCH Publishers Inc., 1993. P. 187–203.
Курьянов В.О. Глюкозаминиды: синтез, структура, свойства: дис. … докт. хим. наук. Донецк, 2013. 351 с.
Курьянов В.О. Чупахина Т.А., Земляков А.Е., Чирва В.Я., Шишкин О.В., Шишкина С.В., Котляр С.А., Камалов Г.Л. // Журн. органічн. та фарм. хімії. 2006. Т. 4. С. 37–41.
Курьянов В.О. Токарев М.К., Чупахина Т.А., Чирва В.Я. // Биоорган. химия. 2011. Т. 37. С. 672–678. [Kur’yanov V.O., Tokarev M.K., Chupakhina T.A., Chirva V.Ya. // Russ. J. Bioorg. Chem.. 2011. V. 37. P. 602–608.] https://doi.org/10.1134/S1068162011050104
El Ashry el S.H., Aly A.A., Aouad M.R., Amer M.R. // Nucleosides Nucleotides Nucleic Acids. 2010. V. 29. P. 698–706. https://doi.org/10.1080/15257770.2010.501777
Курьянов В.О. Чупахина Т.А., Земляков А.Е., Чирва В.Я., Шишкин О.В., Шишкина С.В., Котляр С.А., Камалов Г.Л. // Биоорган. химия. 2005. Т. 31. С. 511–518. [Kur’yanov V.O., Chupakhina T.A., Zemlyakov A.E., Chirva V.Ya., Shishkin О.V., Shishkina S.V., Kotlyar S.A., Kamalov G.L. // Russ. J. Bioorg. Chem. 2005. V. 31. P. 460–466.] https://doi.org/10.1134/S106816201204005X
Чупахина Т.А. Синтез и медико-биологические свойства O-, S- и N-гликозидов N-ацетилглюкозамина и их производных: дис. … канд. хим. наук. Одесса, 2008. 245 с.
Хираока М. // Краун-соединения. Свойства и применение / Ред. Эммануэль Н.М. М.: Мир, 1986. С. 258–260.
Курьянов В.О., Прискока У.С., Чупахина Т.А., Чирва В.Я. // Биоорган. химия. 2005. Т. 31. С. 335–336. [Kur’yanov V.O., Priskoka U.S., Chupakhina T.A., Chirva V.Ya. // Russ. J. Bioorg. Chem. 2005. V. 31. P. 300–301.] https://doi.org/10.1007/s11171-005-0042-4
Pistia-Brueggeman G., Hollingsworth R.I. // Carbohydr. Res. 2003. V. 338. P. 455–458.
Roy R., Tropper F. // Synth. Comm. 1990. V. 20. P. 2097–2102.
Flitsch S., Guilber B. Regioselective Sulfation: US Patent 5874548. 1999. http://patft.uspto.gov/netahtml/search-bool.html.
Лихошерстов Л.М., Новикова О.С., Деревицкая В.А., Кочетков Н.К. // Изв. АН СССР. Сер. хим. 1986. С. 1663–1669. [Likhosherstov L.M., Novikova O.S., Derevitskaya V.A., Kochetkov N.K. // Russ. Chem. Bull. 1986. V. 35. P. 1512–1517.] https://doi.org/10.1007/BF00954837
Хортон Д. // Методы исследования углеводов / Ред. Хорлин А.Я. М.: Мир, 1975. С. 221–224.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия