

Биоорганическая химия, 2020, T. 46, № 6, стр. 708-718
Изучение взаимодействия флуоресцентно-меченого цитохрома с с липосомами, митохондриями и митопластами методом флуоресцентной корреляционной спектроскопии
И. Д. Гусев 1, А. М. Фирсов 2, Р. В. Черткова 1, 3, *, Е. А. Котова 2, Д. А. Долгих 1, 4, М. П. Кирпичников 1, 4, Ю. Н. Антоненко 2, **
1 ФГБУН Институт биоорганической химии имени академиков М.М. Шемякина и Ю.А. Овчинникова РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
2 НИИ физико-химической биологии имени А.Н. Белозерского, МГУ имени М.В. Ломоносова
119991 Москва, Ленинские горы, 1/40, Россия
3 НИУ Высшая школа экономики, факультет биологии и биотехнологии
117312 Москва, ул. Вавилова, 7, Россия
4 МГУ имени М.В. Ломоносова, биологический факультет
119991 Москва, Ленинские горы, 1/12, Россия
* E-mail: cherita@inbox.ru
** E-mail: antonen@belozersky.msu.ru
Поступила в редакцию 30.04.2020
После доработки 10.05.2020
Принята к публикации 12.05.2020
Аннотация
Исследовано взаимодействие меченного флуоресцентным красителем сульфоцианином-3 Cys-замещенного (G56C) цитохрома с (фЦит) с искусственными и природными липидными мембранами методом флуоресцентной корреляционной спектроскопии. Показано, что по сравнению с митохондриями митопласты характеризуются большим количеством сайтов связывания фЦит, при этом обладающих меньшим сродством к этому белку. Показано также, что сродство фЦит к кардиолипин-содержащим липосомам зависит от содержания кардиолипина в липосомах и снижается при повышении ионной силы раствора. Высокая константа связывания фЦит с митохондриями может объясняться наличием на поверхности внешней мембраны специфических сайтов связывания этого белка, отличных от сайтов, характерных для митопластов и липосом. Этот вывод подтверждается тем наблюдением, что фЦит вытесняется с поверхности митохондрий немеченым мутантным вариантом K8T эффективнее, чем цитохромом с дикого типа, тогда как в случае митопластов и липосом цитохром с вытесняет фЦит эффективнее, чем вариант K8T.
ВВЕДЕНИЕ
Гемсодержащий белок цитохром с, один из важнейших компонентов электрон-транспортной или дыхательной цепи (ЭТЦ) митохондрий, переносит электроны от убихинол-цитохром с-оксидоредуктазы (комплекс III) к цитохром с-оксидазе (комплекс IV). Наряду с участием в электронном транспорте, другой его важнейшей функцией является инициация клеточного апоптоза путем участия в формировании апоптосомы [1]. В настоящий момент значительные усилия направлены на исследование цитохрома с в качестве триггера и посредника апоптотических событий.
Программируемая клеточная гибель, или апоптоз, имеет исключительную важность для нормальной жизнедеятельности организма в целом. Во время активации каскада событий, приводящих к апоптозу клетки, цитохром с транслоцируется через внешнюю мембрану митохондрии в цитозоль, где он либо усиливает внешний апоптотический сигнал, либо инициирует активацию каспазного каскада по собственному (цитохром с-зависимому) апоптотическому пути. Ключевым событием в цитохром с-зависимом апоптотическом пути является пермеабилизация внешней митохондриальной мембраны, в результате чего в цитозоль выходят различные апоптогенные факторы, в том числе и цитохром с. В связи с этим большой интерес вызывает изучение механизмов взаимодействия цитохрома с с липидными мембранами. Взаимодействие цитохрома с с кардиолипином – липидом, входящим в состав внутренней мемебраны митохондрии, – значительно усиливает его пероксидазную активность [2, 3]. Предполагается, что посредством запуска перекисного окисления мембранных липидов именно пероксидазная активность способствует транслокации цитохрома с через внешнюю мембрану митохондрий [4]. Следует отметить также, что кардиолипин, сам по себе [5] или в комплексе с цитохром с-оксидазой [6], формирует мембранный сайт связывания цитохрома с на внешней поверхности внутренней мембраны митохондрий и в условиях гомеостаза.
Известно, что связывание цитохрома с с кардиолипином на первых стадиях обратимо, зависит от pH [5] и приводит к изменению третичной и вторичной структуры молекулы цитохрома с [7]. Фактором, влияющим на стабильность комплекса цитохром с/кардиолипин, является окисление кардиолипина, так как цитохром с имеет пониженное сродство к окисленному липиду [8]. Вместе с тем, механизмы взаимодействия цитохрома с с кардиолипином в митохондриальной мембране остаются не до конца изученными и привлекают пристальное внимание исследователей [9–14].
При помощи флуоресцентно-меченого варианта лошадиного цитохрома с методом флуоресцентной корреляционной спектроскопии (ФКС) исследовано взаимодействие экзогенного цитохрома с с природными (митохондрии и митопласты) и искусственными (кардиолипин-содержащими липосомами) липидными мембранами, а также влияние на этот процесс ионной силы и содержания кардиолипина в липидной мембране.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Получение флуоресцентно-меченого цитохрома с. В качестве стратегии мечения цитохрома с нами была выбрана конъюгация молекул белка и малеимидного производного сульфоцианина 3 через остаток Cys, введенный в цитохром с с помощью сайт-направленного мутагенеза. Реакция тиолов с малеимидами широко используется для биоконъюгации и мечения биомолекул, в том числе белков и пептидов [15]; данную реакцию можно проводить в водной среде, что существенно облегчает процедуру.
Введение в молекулу цитохрома с тиоловой группы может быть осуществлено путем единичной замены в ходе мутагенеза поверхностного или концевого аминокислотного остатка на остаток Cys. Очевидно, что остаток, заменяемый на остаток Cys, не должен быть вовлечен во взаимодействия с кардиолипином и белками-партнерами цитохрома с, а также быть значимым для структурно-функциональной целостности белка.
Известно, что на поверхности цитохрома с имеется три участка связывания с кардиолипином: сайт связывания A, состоящий из остатков K72, K73, K86, K87 и R91 [16]; сайт связывания L, состоящий из остатков K22, K25, K27, а также H33, который принимает участие в связывании цитохрома с с кардиолипином при рН < 7.0; сайт связывания С, содержащий остатки N52, H26 и P44 [17]. Кроме того, известны участки связывания цитохрома с c цитохром с-оксидазой (а.к.о. К8, К13, К72, K86, и К87) [18], с убихинол-цитохром с-оксидоредуктазой (а.к.о. K7, K8, K13, K39, K72, K79, K86, K87) [19] и цитохром с-пероксидазой (а.к.о. K13, K27, K72, K86, K87, Q12 и Q16) [20, 21].
Для введения флуоресцентной метки в молекулу цитохрома с нами был сконструирован и получен мутантный вариант цитохрома с с заменой остатка G56 на остаток Cys, так как функциональная группа остатка G56 расположена на поверхности белковой глобулы и не входит ни в один из известных сайтов связывания белка.
Реакцию мечения, а также очистку и анализ продуктов реакции проводили согласно методике, описанной в экспериментальной части. Фракции, в которых соотношение пиков цитохрома с и сульфоцианина было сопоставимым (рис. 1), объединяли и обессоливали на этой же колонке, уравновешенной бессолевым буфером, после чего анализировали спектрофотометрически.
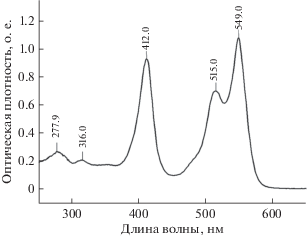
Рис. 1.
Спектр флуоресцентно-меченого цитохрома с с заменой G56C. Поглощение при 412 нм соответствует максимуму поглощения цитохрома с, при 549 нм – максимуму поглощения сульфоцианина.
Исследование взаимодействия флуоресцентно-меченого цитохрома с с липидными мембранами осуществляли при помощи ФКС. Этот метод основан на регистрации флуктуаций флуоресценции отдельных частиц, возникающих в результате броуновского движения через освещенный лазером малый объем пространства (конфокальный объем). Традиционно ФКС используется для исследования процессов, связанных с изменением подвижности молекул и комплексов: агрегации частиц, взаимодействия флуоресцирующих молекул с надмолекулярными комплексами, липидными везикулами, при этом диапазон размеров изучаемых объектов очень широк – от молекул красителей до частиц с размерами, составляющими сотни нанометров [22].
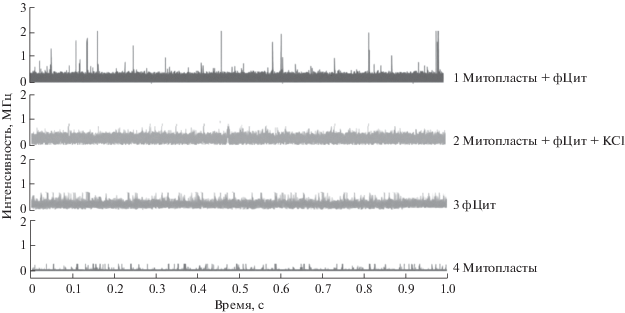
В ходе работы аналитическим сигналом для идентификации связывания являлось изменение количества пиков, регистрируемых в конфокальном объеме. Пики сигнала флуоресценции связаны с перемещением флуоресцирующих частиц через измеряемый объем при перемешивании суспензии частиц. Это могут быть как свободные молекулы меченого белка, так и намного ярче светящиеся частицы мембран со связавшимися с ними несколькими молекулами белка. На рис. 2 представлены типичные записи сигнала для суспензии митопластов и раствора меченого цитохрома с (фЦит). Связывание фЦит с митопластами характеризовалось большим количеством пиков с высокой интенсивностью флуоресценции (1), тогда как в растворе несвязанного флуоресцентно-меченого цитохрома с (3) регистрировались фоновые пики небольшой интенсивности. В суспензии митопластов в отсутствие меченого белка практически отсутствовали характерные для комплексов пики (4), так же, как и в случае дестабилизированного повышением ионной силы комплекса фЦит/митопласты (2).
Рис. 2.
Временные зависимости интенсивности пиков флуоресценции суспензии митопластов (4), раствора меченого цитохрома с (3), суспензии митопластов в присутствии меченого цитохрома с (1), суспензии митопластов в присутствии меченого цитохрома с и 50 мМ KCl (2). Измерения проводили в сахарозном буфере (180 мМ сахарозы, 10 мМ KCl, 10 мМ Tris, 10 мМ MES, pH 7.4). Конечная концентрация митопластов составила 0.1 мг/мл, концентрация меченого цитохрома с 0.26 мкМ, время сбора данных составляло 30 с.
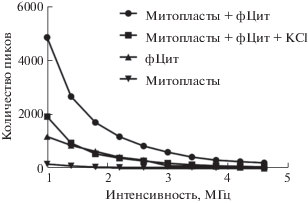
На рис. 3 представлено типичное распределение пиков флуоресценциии по их интенсивности после обработки экспериментальных данных в программе Saligat. Для идентификации связывания нами был выбран феноменологический порог интенсивности в 1.4 МГц. Использование большей величины порога слишком сильно обедняло статистику по пикам, в то время как меньшая величина порога была слишком близка к шуму усилителя, что вносило искажения в регистрируемую статистику.
Рис. 3.
Распределение регистрируемых пиков флуоресценции по интенсивности в суспензии митопластов и растворе меченого цитохрома с, а также при взаимодействии меченого цитохрома с с митопластами, в том числе в условиях повышенной ионной силы 50 мM KCl. Условия измерений даны в подписи к рис. 2.
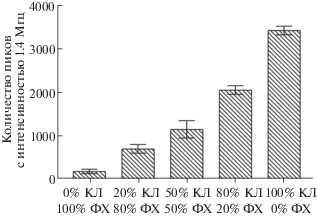
Роль кардиолипина в связывании цитохрома с с липосомами. На рис. 4 представлена зависимость эффективности связывания меченого цитохрома с с липосомами, содержащими различное количество кардиолипина (от 0 до 100%). Анализируя полученные результаты, можно сделать заключение, что эффективность связывания фЦит с липидной мембраной возрастает при увеличении содержания в ней кардиолипина и это, по-видимому, связано с тем, что количество сайтов связывания цитохрома с на поверхности липосом напрямую зависит от процентного содержания в них кардиолипина и достигает максимального значения при взаимодействии с липосомами, состоящими на 100% из кардиолипина.
Рис. 4.
Влияние содержания (%) кардиолипина в составе липосом на сродство цитохрома с к липосомам, выраженное в количестве пиков с интенсивностью 1.4 МГц. Данные приведены для липосом состава “яичный фосфатидилхолин–кардиолипин” в различных соотношениях. Измерения проводили в сахарозном буфере (180 мМ сахарозы, 10 мМ KCl, 10 мМ Tris, 10 мМ MES, pH 7.4). Конечная концентрация липосом различного состава ‒ 0.5 мг/мл, концентрация меченого цитохрома с ‒ 0.26 мкМ.
Определение кажущихся констант вытеснения цитохрома с с различными мембранами. Для определения кажущихся констант вытеснения цитохрома с из комплекса с липидными мембранами был использован метод вытеснения фЦит из комплекса с мембранами при помощи немеченого цитохрома с с подсчетом количества пиков флуоресценции с интенсивностью 1.4 МГц (рис. 5). Метод вытеснения основывается на понижении концентрации флуоресцентно-меченого компонента комплекса путем замещения немеченым белком при повышении концентрации последнего в растворе. При этом наблюдаемое уменьшение количества пиков было вызвано вытеснением флуоресцентно-меченого цитохрома с из комплекса с исследуемой липидной мембраной по мере увеличения в среде концентрации немеченого цитохрома с от 10 нМ до 100 мкМ. Количество пиков подсчитывали при помощи программы Saligat, кривые вытеснения анализировали при помощи программы GraphPrism 8 на основании уравнения y = (Bmax XH)/($K_{{\text{b}}}^{H}$ + X H), и определяли основные характеристики связывания (см. Экспериментальную часть): максимальное количество пиков флуоресценции (Bmax), константу вытеснения (Kb) и коэффициент кооперативности связывания (коэффициент Хилла, H). Максимальное количество пиков флуоресценции Bmax отражает концентрацию флуоресцирующих частиц в системе, а также зависит от количества сайтов связывания фЦит на одной частице. В этом уравнении X обозначает концентрацию цитохрома с. Константа вытеснения отражает усредненное значение константы диссоциации всех сайтов связывания, с которыми мог связаться меченый цитохром с. Таким образом, различия констант вытеснения обусловлены различием сродства липидных мембран к цитохрому с. Коэффициент Хилла отражает влияние повышения (положительная кооперативность) или понижения (отрицательная кооперативность) сродства сайтов связывания на липидных мембранах при протекании процесса связывания с ними белка. Следует сказать, что данное уравнение применимо для анализа при выполнении двух условий: (1) концентрация фЦит должна быть много больше, чем концентрация центров связывания; (2) при выбранных концентрациях фЦит и мембран должно быть обеспечено насыщение связывания. При проведении наших опытов данные условия соблюдались лишь частично, поэтому вместо истинных констант связывания в опытах оценивались кажущиеся константы связывания. Кажущаяся константа вытеснения в случае комплекса фЦит с митопластами существенно превышает таковые для комплексов с митохондриями и составляет 25 мкМ (табл. 1). При этом максимальное количество регистрируемых пиков флуоресценции в случае комплекса с митопластами больше, чем для комплексов с митохондриями. Поскольку концентрация частиц митохондрий и митопластов в наших опытах была одинакова, то эти данные свидетельствуют о том, что на мембранах митопластов имеется существенно большее количество сайтов связывания фЦит. Однако различия в величинах Kb говорит о том, что сродство к белку для сайтов связывания на митопластах примерно в десять раз ниже, чем сродство аналогичных сайтов для митохондрий.
Рис. 5.
Вытеснение меченого цитохрома с из комплекса с митопластами, митохондриями и липосомами (20% кардиолипина) немеченым цитохромом с. Конечная концентрация митопластов ‒ 0.1 мг/мл, митохондрий ‒ 0.1 мг/мл, липосом ‒ 0.5 мг/мл, меченого цитохрома с ‒ 0.26 мкМ. Измерения проводили в сахарозном буфере (180 мМ сахарозы, 10 мМ KCl, 10 мМ Tris, 10 мМ MES, pH 7.4).
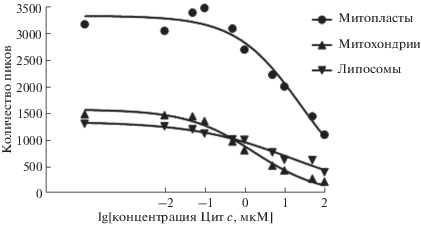
Таблица 1.
Значения максимального количества пиков (Bmax), коэффициента Хилла (H) и кажущейся константы вытеснения (Kb) немеченным цитохромом с дикого типа (WT), полученные после обработки кривых титрования для каждого типа изучаемых липидных мембран в присутствии и в отсутствие 50 мМ KCl
| Липидная мембрана | Количество пиков, Bmax | Коэффициент Хилла, H | Константа вытеснения, Kb, мкМ |
|---|---|---|---|
| Митопласты | 3200 ± 100 | –0.5 ± 0.1 | 25 ± 5 |
| Митопласты + KCl | 2220 ± 30 | –0.6 ± 0.1 | 67 ± 15 |
| Липосомы (20% кардиолипина) | 1700 ± 200 | –0.47 ± 0.15 | 17 ± 2 |
| Липосомы (20% кардиолипина) + KCl | 1240 ± 50 | –6 ± 0.1 | 88 ± 20 |
| Митохондрии | 1750 ± 150 | –0.4 ± 0.1 | 2 ± 0.5 |
| Митохондрии + KCl | 1800 ± 50 | –0.15 ± 0.05 | 29 ± 5 |
Также было изучено влияние ионной силы на кажущуюся константу вытеснения (Kb) и кооперативность связывания (H, табл. 1). Анализ полученных данных позволяет заключить, что в присутствии KCl кажущаяся константа вытеснения увеличивалась, что говорит о снижении сродства цитохрома с к липидным мембранам. Количество пиков с интенсивностью >1.4 MHz уменьшалось с ростом ионной силы раствора. В случае митохондрий и липосом при добавлении 50 мМ KCl наблюдалось многократное увеличение кажущейся константы вытеснения Kb с 2 до 29 мкМ (митохондрии) и с 17 до 88 мкМ (липосомы, табл. 1). Влияние KCl на Kb было значительно слабее в случае митопластов. Снижение связывания фЦит с кардиолипином в составе липосом при повышении ионной силы соответствует имеющимся литературным данным [22].
Определение кажущихся констант вытеснения мутантных форм цитохрома с с различными мембранами. Проведен эксперимент по вытеснению флуоресцентно-меченого цитохрома с из комплексов с липидными мембранами при помощи немеченых мутантных вариантов цитохрома с с единичной заменой K8T и множественными заменами K72E/E69K/K86E/K87E (4Mut), полученными в рамках наших предыдущих работ [23, 24]. Эксперимент проводили аналогично предыдущим экспериментам с титрованием комплекса фЦит/ липидная мембрана немеченым белком в концентрациях от 10 нМ до 100 мкМ, при этом наблюдали характерное падение количества пиков (рис. 6).
Рис. 6.
Вытеснение флуоресцентно-меченого цитохрома с под действием немеченых мутантных вариантов K8T и 4Мut из комплексов с: (а) липосомами (20% кардиолипин, 80% яичный лецитин), (б) митопластами, (в) митохондриями. Графики нормированы на максимальное связывание. Измерения проводили в сахарозном буфере (180 мМ сахарозы, 10 мМ KCl, 10 мМ Tris, 10 мМ MES, pH 7.4). Концентрация митопластов ‒ 0.1 мг/мл, митохондрий ‒ 0.1 мг/мл, липосом ‒ 0.5 мг/мл, меченого цитохрома с ‒ 0.26 мкМ.
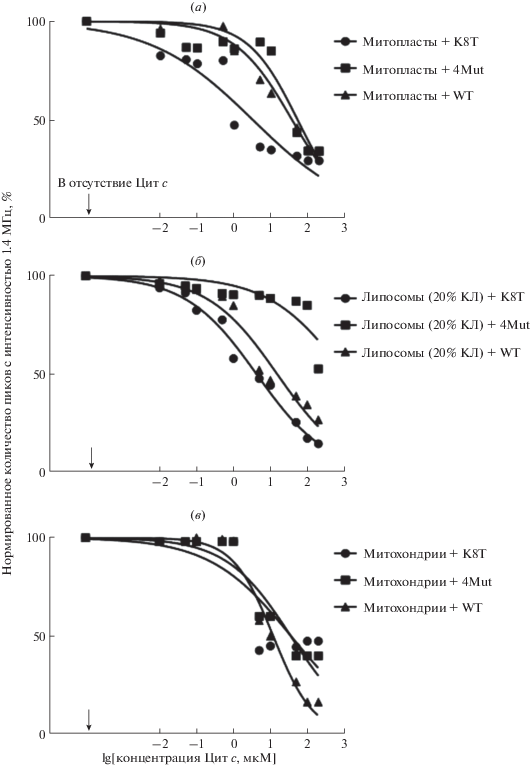
О различиях в эффективности вытеснения меченого цитохрома с из комплекса фЦит/липосома (20% кардиолипин, 80% яичный лецитин) под действием немеченых мутантных вариантов цитохрома с свидетельствуют отличающиеся кажущиеся константы вытеснения, при том, что коэффициент Хилла примерно одинаков для всех вариантов цитохрома с. Основываясь на данных, представленных в табл. 2, можно сделать вывод, что мутантный цитохром с с заменой K8T, обладая повышенным сродством к кардиолипину, наиболее эффективно вытеснял флуоресцентно-меченый цитохром с из комплексов с кардиолипин-содержащими липосомами. В случае митопластов наблюдалась аналогичная картина. В то же время, этот белок заметно слабее связывался с митохондриями по сравнению с диким типом. Мутантный вариант K72E/E69K/K86E/K87E (4Мut) благодаря введению отрицательно-заряженных а.к.о. на позиции трех остатков Lys связывался хуже цитохрома с дикого типа (WT) в случае всех изучаемых типов мембран.
Таблица 2.
Значения коэффициента Хилла (H) и кажущейся константы вытеснения (Kb), полученные после обработки нормированных кривых титрования в программе Prism для немеченых мутантных цитохромов с
| Цитохром с | Липосомы | Митопласты | Митохондрии | |||
|---|---|---|---|---|---|---|
| коэффициент Хилла, H | Kb, мкМ | коэффициент Хилла, H | Kb, мкМ | коэффициент Хилла, H | Kb, мкМ | |
| K8T | –0.45 ± 0.15 | 4.3 ± 1 | –0.3 ± 0.15 | 3 ± 1 | –0.4 ± 0.1 | 36 ± 5 |
| K72E/E69K/K86E/K87E | –0.42 ± 0.15 | 1088 ± 10 | –0.45 ± 0.15 | 52 ± 10 | –0.5 ± 0.1 | 35 ± 5 |
Согласно полученным данным, большей эффективностью связывания с митохондриями обладает цитохром с WT, тогда как эффективность связывания для 4Mut и K8T совпадала и была заметно ниже по сравнению с WT. Данный факт может свидетельствовать о том, что связывание цитохрома с с митохондриями не зависит от эффективности связывания цитохрома с с кардиолипином и осуществляется за счет других взаимодействий, нежели с митопластами и липосомами.
При помощи метода ФКС нами изучено взаимодействие флуоресцентно-меченого цитохрома с лошади с тремя видами липидных мембран: кардиолипин-содержащими липосомами, митопластами и митохондриями, полученными из препаратов печени крысы. Показано, что взаимодействие цитохрома с с митопластами характеризуется наибольшим количеством сайтов связывания, которые обладают, однако, меньшим сродством в сравнении с митохондриями. Поверхность мембраны митопластов состоит по большей части из внутренней митохондриальной мембраны, в липидный состав которой входит кардиолипин. Кроме того, внутренняя мембрана митохондрий содержит интегрированные в нее белковые комплексы – убихинол-цитохром с-оксидоредуктазу (комплекс III) и цитохром с-оксидазу (комплекс IV), являющиеся белками-партнерами цитохрома с по ЭТЦ, с которыми цитохром с образует временные функциональные комплексы в процессе переноса электрона. Таким образом, на митопластах имеются сайты связывания цитохрома с как минимум двух принципиально разных типов: 1) кардиолипин, с которым реализуется белок-липидное связывание; 2) III и IV комплексы ЭТЦ, с которыми реализуется белок-белковое связывание. По-видимому, именно этим объясняется наблюдаемое нами повышенное количество сайтов связывания на митопластах по сравнению с митохондриями. Оба рассматриваемых типа связывания цитохрома с реализуются с участием электростатических взаимодействий; с этим согласуются наши экспериментальные наблюдения уменьшения сродства цитохрома с при повышении ионной силы в среде измерения в случае комплексов с липосомами или митопластами.
Монотонное увеличение сродства фЦит к липосомам при увеличении в них процентного содержания кардиолипина свидетельствует об определяющей роли взаимодействия фЦит с кардиолипином в процессе связывания белка с кардиолипин-содержащей липосомальной мембраной.
Характер связывания фЦит с внешней митохондриальной мембраной (митохондрии) отличался от связывания с митопластами и кардиолипин-содержащими липосомами. Тем не менее, несмотря на отсутствие в составе внешней мембраны митохондрий кардиолипина и белков-партнеров ЭТЦ, связывающих цитохром с, в наших экспериментах наблюдалось устойчивое связывание. Таким образом, полученные нами экспериментальные данные могут свидетельствовать о наличии на внешней митохондриальной мембране специфических сайтов связывания цитохрома с.
Результаты эксперимента по вытеснению флуоресцентно-меченого цитохрома с из комплекса с липидными мембранами мутантными вариантами белка не противоречат предположению о том, что на внешней мембране митохондрий имеются специфические сайты связывания цитохрома с. Оба мутантных варианта одинаково эффективно (но заметно хуже по сравнению с диким типом цитохрома с) вытесняли флуоресцентно-меченый цитохром с из комплекса с митохондриями. В то же время, в случае комплексов как с липосомами, так и с митопластами вариант с заменой K8T, имеющий более высокое сродство к кардиолипину, вытеснял меченый цитохром с эффективнее варианта K72E/E69K/K86E/K87E и белка дикого типа.
Полученные нами данные представляют интерес с точки зрения понимания фундаментальных механизмов взаимодействия цитохрома с с липидными мембранами, а также влияния на эти механизмы различных внешних факторов. Понимание механизмов, в свою очередь, имеет практическую значимость, поскольку открывает широкие возможности для рационального дизайна биомедицинских препаратов, обладающих про- или антиапоптотическими свойствами.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали компоненты для культуральных сред и буферных растворов для хроматографии и электрофореза фирмы AppliChem (Германия); ампициллин, эндонуклеазу рестрикции DpnI фирмы Fermentas (Литва), Pfu ДНК-полимеразу, кардиолипин бычьего сердца (Sigma, США), фосфатидилхолин яичного желтка (Avanti Polar Lipids, США), флуоресцентные красители сульфо-цианин и BDP TMR в малеимидной форме (Lumiprobe GmbH, Германия). Дистиллированную воду дополнительно очищали с помощью системы Milli-Q (Millipore, США).
Конструирование мутантных генов цитохрома с. Введение мутации в ген лошадиного цитохрома с в составе экспрессионного плазмидного вектора pBP(CYC1) осуществляли с помощью сайт-направленного мутагенеза по методу QuikChangeTM Mutagenesis Kit (Stratagene, США) [23]. После завершения реакции к смеси добавляли 10 ед. акт. рестриктазы DpnI и инкубировали в течение 60 мин при 37°С. Далее аликвоты полученной смеси использовали для трансформации компетентных клеток E. coli XL-1 Blue по стандартной методике. Наработку мутантной ДНК в ходе мутагенеза анализировали при помощи электрофореза в 1% агарозном геле. Определение нуклеотидной последовательности мутантных генов в составе плазмидной ДНК осуществляли с помощью автоматического секвенатора ABI Prism 3100-Avant Genetic Analyzer (Applied Biosystems, США). Отобранные мутантные гены переклонировали в экспрессионный вектор pBP(CYC1) [25], модифицированный для экспрессии генов лошадиного цитохрома c [24].
Экспрессию мутантных генов цитохрома с осуществляли в штамме E. coli JM109 в жидкой питательной среде SB с ампициллином (конечная концентрация 100 мкг/мл) без добавления индуктора при 37°С и интенсивном перемешивании в течение 22–24 ч [26]. По окончании роста клетки E. coli осаждали центрифугированием при 4000 g и 4°С в течение 20 мин. Полученный клеточный осадок ресуспендировали в буфере 25 мМ Na–Pi, pH 6,0, 1 мМ NaN3. Гомогенизацию клеток осуществляли продавливанием под высоким давлением на установке “French Press” (Spectronic Instruments, Inc., США) с последующим центрифугированием при 95 000 g в течение 20 мин [27].
Выделение и очистку целевых белков проводили с использованием жидкостной хроматографической системы “АКТА FPLC” (GE-HEALTHCARE, США), согласно схеме, разработанной ранее [25, 27]. Клеточный экстракт наносили на катионообменную колонку MP HS 10/10 (BIO-RAD, США), уравновешенную буфером, содержащем 25 мМ Na–Pi, pH 6.0, 1 мМ NaN3. Цитохром с элюировали линейным градиентом 1 М NaCl в том же буфере со скоростью 3 мл/мин. Фракции, полученные после очистки на MP HS, анализировали спектрофотометрически и при помощи электрофореза в 12% SDS-ПААГ, затем диализовали против буфера для адсорбционной хроматографии (10 мМ Na–Pi, pH 7.0, 1 мМ NaN3) и наносили на колонку с гидроксиапатитом CHT-I (BIO-RAD, США). Цитохром с элюировали линейным градиентом 500 мМ Na–Pi, рН 7.0 со скоростью 2 мл/мин. Степень очистки и концентрацию цитохрома с в полученных фракциях определяли спектрофотометрически и при помощи электрофореза в 12% SDS-ПААГ. Степень очистки цитохрома с в полученных фракциях определяли по отношению оптической плотности в максимуме поглощения в области Соре к оптической плотности при 280 нм. Фракции с отношением, равным 4.5–5.0 (что соответствует чистоте коммерческого препарата ≥95%, Sigma, США), объединяли, дважды диализовали против 10 мМ аммоний-карбонатного буфера, рН 7.9, и лиофилизовали на установке ALPHA I-5.
Получение флуоресцентно-меченого цитохрома с. Лиофилизованный препарат мутантного варианта цитохрома с с заменой G56C, растворяли в реакционном буфере (10 мМ NaH2PO4, 200 мМ NaCl, pH 7.0) до концентрации 1–2 мг/мл. Флуоресцентный краситель – водорастворимый сульфоцианин (Sulfo-Cyanine3) в виде малеимида – растворяли в ДМСО до концентрации (0.01 мг/мл). Раствор красителя добавляли к раствору белка в соотношении 1 : 5, перемешивали и инкубировали при комнатной температуре в течение 12–14 ч. Продукт реакции очищали на гель-фильтрационной колонке (Sephadex G-25), уравновешенной реакционным буфером. По окончании гель-фильтрации фракции, содержащие флуоресцентно-меченый белок, обессоливали на этой же колонке, уравновешенной бессолевым буфером (10 мМ NaH2PO4, pH 7.0). Количественные и качественные характеристики модифицированного цитохрома с определяли спектрофотометрически на спектрофотометре Cary50-Bio (Varian, США) в максимумах поглощения для цитохрома с (412 нм), Sulfo-Cyanine3 (549 нм).
Выделение препаратов митохондрий и митопластов из печени крыс. Митохондрии печени крыс выделяли при помощи дифференциального центрифугирования [28] в буфере, содержащем 250 мМ сахарозы, 10 мМ MOPS, 1 мМ ЭГТА и 0.1 мг/мл бычьего сывороточного альбумина (БСА), рН 7.4. После центрифугирования препарат митохондрий промывали в том же буфере. Митопласты получали в процессе инкубирования митохондрий печени крысы в течение 10 мин в буфере (MOPS 10 мM, ЭДТА 1 мM, Tris 10 мM, pH 7.4), содержащем 25 мM KCl. После инкубирования суспензию центрифугировали при 4200 g в течение 5 мин, удаляли супернатант, а осадок суспендировали в сахарозном буфере (180 мМ сахарозы, 10 мМ KCl, 10 мМ Tris, 10 мМ MES, pH 7.4), содержащим 1 мM ЭДТА. Выравнивание концентраций частиц митопластов и митохондрий в наших опытах достигалась тем, что при приготовлении митопластов одновременно в другую пробирку отбиралось такое же количество белка митохондрий, которое суспендировалось в том же объеме буфера, который добавлялся при последнем осаждении митопластов.
Получение кардиолипин-содержащих мультиламеллярных липосом. Для приготовления липосом 5 мг липида (яичный лецитин, кардиолипин или их смесь) растворяли в 250 мкл хлороформа. После выпаривания хлороформа в атмосфере азота добавляли 0.5 мл буфера (Tris 10 мМ, MES 10 мМ, KCl 100 мМ, pH 7.4). Полученную смесь встряхивали, после чего подвергали пяти циклам замораживания-оттаивания. По приведенной схеме были получены мультиламеллярные липосомы с содержанием кардиолипина от 1 до 5 мг.
Флуоресцентная корреляционная спектроскопия. В настоящей работе был использован прибор, сконструированный на основе инвертированного флуоресцентного микроскопа Olympus IMT-2 (Artisan Technology Group, США) [28]. Для возбуждения красителя использовался Nd:YAG твердотельный лазер 532 нм, сопряженный с вышеупомянутым микроскопом с водноиммерсионным объективом 40×, 1.2 NA (“Carl Zeiss”, Германия). Флуоресцентный сигнал проходил через соответствующий дихроический разделитель и проецировался на сердцевину световода (50 мкм), связанного с лавинным фотодиодом (SPCM-AQR-13-FC, “PerkinElmer Optoelectronics”, Канада). Сигнал преобразовывался с помощью интерфейсной карты Flex02-01D/C (“Correlator.com”, США). Для корректной работы прибора в начале эксперимента осуществлялась настройка положения световода относительно оптической оси. Все измерения проводили при перемешивании раствора лопастевидным 3-мм пластиковым стержнем, вращающимся со скоростью 600 об./мин. Для измерения к 60 мкл сахарозного буфера (180 мМ сахарозы, 10 мМ KCl, 10 мМ Tris, 10 мМ MES, pH 7.4) добавляли БСА до конечной концентрации 0.1 мг/мл и 1–3 мкл суспензии митохондрий, митопластов или липосом. Конечная концентрация митохондриального белка в экспериментах составляла 0.2 мг/мл, а концентрация липида в случае липосом – 0.5 мг/мл. Измеряли флуктуации флуоресценции окрашенных частиц до и после добавления флуоресцентно-меченого цитохрома с до конечной концентрации 0.26 мкМ. Время сбора данных составляло 30 с. Флуоресценцию регистрировали из конфокального объема, расположенного примерно на 50 мкм выше поверхности покровного стекла. Перед измерениями смесь меченого белка с митохондриями (а также митопластами или липосомами) инкубировали в течение 5 мин.
В эксперименте по вытеснению флуоресцентно-меченого цитохрома с немеченым цитохромом с повторяли все вышеописанные действия, дополнительно измеряя флуктуации флуоресценции каждый раз после добавления немеченого белка (концентрации варьировались от 10 нМ до 100 мкМ).
Для количественной оценки связывания белка с липосомами и митохондриями подсчитывали количество пиков сигнала флуоресценции (n(F > F0)) с интенсивностью выше некоего порога (F0) [22] при помощи программы WinEDR Strathclyde Electrophysiology Software, разработанной Д. Демпстером из Университета Страфклида (J. Dempster, University of Strathclyde, Glasgow, UK) или аналогичной программой Saligat, разработанной В. Козловским (НИИ физико-химической биологии им. А.Н. Белозерского МГУ). В настоящей работе для оценки связывания цитохрома с использовался порог F0 равный 1.4 MГц. В типичном опыте такая величина порога позволяла регистрировать около 2000 пиков, что обеспечивало достаточную статистику и хорошую воспроизводимость измерений.
Константу вытеснения и значения коэффициента Хилла рассчитывали, решая параметрическое уравнение вида: y = (Bmax X H)/($K_{{\text{b}}}^{H}$ + X H), где Bmax – максимальное количество пиков, нормированное для всех измерений, X – концентрация немеченого цитохрома с, Kb – константа вытеснения, H – коэффициент Хилла. Для фиттирования данных этим уравнением использовали программу GraphPad Prism 8 (GraphPad Software, Inc., США). Все данные были получены в результате четырех независимых экспериментов и статистически обработаны в программе GraphPad Prism 8 (Annova test), уровень значимости P был не менее 0.05.
Список литературы
Liu X., Kim C.N., Yang J., Jemmerson R., Wang X. // Cell. 1996. V. 86. P. 147–157. https://doi.org/10.1016/S0092-8674(00)80085-9
Kagan V.E., Tyurin V.A., Jiang J., Tyurina, Y.Y., Ritov, V.B., Amoscato A.A., Osipov A.N., Belikova N.A., Kapralov A.A., Kini V. // Nature Chem. Biol. 2005. V. 1. P. 223–232. https://doi.org/10.1038/nchembio727
Kagan V.E., Borisenko G.G., Tyurina Y.Y., Tyurin V.A., J-iang J., Potapovich A.I., Kini V., Amoscato A.A., Fujii Y. // Free Radical Biology and Medicine. 2004. V. 37. P. 1963–1985. https://doi.org/10.1016/j.freeradbiomed.2004.08.016
Kagan V.E., Bayir H.A., Belikova N.A., Kapralov O., Tyurina Y.Y., Tyurin V.A., Jiang J., Stoyanovsky D.A., Wipf P., Kochanek P.M. // Free Radical Biology and Medicine. 2009. V. 46. P. 1439–1453. https://doi.org/10.1016/j.freeradbiomed.2009.03.004
Mustonen P., Virtanen J.A., Somerharju P.J., Kinnunen P.K.J. // Biochemistry. 1987. V. 26. P. 2991–2997. https://doi.org/10.1021/bi00385a006
Zdzislaw S., Gordon T. // Biophys. J. 1996. V. 71. P. 848–857. https://doi.org/10.1016/S0006-3495(96)79286-X
Bradley J.M., Silkstone G., Wilson M.T., Cheesman M.R., Butt J.N. // J. Am. Chem. Soci. 2011. V. 133. P. 19 676–19 679. https://doi.org/10.1021/ja209144h
Shidoji Y., Hayashi K., Komura S., Ohishi N., Yagi K. // Biochem. Biophys. Res. Commun. 1999. V. 264. P. 343–347. https://doi.org/10.1006/bbrc.1999.1410
Cuiping L., Stonestrom A.J., Christian T., Yong J., Takase R., Hou Y., Yang X. // J. Biol. Chem. 2016. V. 291. P. 10 426–10 436. https://doi.org/10.1074/jbc.M115.697789
Capdevila D.A., Oviedo Rouco S., Tomasina F., Tórtora V., Demicheli V., Radi R., Murgida D.H. // Biochemistry. 2015. V. 54. P. 7491–7504. https://doi.org/10.1021/acs.biochem.5b00922
Kalanxhi E., Wallace C. // Biochem. J. 2007. V. 407. P. 179–187. https://doi.org/10.1042/BJ20070459
Hanske J., Toffey J.R., Morenz A.M., Bonilla A.J., Schiavoni K.H., Pletneva E.V. // Proceedings of the National Academy of Sciences. 2012. V. 109. P. 125–130. https://doi.org/10.1073/pnas.1112312108
O’Brien E.S., Nucci N.V., Fuglestad B., Tommos C., Wand A.J. // J. Biol. Chem. 2015. V. 290. P. 30 879–30 887. https://doi.org/10.1074/jbc.M115.689406
Gorbenko G.P., Molotkovsky J.G., Kinnunen P.K.J. // Biophys. J. 2006. V. 90. P. 4093–4103. https://doi.org/10.1529/biophysj.105.080150
Younggyu Kim Sam O Ho Gassman N.R., Korlann Y., Landorf E.V., Collart F.R., Weiss S. // Bioconjugate Chem. 2008. V. 19. P. 786–791. https://doi.org/10.1021/bc7002499
Belikova N.A., Vladimirov Y.A., Osipov A.N., Karpalov A.A., Tyurin V.A., Potapovich M.V., Basova L.V., Peterson J., Kurnikov I.V., Kagan V.E. // Biochemistry. 2006. V. 45. P. 4998–5009. https://doi.org/10.1021/bi0525573
Abe M., Niibayashi R., Koubori S., Moriyama I., Miyoshi H. // Biochemistry. 2011. V. 50. P. 8383–8391. https://doi.org/10.1021/bi2010202
Shimada S., Shinzawa-Itoh K., Baba J., Aoe S., Shimada A., Yamashita E., Kang J., Tateno M., Yoshikawa S., Tsukihara T. // The EMBO J. 2017. V. 36. P. 291–300. https://doi.org/10.15252/embj.201695021
Lange C., Hunte C. // Proceedings of the National Academy of Sciences. 2002. V. 99. P. 2800–2805. https://doi.org/10.1073/pnas.052704699
Volkov A.N., Nicholls P., Worrall J.A. // Biochim. et Biophys. Acta (BBA)-Bioenergetics. 2011. V. 1807. P. 1482–1503. https://doi.org/10.1016/j.bbabio.2011.07.010
Sinibaldi F., Fiorucci L., Patriarca A., Lauceri R., Ferri T., Coletta M., Santucci R. // Biochemistry. 2008. V. 47. P. 6928–6935. https://doi.org/10.1021/bi800048v
Antonenko Y.N., Lapashina A.S., Kotova E.A., Ramonova A.A., Moisenovich M.M., Agapov. I.I. // The J. Memb. Biol. 2017. V. 250. P. 77–87. https://doi.org/10.1007/s00232-016-9938-6
Пепелина Т.Ю., Черткова Р.В., Долгих Д.А., Кирпичников М.П. // Биоорган. химия. 2010. Т. 36. С. 98–104. [Pepelina T.Y., Chertkova R.V., Dolgikh D.A., Kirpichnikov M.P. // Russ. J. Bioorg. Chem. 2010. V. 36. P. 90–96. https://doi.org/10.1134/S1068162010010097
Pepelina T.Y., Chertkova R. V., Ostroverkhova T.V., Dolgikh D.A., Kirpichnikov M.P., Grivennikova V.G., Vinogradov A.D. // Biochemistry (Mosc.). 2009. V. 74. P. 625–632. https://doi.org/10.1134/S000629790906006
Chertkova R.V., Brazhe N.A., Bryantseva T.V., Nekrasov A.N., Dolgikh D.A., Yusipovich A.I., Sosnovtseva O., Maksimov G.V., Rubin A.B., Kirpichnikov M.P. // PLoS One. 2017. V. 12. P. e0178 280. https://doi.org/10.1371/journal.pone.0178280
Долгих Д.А., Латыпов Р.Ф., Абдуллаев З.X., Колон В., Родер X., Кирпичников М.П. // Биоорган. химия. 1998. Т. 24. С. 756–759. [ Dolgikh D.A., Latypov R. F., Abdullaev Z. K., Kolov V., Roder H., Kirpichnikov M.P. // Russ. J. Bioorg. Chem. V. 24. P. 672–675.]https://doi.org/10.1006/prep.2001.1438
Chertkova R.V., Sharonov G.V., Feofanov A.V., Bocharova O.V., Latypov R.F., Chernyak B.V., Arseniev A.S., Dolgikh D.A., Kirpichnikov M.P. // Mol. Cell. Biochem. 2008. V. 314. P. 85. https://doi.org/10.1007/s11010-008-9768-7
Johnson D., Lardy H. Methods in Enzymology. Academic Press, 1967. V. 10. P. 94–96.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия