

Биоорганическая химия, 2021, T. 47, № 1, стр. 128-134
Превращения хромофора при созревании хромобелка из Actinia equina
А. А. Пахомов 1, 2, *, А. А. Пастухова 1, Г. В. Тишкин 1, В. И. Мартынов 1
1 ФГБУН “Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова” РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
2 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119334 Москва, ул. Вавилова, 28, Россия
* E-mail: alpah@mail.ru
Поступила в редакцию 17.06.2020
После доработки 28.06.2020
Принята к публикации 30.06.2020
Аннотация
В настоящее время считается, что синтез хромофора красных флуоресцентных белков проходит через промежуточную “синюю” форму с максимумом поглощения в районе 400 нм, непосредственно превращающуюся в “красную” через образование двойной связи в боковой цепи хромофоробразующего остатка Tyr. Показано, что в хромобелке из Actinia equina (aeCP), как и в большинстве красных флуоресцентных белков, синтез хромофора проходит путем дегидратации и двух стадий окисления. Однако на промежуточной стадии наблюдается появление дополнительной “зеленой” формы с максимумом поглощения в районе 530 нм.
ВВЕДЕНИЕ
Флуоресцентные белки семейства GFP (зеленого флуоресцентного белка из Aequorea victoria) нашли широкое применение в молекулярной и клеточной биологии [1–3]. Среди них особенно привлекательны – красные флуоресцентные белки (RFP), т.к. они позволяют работать в диапазоне с минимальной клеточной автофлуоресценцией [4, 5]. С увеличением длины волны прозрачность тканей животных возрастает за счет уменьшения рассеивания, а в диапазоне длин волн >650 нм проницаемость тканей для света дополнительно возрастает за счет минимизации вклада поглощения гемоглобина. Это позволяет использовать RFP для визуализации процессов в рамках целого организма [6, 7].
В настоящее время общепринята модель, согласно которой хромофор RFP синтезируется из промежуточной “синей” формы [8–10], которая образуется в результате посттрансляционных модификаций (циклизации, окисления и дегидратации) трех аминокислотных остатков внутри белка и поглощает в области 400 нм. Также известно, что в RFP, хромофор которых синтезируется из аминокислотной последовательности -Asp-Tyr-Gly-, на промежуточной стадии образуется “зеленая” форма (максимум поглощения ~500 нм), превращающаяся в “красную” по пути окислительного декарбоксилирования первого хромофоробразующего остатка Asp [11–15]. В настоящей работе на примере хромобелка из Actinia equina (aeCP) показано, что на промежуточной стадии синтеза хромофора RFP может образовываться форма, поглощающая в области, характерной для зеленых и желтых флуоресцентных белков, даже в случае, если хромофор синтезируется не из аминокислотной последовательности -Asp-Tyr-Gly-.
Хромобелок aeCP имеет максимум поглощения при 593 нм и в исходном виде не флуоресцирует. Однако ранее на его основе, путем внесения нескольких точечных замен в аминокислотное окружение хромофора, были получены красные флуоресцентные белки AQ14 и AQ143 с максимумами эмиссии в районе 660 нм [16]. Ключевыми заменами, приводящими к повышению квантового выхода в белках AQ14 и AQ143 по сравнению с aeCP, стали C143S и S158A, отвечающие за цис-транс-изомерию хромофора [17, 18]. Введением аналогичных замен в окружение хромофора были получены флуоресцентные варианты и других хромобелков [18, 19]. Высокий квантовый выход флуоресценции определяется в основном планарностью и конформационной неподвижностью хромофора внутри белка, при этом химическая структура хромофора в разных мутантных вариантах остается одной и той же [20]. Таким образом, отнесение белка к RFP или к хромобелку с точки зрения структуры хромофора носит скорее номенклатурный характер, т.к. известные на сегодня природные красные флуоресцентные белки и хромобелки имеют такую же химическую структуру хромофора, как RFP, клонированный из Discosoma sp. (DsRed) [21, 22].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
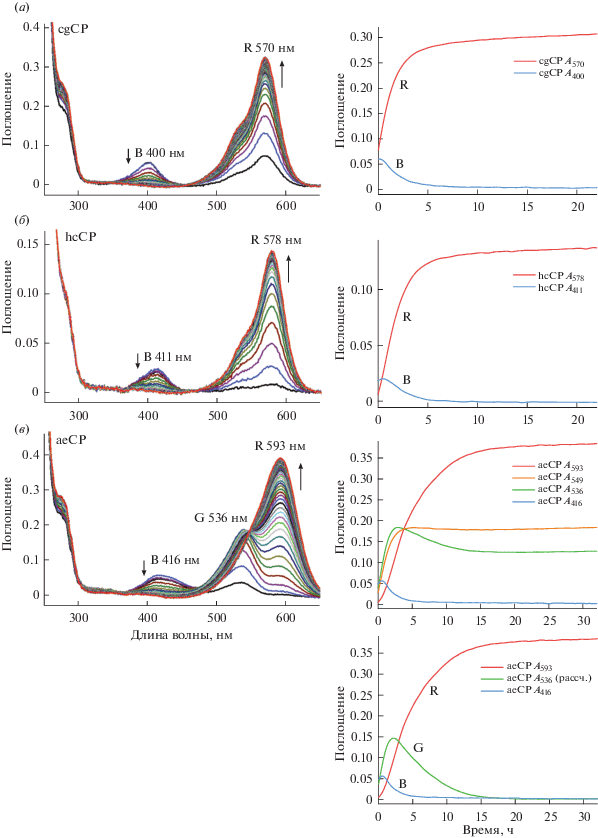
При изучении кинетики созревания ряда хромобелков, поглощающих в дальнем красном диапазоне (aeCP, hcCP (хромобелок из Heteractis crispa) и cgCP (хромобелок из Condylactis gigantea)), нами было обнаружено, что на промежуточной стадии образуется “синяя” форма с максимумом поглощения в районе 400 нм (рис. 1), что согласуется с данными, опубликованными ранее [23]. Аналогичное поведение было обнаружено также у хромобелка из Anemonia sulcata (asCP) [24] и других красных флуоресцентных белков, в частности, в мономерном варианте RFP из Entacmaea quadricolor – tagRFP [25]. Однако в случае aeCP не наблюдалось прямого перехода из “синей” формы в конечное “красное” состояние, а появлялась еще и промежуточная форма с максимумом поглощения при 536 нм (рис. 1в). Такое поглощение характерно для желтых и зеленых флуоресцентных белков, особенно если принимать во внимание значительный батохромный сдвиг максимума поглощения созревшего aeCP по сравнению с другими RFP типа DsRed. По прошествии 5 ч от начала эксперимента наблюдается переход из “зеленой” формы 536 нм в конечное “красное” состояние с изобестической точкой при 549 нм (рис. 1в). Это свидетельствует о непосредственном превращении “зеленой” формы в “красную” при созревании aeCP без промежуточных долгоживущих интермедиатов. Чтобы получить кинетическую кривую для промежуточной “зеленой” формы, из общего спектра поглощения вычитали спектр полностью созревшего белка, нормированный на значение поглощения при 593 нм (рис. 1в, форма G). После такого преобразования становится четко видно, что “синяя” форма образуется раньше “зеленой”, которая затем превращается в “красную”.
Рис. 1.
Изменение спектров поглощения хромобелков cgСР (а), hcСР (б) и аеСР (в) в процессе созревания белка. Спектры снимали с интервалом 30 мин при комнатной температуре. Кинетические кривые созревания при разных длинах волн показаны справа. Поглощение при 549 нм для aeCP соответствует изобестической точке спектрального перехода из формы G в форму R. Кинетические кривые созревания аеСР с учетом вычитания “красной” компоненты для промежуточной формы G приведены внизу справа.
Ранее методами рентгеноструктурного анализа было показано, что “синяя” форма хромофора образуется в результате посттрансляционных модификаций трех аминокислотных остатков внутри белка с образованием ацилиминного заместителя возле первого хромофоробразующего остатка (рис. 2, структура B). C помощью мутагенеза аминокислотных остатков в окружении хромофора красного флуоресцентного белка tagRFP удалось получить белок tagBFP, в котором синтез хромофора останавливался на стадии “синей” формы [25, 26]. В этом ключе образование “зеленой” формы хромофора в aeCP можно было бы объяснить таутомеризацией BFP-хромофора с образованием двойной связи в боковой цепи второго хромофоробразующего остатка Tyr (рис. 2, структура G). Такая структура – энергетически более выгодна, т.к. приводит к сопряжению двух ароматических колец, и образуется в большинстве зеленых флуоресцентных белков. В tagBFP такая таутомеризация, вероятно, не происходит, поскольку фенольная группа хромофоробразующего остатка Tyr ориентирована под большим углом к плоскости имидазолонового цикла, из-за чего сопряжение ароматических колец является проблематичным [27].

Превращение из “зеленого” состояния в конечное “красное” возможно за счет окисления структуры G (рис. 2) с образованием двойной связи в боковой цепи первого хромофоробразующего остатка. Мы проанализировали структуру конечного состояния хромофора при помощи масс-спектрометрии выделенного из белка короткого пептида, содержащего хромофор. Для этого белок денатурировали и подвергали протеолитическому расщеплению пепсином. Далее из протеолитического гидролизата при помощи ВЭЖХ выделяли короткий пептид, содержащий хромофор. Масса выделенного хромопептида составила 1037.4 Да (m/z 1038.4). Это на 22 Да меньше расчетной массы немодифицированного пептида A59PCCMYGSKT (1059.4 Да), что соответствует одной стадии дегидратации (–18 Да) и двум стадиям окисления (–4 Да) при синтезе хромофора и наблюдается в большинстве красных белков. Таким образом, в конечном состоянии aeCP имеет хромофор, типичный для RFP (рис. 2, соединение R).
Отметим, что ранее были получены мутантные варианты DsRed [28], asCP [24] и c-gCP [29], синтез хромофора которых частично не доходит до “красной” формы, а останавливается на стадии “зеленого” хромофора с максимумом поглощения в районе 500 нм. В зрелом DsRed дикого типа значительная часть белка также содержит “зеленый” хромофор [28]. В отличие от aeCP, “зеленая” форма в этих белках является конечной и не способна превращаться в “красную” [23].
Чтобы попытаться понять причины отличий в кинетике созревания aeCP и других белков, мы проанализировали аминокислотное окружение хромофора aeCP в сравнении с гомологами. Пространственная структура aeCP и cgCP к настоящему времени не установлена, так же как и для hcCP, однако установлена структура мутантного варианта hcCP – HcRed, флуоресцирующего в красной области спектра. Ранее было показано, что гомологичное моделирование можно использовать для оценки аминокислотного окружения флуоресцентных белков и объяснения их свойств [30, 31]. В настоящей работе при помощи гомологичного моделирования была построена модель пространственной структуры aeCP с использованием хромобелка из Cnidopus japonicus (cjBlue, степень гомологии 65%) [32] в качестве белка-шаблона. Модель cgCP и hcCP строилась на основе HcRed [33]. Согласно полученным данным, хромофор aeCP вовлечен в стекинг-взаимодействие с остатком His197 (Н197) (рис. 3а), что, вероятно, вносит значительный вклад в батохромный сдвиг. Анализ ближайшего аминокислотного окружения хромофора aeCP не обнаружил принципиальных отличий в сравнении с другими GFP-подобными белками. В целом ближайшее окружение хромофора aeCP очень похоже на таковое у других гомологов, включая каталитические остатки Glu215 (Е215) и Arg92 (R92) (рис. 3а). Анализ более отдаленных участков показал, что в aeCP могут присутствовать полости, отсутствующие в других белках (рис. 3б). Это может сказываться как на общей подвижности боковых цепей внутри β-бочонка, так и на прохождении молекул растворителя либо кислорода снаружи белка к хромофору. Недавно было показано, что на протекание реакции зелено-красной конверсии в RFP из Zoanthus sp. (z-oan2RFP) может оказывать влияние даже подвижность аминокислотных остатков, боковые цепи которых ориентированы в растворитель, а не внутрь β-бочонка [14]. При этом вклад таких остатков может быть существенно большим, чем остатков, участвующих во взаимодействиях между субъединицами мономера в тетрамерном комплексе белка. Таким образом, анализ модели aeCP не выявил принципиальных отличий в структуре белка относительно других гомологов, и необходимо проведение дополнительных исследований для более точного определения причин, по которым при синтезе хромофора aeCP промежуточная “зеленая” форма является, с одной стороны, довольно долгоживущей, а с другой стороны, способна полностью превращаться в “красное” состояние.
Рис. 3.
Рассчитанная пространственная структура хромобелка aeCP в сравнении с гомологами. (а) – Аминокислотное окружение хромофора в белках aeCP, cgCP, hcCP и tagRFP (PDB ID: 3M22), подписаны аминокислотные остатки aeCP; (б) – полости внутри β-бочонка для этих же белков. Стрелки указывают на полости в aeCP, не обнаруженные в других гомологах.

ЗАКЛЮЧЕНИЕ
На примере хромобелка aeCP показано, что механизм синтеза хромофора “красных” белков семейства GFP может протекать через последовательные промежуточные “синее” и “зеленое” состояния. Наиболее вероятным механизмом выступает образование сначала “синего” хромофора типа tagBFP, который таутомеризуется в “зеленый” хромофор GFP-типа. “Красный” хромофор DsRed-типа образуется, вероятно, в результате последующего окисления GFP-хромофора. Потенциально такой сине-зелено-красный переход можно использовать для разработки флуоресцентных таймеров на основе aeCP.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Получение и очистка белков. Клетки Escherichia сoli штамма JM-109 трансформировали плазмидой pQE-30, несущей ген целевого белка, и выращивали в среде LB, содержавшей ампициллин в концентрации 100 мкг/мл. Для получения полностью созревшего белка клеточную культуру выращивали при постоянном перемешивании (250 об./мин) и температуре 37°С в течение ночи, затем температуру понижали до 22°С и продолжали перемешивание клеточной суспензии в течение суток для максимального созревания белка. Для получения белка в незрелом состоянии клетки выращивали при 37°С в течение ~12 ч и сразу же выделяли из них белок. Клеточную биомассу осаждали центрифугированием (3000 g, 15 мин при 4°С), затем осадок ресуспендировали в промывочном буфере (50 мМ Tris-HCl, 300 мМ NaCl, 20 мМ имидазол, pH 8.0) и охлаждали во льду. После этого клетки лизировали при помощи ультразвукового дезинтегратора Sonopuls HD-3100 (Bandelin, Германия), получившийся клеточный лизат центрифугировали при 12 000 g в течение 10 мин при 4 °С. Поскольку целевые белки на своем N-конце содержали гексагистидиновую последовательность, их выделение осуществляли методом металл-хелатной хроматографии на Ni-NTA-агарозе (Quiagen, США). Клеточный лизат наносили на хроматографическую колонку, предварительно уравновешенную промывочным буфером, промывку проводили 4‑кратным объемом того же буфера, после чего осуществляли элюирование белков буфером, содержавшим 50 мМ Tris-HCl, 300 мМ NaCl, 250 мМ имидазол, pH 8.0.
Спектры поглощения измеряли на спектрофотометре Cary 50 Bio (Varian, США). Для наблюдения за кинетикой созревания белков образцы сразу после выделения на Ni-NTA-агарозе переносили в кювету и снимали спектры поглощения через каждые 30 мин при комнатной температуре (22°C).
Выделение хромофорсодержащих пептидов. Раствор белка титровали до рН 2.8 при помощи уксусной кислоты для денатурации, после чего к раствору добавляли пепсин (Sigma, США) в соотношении 1 : 30 и оставляли на ночь. Полученный гидролизат белка наносили на ВЭЖХ-колонку с обращенной фазой (Ultrasphere ODS, BD Biosciences, США), уравновешенную 10 мМ натрий-фосфатным буфером. Пептиды элюировали с помощью линейного градиента ацетонитрила в том же буфере. Детекцию пептидных фракций проводили одновременно при 220 и 380 нм. Основную фракцию, поглощающую при 380 нм, анализировали при помощи масс-спектроскопии.
MALDI-TOF-масс-спектрометрический анализ хромопептидов проводили на масс-спектрометре Ultraflex MALDI-TOF (Bruker Daltonics, Германия) в режиме рефлектрона. В качестве матрицы использовали 2,5-дигидроксибензойную кислоту.
Гомологичное моделирование. Парное выравнивание последовательностей и последующее гомологичное моделирование осуществляли при помощи программы MODELLER v9.24 [34]. При моделировании хромофор был введен как жесткое тело в режиме, учитывающем гетероатомные остатки (с параметром env.io.hetatm = True). Для моделирования пространственной структуры aeCP в качестве шаблона использовали ближайший гомолог с известной пространственной структурой – cjBlue (PDB ID: 2IB5) [32]. Структуры cgCP и hcCP были получены с использованием белка HcRed (PDB ID: 1YZW) [33] в качестве шаблона. Анализ и сравнение структур белков проводили с использованием программы PyMOL [35].
Список литературы
Chudakov D.M., Matz M.V., Lukyanov S., Lukyanov K.A. // Physiol. Rev. 2010. V. 90. P. 1103–1163. https://doi.org/10.1152/physrev.00038.2009
Martynov V.I., Pakhomov, A.A., Deyev I.E., Petrenko A.G. // Biochim. Biophys. Acta Gen. Subj. 2018. V. 1862. P. 2924–2939.https://doi.org/10.1016/j.bbagen.2018.09.013
Pakhomov A.A., Martynov V.I., Orsa A.N., Bondarenko A.A., Chertkova R.V., Lukyanov K.A., Petrenko A.G., Deyev I.E. // Biochem. Biophys. Res. Commun. 2017. V. 493. P. 1518–1521. https://doi.org/10.1016/j.bbrc.2017.09.170
Miyawaki A., Shcherbakova D.M., Verkhusha V.V. // Curr. Opin. Struct. Biol. 2012. V. 22. P. 679–688. https://doi.org/10.1016/j.sbi.2012.09.002
Fabritius A., Ng D., Kist A.M., Erdogan M., Portugues R., Griesbeck O. // Cell Chem. Biol. 2018. V. 25. P. 1554e8–1561e8. https://doi.org/10.1016/j.chembiol.2018.08.008
Shcherbo D., Merzlyak E.M., Chepurnykh T.V., Fradkov A.F., Ermakova G.V, Solovieva E.A., Lukyanov K.A., Bogdanova E.A., Zaraisky A.G., Lukyanov S., Chudakov D.M. // Nat. Methods. 2007. V. 4. P. 741–746. https://doi.org/10.1038/nmeth1083
Luker K.E., Pata P., Shemiakina I.I., Pereverzeva A., Stacer A.C., Shcherbo D.S., Pletnev V.Z., Skolnaja M., Lukyanov K.A., Luker G.D., Pata I., Chudakov D.M. // Sci. Rep. 2015. V. 5. P. 10 332. https://doi.org/10.1038/srep10332
Shcherbakova D.M., Subach O.M., Verkhusha V.V. // Angew Chem. Int. Ed. Engl. 2012. https://doi.org/10.1002/anie.201200408
Subach F.V., Verkhusha V.V. // Chem. Rev. 2012. V. 112. P. 4308–4327. https://doi.org/10.1021/cr2001965
Плетнева Н.В., Горячева Е.А., Артемьев И.В., Архипова С.Ф., Плетнев В.З. // Биоорг. химия. 2019. Т. 45 С. 339–347. [Pletneva N.V., Goryacheva E.A., Artemyev I.V., Arkhipova S.F., Pletnev V.Z. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 187–194.] https://doi.org/10.1134/S106816201903004X
Pakhomov A.A., Martynov V.I. // Biochemistry. 2007. V. 46. P. 11 528–11 535. https://doi.org/10.1021/bi700721x
Pletneva N., Pletnev V., Tikhonova T., Pakhomov A.A., Popov V., Martynov V.I., Wlodawer A., Dauter Z. // Acta Crystallogr. D Biol. Crystallogr. 2007. V. 63. P. 1082–1093. https://doi.org/10.1107/S0907444907042461
Pletneva N., Pletnev S., Tikhonova T., Popov V., Martynov V., Pletnev V. // Acta Crystallogr. D. Biol. Crystallogr. 2006. V. 62. P. 527–532. https://doi.org/10.1107/S0907444906007852
Kim S.E., Hwang K.Y., Nam K.H. // Protein Sci. 2019. V. 28. P. 375–381. https://doi.org/10.1002/pro.3540
Pakhomov A.A., Frolova A.Y., Tabakmakher V.M., Chugunov A.O., Efremov R.G., Martynov V.I. // J. Photochem. Photobiol. B Biol. 2020. V. 206. P. 111 853. https://doi.org/10.1016/j.jphotobiol.2020.111853
Shkrob M.A, Yanushevich Y.G., Chudakov D.M., Gurskaya N.G., Labas Y.A, Poponov S.Y., Mudrik N.N., Lukyanov S., Lukyanov K.A. // Biochem. J. 2005. V. 392. P. 649–654. https://doi.org/10.1042/BJ20051314
Bulina M.E., Chudakov D.M., Mudrik N.N., Lukyanov K.A. // BMC Biochem. 2002. V. 3. P. 1–8. https://doi.org/10.1186/1471-2091-3-7
Chudakov D.M., Feofanov A.V., Mudrik N.N., Lukyanov S., Lukyanov K.A. // J. Biol. Chem. 2003. V. 278. P. 7215–7219. https://doi.org/10.1074/jbc.M211988200
Gurskaya N.G., Fradkov A.F., Terskikh A., Matz M.V., Labas Y.A., Martynov V.I., Yanushevich Y.G., Lukyanov K.A., Lukyanov S.A. // FEBS Lett. 2001. V. 507. P. 16–20. https://doi.org/10.1016/S0014-5793(01)02930-1
Andresen M., Wahl M.C., Stiel A.C., Grater F., Schafer L.V., Trowitzsch S., Weber G., Eggeling C., Grubmuller H., Hell S.W., Jakobs S. // Proc. Natl. Acad. Sci. USA. 2005. V. 102. P. 13 070–13 074. https://doi.org/10.1073/pnas.0502772102
Gross L.A., Baird G.S., Hoffman R.C., Baldridge K.K., Tsien R.Y. // Proc. Natl. Acad. Sci. USA. 2000. V. 97. P. 11 990–11 995. https://doi.org/10.1073/pnas.97.22.11990
Yarbrough D., Wachter R.M., Kallio K., Matz M.V., Remington S.J. // Proc. Natl. Acad. Sci. USA. 2001. V. 98. P. 462–467. https://doi.org/10.1073/pnas.98.2.462
Verkhusha V.V., Chudakov D.M., Gurskaya N.G., Lukyanov S., Lukyanov K.A. // Chem. Biol. 2004. V. 11. P. 845–854. https://doi.org/10.1016/j.chembiol.2004.04.007
Пахомов А.А., Третьякова Ю.А., Мартынов В.И. // Биоорг. химия. 2010. Т. 36. С. 117–121. [Pakhomov A.A., Tretyakova Y.A., Martynov V.I. // Russ. J. Bioorg. Chem. 2010. V. 36. P. 109–113.] https://doi.org/10.1134/S1068162010010127
Subach O.M., Malashkevich V.N., Zencheck W.D., Morozova K.S., Piatkevich K.D., Almo S.C., Verkhusha V.V. // Chem. Biol. 2010. V. 17. P. 333–341. https://doi.org/10.1016/j.chembiol.2010.03.005
Subach O.M., Gundorov I.S., Yoshimura M., Subach F.V., Zhang J., Grüenwald D., Souslova E.A., Chudakov D.M., Verkhusha V.V. // Chem. Biol. 2008. V. 15. P. 1116–1124. https://doi.org/10.1016/j.chembiol.2008.08.006
Pletnev S., Subach F.V., Dauter Z., Wlodawer A., Verkhusha V.V. // J. Am. Chem. Soc. 2010. V. 132. P. 2243–2253. https://doi.org/10.1021/ja908418r
Baird G.S., Zacharias D.A., Tsien R.Y. // Proc. Natl. Acad. Sci. USA. 2000. V. 97. P. 11 984–11 989. https://doi.org/10.1073/pnas.97.22.11984
Pakhomov A.A., Pletneva N.V., Balashova T.A., Martynov V.I. // Biochemistry. 2006. V. 45. P. 7256–7264. https://doi.org/10.1021/bi060207q
Пахомов А.А., Мартынов В.И. // Биоорг. химия. 2011. Т. 37. С. 429–432. [Pakhomov A.A., Martynov V.I. // Russ. J. Bioorg. Chem. 2011. V. 37. P. 383–386.] https://doi.org/10.1134/S1068162011030137
Pakhomov A.A., Martynov V.I. // Biochem. Biophys. Res. Commun. 2011. V. 407. P. 230–235. https://doi.org/10.1016/j.bbrc.2011.03.004
Chan M.C.Y., Karasawa S., Mizuno H., Bosanac I., Ho D., Privé G.G., Miyawaki A., Ikura M. // J. Biol. Chem. 2006. V. 281. P. 37 813–37 819. https://doi.org/10.1074/jbc.M606921200
Wilmann P.G., Petersen J., Pettikiriarachchi A., Buckle A.M., Smith S.C., Olsen S., Perugini M.A., Devenish R.J., Prescott M., Rossjohn J. // J. Mol. Biol. 2005. V. 349. P. 223–237. https://doi.org/10.1016/j.jmb.2005.03.02
Webb B., Sali A. // Curr. Protoc. Bioinformatics. 2016. V. 54. P. 5.6.1–5.6.37. https://doi.org/10.1002/cpbi.3
Rigsby R.E., Parker A.B. // Biochem. Mol. Biol. Educ. 2016. V. 44. P. 433–437. https://doi.org/10.1002/bmb.20966
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия