

Биоорганическая химия, 2021, T. 47, № 1, стр. 111-128
Гетероциклические аналоги ресвератрола – синтез и физиологическая активность. Часть 1. Аналоги, полученные заменой арильных фрагментов на гетероциклические структуры
А. В. Семенов 1, *, Е. В. Семенова 1, О. И. Балакирева 1
1 Национальный исследовательский Мордовский государственный университет им. Н.П. Огарева
430005 Саранск, ул. Большевистская, 68, Россия
* E-mail: salexan@mail.ru
Поступила в редакцию 02.06.2020
После доработки 16.06.2020
Принята к публикации 24.06.2020
Аннотация
Ресвератрол – полифенольный фитоалексин, производное стильбена, исследованию физиологических эффектов которого посвящено большое количество работ, в результате которых для него было показано антиоксидантное, противоопухолевое, нейропротекторное, противовоспалительное, антибактериальное и противовирусное действие. В последние десятилетия большое внимание уделялось созданию производных ресвератрола для улучшения его фармакологической активности и фармакокинетики. Первая часть обзора посвящена синтетическим аналогам ресвератрола, полученным путем замены одного или двух фенильных фрагментов стильбенового остова на биоизостерные гетероциклические структуры. Основное внимание уделено методам синтеза аналогов ресвератрола и проявляемой ими биологической активности.
ВВЕДЕНИЕ
Ресвератрол (3,5,4'-тригидрокистильбен) относится к фитоалексинам полифенольной природы – группе широко распространенных соединений растительного происхождения. Благодаря наличию в структуре двойной связи С=С он может существовать в виде цис- и транс-конфигураций (рис. 1), которые при определенных условиях достаточно легко переходят друг в друга.

В природных условиях преобладает более устойчивый транс-ресвератрол, впервые обнаруженный и выделенный в чистом виде японским исследователем M. Takaoka еще в 1940 г. из одного из видов черемицы (Veratrum grandiflorum) [1]. В дальнейшем этот фитоалексин был идентифицирован во многих природных объектах: коре сосны, орехах (арахис, фисташки), какао-бобах, ягодах (малина, слива, виноград) и др. В настоящее время основными природными источниками ресвератрола являются экстракты кожицы и семян винограда, а также экстракт корней горца японского.
Интерес к ресвератролу резко возрос в 1992 г., когда была выдвинута гипотеза для объяснения кардиопротекторных эффектов красного вина и так называемого “французского парадокса” – относительно низкой частоты сердечно-сосудистых заболеваний в тех регионах Франции, где совместно с высококалорийной диетой с большим содержанием насыщенных жиров употребляется значительное количество красного вина [2]. Последующие многочисленные исследования, обобщенные в ряде обзоров, показали наличие у ресвератрола не только кардиопротекторных [3], но и противоопухолевых [4], нейропротекторных [5] и противовоспалительных свойств [6], а также способность замедлять процессы старения и увеличивать продолжительность жизни, что было показано для ряда организмов (дрожжи, черви, мухи и рыбы) [7, 8]. Многие из этих эффектов опосредованы выраженными антиоксидантными свойствами соединения [9].
Однако мультитаргетность действия ресвератрола на практике ассоциирована с относительно невысокой клинической эффективностью. Недостаточная терапевтическая эффективность препарата у человека многими авторами объясняется низкой биодоступностью, связанной с очень быстрой пресистемной элиминацией в кишечнике и печени [10, 11]. В связи с этим проводится активный поиск синтетических аналогов ресвератрола в направлении повышения фармакологической активности или селективности действия, а также оптимизации фармакокинетики. Представленные в литературе модификации структуры нативного соединения, в том числе с учетом концепции биоизостерных замен, можно разделить на три ключевых направления: 1) варьирование типа, числа и количества заместителей в стильбеновом остове ресвератрола; 2) замена арильных фрагментов на гетероциклические структуры; 3) модификация линкера, соединяющего бензольные кольца.
Первая часть обзора посвящена второму варианту трансформации нативной структуры. Каждая серия аналогов будет рассмотрена с точки зрения подходов к синтезу и их биологической активности.
ЗАМЕНА АРИЛЬНЫХ ФРАГМЕНТОВ В РЕСВЕРАТРОЛЕ НА БИОИЗОСТЕРНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СТРУКТУРЫ
Общая стратегия синтеза аналогов этого типа обычно базируется на формировании непредельного линкера, соединяющего циклические фрагменты независимо от их природы. Для этого используются как различные модификации реакции Виттига, так и процессы кросс-сочетания в присутствии металлокомплексных катализаторов.
Аналоги с антиоксидантной активностью
Антиоксидантные свойства ресвератрола во многих случаях рассматриваются в качестве базовой основы проявляемых им физиологических эффектов. Поэтому множество работ посвящены созданию аналогов ресвератрола с улучшенной способностью подавлять негативные эффекты, вызываемые ROS. С другой стороны, показанная в ряде исследований прооксидантная активность соединения также представляет несомненный интерес. В случае гетероциклических аналогов модулирование данных видов активностей в основном построено на создании гибридных молекул за счет объединения в одной структуре нескольких фармакофоров. В последнее время такой подход в медицинской химии привлекает большое внимание. Примером может служить синтез производных ресвератрола с включением в структуру фрагмента витамина Е [12]. Ключевой стадией получения структур (1) и (2) стала реакция Виттига–Хорнера между предварительно синтезированным альдегидом с хромановым остовом и соответствующей серии диэтилфосфонатов (схема 1 ).
Кинетические исследования позволили установить, что антиоксидантное действие синтезированных молекул согласуется с механизмом отрыва протона с последующим переносом электрона Sequential proton-loss electron transfer (SPLET), и при этом фиксируется существенный рост антирадикальной активности по сравнению с родительскими структурами.
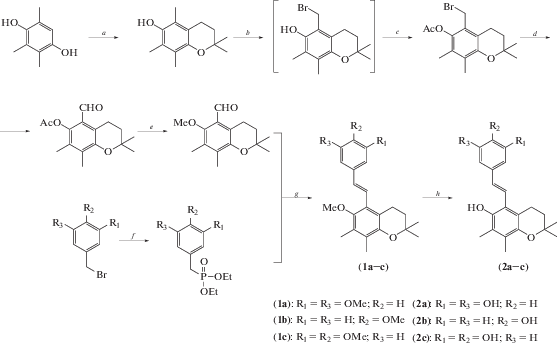
Схема 1 . Синтез соединений (1) и (2). Реагенты и условия реакций: a – 2-метилбут-3-ен-2-ол, TFA; b – Br2; c – Ac2O, AcOH, H2SO4; d – N-метилморфолин-N-оксид; e – 1. NH4OAc, 2. MeI, K2CO3; f – (EtO)3P; g – NaH; h – NaH, EtSH.
В своих исследованиях [13, 14] мы также обратились к созданию гибридных антиоксидантов, включив в остов ресвератрола 3-гидроксипиридиновый фрагмент – структурную основу таких препаратов, как эмоксипин и мексидол. Синтез аналогов проведен по схеме 2 и включает получение четвертичных пиридиниевых солей с последующей их конденсацией в условиях реакции Кнёвенагеля с коммерчески доступными альдегидами.

Схема 2 . Синтез соединений (3–6). Реагенты и условия реакций: a – 1. CH3ONa, CH3OH, MW. 2. CH3I, MW или 1. CH2N2, Et2O. 2. CH3I, CHCl3, MW; b – ArCHO, BuOH, пиперидин; c – Py ⋅ HCl.
Большинство полученных производных показало высокую антирадикальную (в тесте ДФПГ) и антиоксидантную (на модели железо-аскорбат индуцированного окисления липидных мембран митохондрий) активность, превышающую параметры прототипа, при низкой токсичности. Наилучшими характеристиками обладали производные (5b, c) и (6b, c).
Еще одним примером может служить синтез гибридов ресвератрола и известного хелатирующего агента – деферипрона [15]. Синтетическая схема 3 базируется на реакции Виттига, в рамках которой илиды на основе койевой кислоты вводились во взаимодействие с доступными замещенными бензальдегидами.

Схема 3 . Синтез соединений (7–11). Реагенты и условия реакций: a – BnCl, MeOH, кипячение, 16 ч; b – SOCl2, 2 ч; c – PPh3, CHCl3, 24 ч; d – NaOH; e – NH3 · H2O; f – MeNH2; g – 6 н HCl или BBr3.
Оценка in vitro показала, что большинство полученных соединений одновременно проявляют высокую антиоксидантную и металлхелатирующую активность. Наиболее перспективными оказались производные (10i) и (11f).
На основе реакции кросс-сочетания (схема 4 ) были получены новые аналоги ресвератрола, содержащие пяти- и шестичленные нитроксиды и изоиндолиновые нитроксиды [16].

Схема 4 . Синтез соединений (12–14). Реагенты и условия реакций: a – K2CO3, Pd(OAc)2, Bu4NBr, DMF; b – 1. аскорбиновая кислота, N2, диоксан, H2O; 2. Et3N, AcCl; c – Fe, AcOH; d – 1. KOAc, Pd(OAc)2, Bu4NBr, DMF, 2. MeONa, MeOH.
К сожалению, их способность к перехвату свободных радикалов не превзошла наблюдаемую для прототипа.
Аналоги с противоопухолевой активностью
Согласно имеющимся в литературе сведениям противоопухолевые эффекты ресвератрола могут реализовываться различными механизмами, одним из которых является антимитотическое действе путем воздействия на различные фазы клеточного цикла. В частности, у ресвератрола выявлена способность ингибировать функции микротрубочек. При этом установлено, что более эффективным воздействием на тубулин обладают его метоксилированные производные, например, (E)‑3,4,5,4ꞌ-тетраметоксистильбен (DMU-212) [17], имеющий структурное сходство с известным природным антимитотическим агентом комбретастатином А-4 (рис. 2).

Учитывая вероятную ключевую роль триметоксистирильного фрагмента в проявляемой активности, был синтезирован ряд аналогов DMU-212, в которых 4-метоксифенильный фрагмент был заменен на индольный, бензофурановый, бензотиазольный или бензотиофеновый остатки [18]. В основу синтеза была положена реакция Виттига между соответствующими гетероциклическими альдегидами и трифенилфосфониевыми солями (схема 5 ).

Схема 5 . Синтез соединений (15).
Было установлено, что именно аналоги, содержащие транс-3,4,5-триметоксистирильный фрагмент (соединения (15a), (15d), (15f) и (15g) соответственно), продемонстрировали мощный ингибирующий эффект (со значениями GI50 < 1 мкМ) в 85% из 60 исследованных линий раковых клеток человека. При этом 3,4- и 3,5-диметоксизамещенные производные показали значительно меньшее ингибирование роста. Проведенное молекулярное моделирование выявило, что четыре наиболее активные соединения (15a), (15d), (15f) и (15g) эффективно взаимодействуют с колхициновым сайтом связывания тубулина.
Также была предпринята попытка создания гибридной молекулы (схема 6 ) на основе ресвератрола и известного ингибитора полимеризации тубулина – 2-анилинопиридин сульфонамида (E7010) [19].
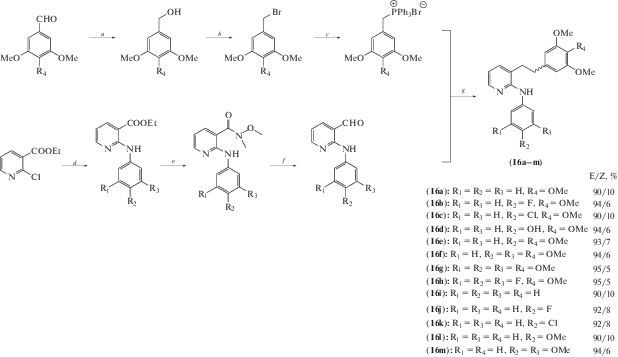
Схема 6 . Синтез соединений (16). Реагенты и условия реакций: a – NaBH4, MeOH; b – PBr3, CH2Cl2; c – PPh3, толуол; d – ArNH2, этиленгликоль; e – N,O-диметилгидроксиламин, AlMe3, CH2Cl2; f – DIBAL-H, CH2Cl2; g – NaH, THF.
Антипролиферативную активность соединений исследовали на четырех раковых клеточных линиях человека (A549, HepG2, HeLa и Du145). Были получены значения GI50 в диапазоне 2.1–20 мкМ. В отношении клеточной линии HepG2 рака шейки матки человека производные (16a) и (16k) показали в 13 раз более низкие значения GI50 по сравнению с ресвератролом и в 1.1–2 раза более низкие значения GI50 по отношению к E7010. Установлено, что соединения (16a) и (16k) в рассматриваемой клеточной линии активно взаимодействовали с колхициновым сайтом связывания, ингибировали сборку тубулина, деполимеризовали микротрубочки и останавливали клеточный цикл в фазе G2/M. Также они продемонстрировали высокую эффективность и селективность на панели линий раковых клеток человека NCI 60.
Антимитотическая активность оценивалась и для стирилхиназолиновых аналогов [20], синтезированных в условиях реакции Кнёвенагеля по схеме 7 [21].
Подавляющее большинство полученных производных показало ингибирующую активность по отношению к трем раковым клеточным линиям HeLa, HL-60 и A549 более выраженную, чем у ресвератрола. При этом наиболее мощным цитотоксическим эффектом обладало триацетильное производное (17g), проявляющее в концентрациях 2–10 мкМ цитостатический, а в концентрации 10 мкМ – цитоцидный эффект в отношении клеток рака шейки матки человека HeLa. Было установлено, что соединение (17g) ингибирует пролиферацию клеток HeLa за счет остановки клеточного цикла в фазе G2/M, связанной с активизацией экспрессии циклина B1, за счет усиления фосфорилирования циклинзависимой протеинкиназы 1 (Cdk1) и белка Cdc25C, а также путем повышения уровня белка p21CIP1/WAF1 по p53-зависимому механизму. Последующие исследования показали, что в случае клеточной линии лейкемии HL-60 соединение (17g) стимулирует апоптоз Fas-опосредованным каспазо-8-зависимым путем через генерацию ROS и, в меньшей степени, путем активации высвобождения цитохрома с и каспазы-9 [22].
Серия гибридных молекул, содержащих хинолиновый фрагмент, была получена по синтетической схеме 8 , в основу которой положена реакция Виттига [23].

Схема 7 . Синтез соединений (17).
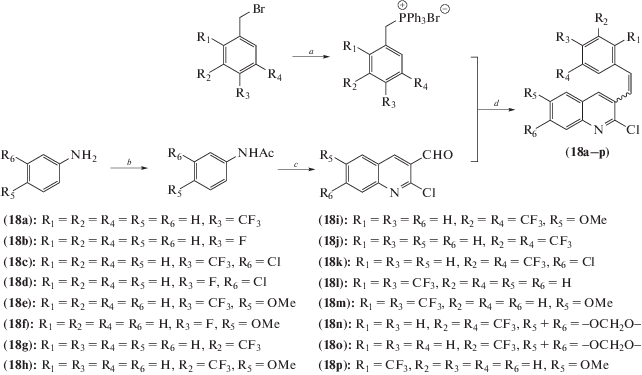
Схема 8 . Синтез соединений (18). Реагенты и условия реакций: a – PPh3, PhMe, кипячение; b – HCl, Ac2O; c – POCl3 : DMF 3 : 1; d – NaOH, DMSO.
Исследования активности, проведенные на четырех клеточных линиях (HeLa, MDA-MB231, MCF7 и MDA-MB468), показали, что три производные (18g), (18h), (18p) в виде Z-изомеров не только обладали достаточно мощным антипролиферативным эффектом (IC50 < 4 мМ), но и показывали приблизительно двукратную селективность в отношении раковых клеток по сравнению с нормальными клетками. Три других соединения (18a), (18i), (18j) в виде Z-изомеров показали сравнительно хорошую активность со значениями IC50 в диапазоне 4–10 мМ. Соединение (18b) в виде Е-изомера проявило высокую активность только в отношении MDA-MB468 (IC50 = 0.12 мМ), вызывая значительное повреждение ДНК и останавливая клеточный цикл в S-фазе. Производное (18g) в виде Z-изомера оказалось эффективным против всех исследованных раковых клеточных линий, вызывая остановку клеточного цикла путем ингибирования полимеризации микротрубочек и, в конечном итоге, приводя к развитию апоптоза. Эффект опосредован взаимодействием соединения с тубулином в сайте связывания, характерным для подофиллотоксина.
Наряду с прямым цитостатическим действием оценивалась противоопухолевая активность ресвератрола, связанная с подавлением опухолевого ангиогенеза [24], в частности, путем влияния на фактор роста эндотелия сосудов (VEGF), сверхэкспрессия производства которого встречается в различных типах опухолевых клеток, способствует их пролиферации и миграции при увеличении проницаемости сосудов. Также сообщалось [25] о влиянии ресвератрола на экспрессию генов, участвующих в активации теломеразы – фермента, экспрессия которого ограничена или отсутствует в нормальных соматических клетках человека, но активирована у 90% злокачественных. С целью повышения эффективности противоопухолевой терапии предпринимаются попытки совмещения высокой цитотоксической активности со способностью подавлять опухолевый ангиогенез и экспрессию генов, связанных с активацией теломеразы. Так, по реакции Хека был синтезирован ряд пиридиновых и пиримидиновых аналогов ресвератрола (схема 9 ) [26].

Cхема 9. Синтез соединений (19) и (20).
Соединения (19c), (19e), (19g), (19h) и (19i) демонстрировали более низкие значения IC50, чем ресвератрол, в отношении опухолевой клеточной линии HT-29 и имели более высокие значения IC50 при исследовании на неопухолевой клеточной линии HEK-293. Наиболее активными оказались метоксизамещенные соединения (19g–i), содержащие 4-пиридиновое кольцо.
Кроме того, соединение (19g) проявило способность снижать образование белка VEGF и экспрессию гена hTERT в концентрациях ниже значения IC50 по отношению к неопухолевым клеткам HEK-293. Для производного (19h) помимо высокой цитотоксичности к HT-29 и снижения продукции VEGF также показано уменьшение экспрессии гена c-Myc. В то же время гидроксизамещенные аналоги (20a–c) не проявили выраженной активности, что также показано в работе [27], где альтернативным путем было получено соединение (20c), которое проявило посредственную цитотоксичность на уровне ресвератрола по отношению к клеточным линиям рака поджелудочной железы AsPC-1, Capan-2 и BxPC-3.
Еще одной перспективной мишенью для создания противоопухолевых средств является лизинспецифическая гистондеметилаза (LSD1) – фермент, играющий важную роль в росте и дифференцировке клеток, избыточная экспрессия которого наблюдается при многих типах злокачественных новообразований и связывается с началом и прогрессированием роста опухоли. Было показано, что ресвератрол проявляет сильное ингибирующее влияние на активность LSD1, более выраженное, чем у известного ингибитора – транс-2-фенилциклопропиламина [28]. На основе структуры ресвератрола были предприняты попытки создания новых обратимых ингибиторов LSD1 [29, 30], синтез которых на основе реакции Виттига–Хорнера представлен на схеме 10 .

Схема 10 . Синтез соединений (21–23). Реагенты и условия реакций: a – t-BuOH, t-BuOK; b – t-BuOK, DMF; c – NH2OH · HCl, Et3N, CH3OH, кипячение, 3–8 ч; d – BBr3, CH2Cl2.
В результате испытаний было выявлено несколько соединений, ингибирующих LSD1, со значениями IC50 в субмикромолярном диапазоне. Соединение (22с) оказалось наиболее сильным обратимым ингибитором LSD1 (IC50 = 283 нМ), способным усиливать экспрессию суррогатного клеточного биомаркера CD86 в клеточной линии лейкемии человека THP-1. Соединение (22c) показало хорошее ингибирование роста клеток THP-1 и MOLM-13 согласно значениям IC50, которые составляли 5.76 и 8.34 мкМ соответственно. Кроме того, соединение (22с) дозозависимо подавляло деление клеток THP-1.
Стильбеновая структура ресвератрола с высокой вероятностью предполагает у него наличие эстроген-модулирующих эффектов, которые изучались на различных клеточных линиях рака молочной железы (MCF-7, T47D, LY2 и S30). Было установлено, что в присутствии эстрадиола ресвератрол действует как антиэстроген и таким образом должен оказывать положительное влияние при химиотерапии рака молочной железы [31].
Были предложены новые структуры (схема 11 ), сочетающие в себе защищенный пиридоксиновый фрагмент и остов ресвератрола [32–34].

Схема 11 . Синтез соединений (24) и (25). Реагенты и условия реакций: a – (CH3)2CO, H+; b – H2O, HCHO, NaOH, 70°C; c – SOCl2, CH2Cl2 или PhCH3, кипячение; d – PPh3, CH3CN, кипячение; e – ArCHO, NaH, CH2Cl2 или THF; f – ArCHO, Et3N, CH2Cl2, кипячение; g – P(OEt)3, кипячение.
Синтезированные соединения были исследованы на наличие противоопухолевой активности и цитотоксические эффекты in vitro. Установлено, что аналоги (E-25b) и (E-25c) не обладали цитотоксичностью как по отношению к опухолевым (MCF-7, SNB-19 и HCT-15), так и нормальным (HEK-293) клеточным линиям, в то время как соединение (E-25a) оказывало более существенное влияние на клетки HEK-293, чем на опухолевые клетки. Наиболее перспективными оказались соединения (E-24b) и (E-24c), продемонстрировавшие высокую селективную эффективность в отношении эстроген-зависимой клеточной линии MCF-7 рака молочной железы (IC50 в диапазоне 1.9–7.9 мкМ). Соединение (E-24b) также показало четкую селективность в отношении эстроген-зависимых клеток рака молочной железы MCF-7 по сравнению с эстроген-негативными клетками рака молочной железы MDA-MB-231 (индекс селективности 10), что свидетельствует о существенной роли рецепторов эстрогена в качестве потенциальной мишени.
Аналоги с нейропротекторной активностью
Во множестве исследований было показано нейропротекторное действе ресвератрола как путем предотвращения гибели нейронов от воздействия АФК благодаря прямому антиоксидантному эффекту и активации сиртуина SIRT1, так и другими путями, например, через подавление роста амилоидных бляшек (за счет преобразования растворимых олигомеров и фибриллярных β-амилоидов в нетоксичную форму), снижения активации микроглии (путем подавления провоспалительных медиаторов или подавлением поляризации М1 и активации поляризации М2), митохондриального биогенеза (за счет стимуляции экспрессии PGC-1α и активации AMPK), профилактики апоптотической гибели клеток в гиппокампе (путем увеличение митохондриальной экспрессии UCP2 через активацию PPAR-γ).
Дизайн гетероциклических аналогов ресвератрола с нейропротекторной активностью обычно основывается на попытках создания многоцелевых лигандов, что продиктовано сложным патогенезом нейродегенеративных заболеваний.
В работе [34] вышеописанные производные (25) изучались в качестве ингибиторов моноаминоксидазы B – потенциальных средств для лечения болезни Паркинсона. Большинство из них проявили сильную ингибирующую активность на МАО-B с высокой селективностью. В частности, соединения (25i) и (25n) наиболее эффективно ингибировали hMAO-B со значениями IC50 в диапазоне 0.01–0.02 мкМ. Дальнейшее исследования показали, что (25n) является обратимым, а (25i) необратимым ингибитором МАО-В. Причины различий и особенности взаимодействия соединений с МАО-B были оценены методом молекулярного докинга. Кроме того, (25i) и (25n) показали хорошую антиоксидантную активность, нейропротекторный эффект в тесте H2O2-индуцированного повреждения клеток PC-12 и способность преодолевать гематоэнцефалический барьер, что свидетельствует о потенциальной ценности данных соединений для лечения болезни Паркинсона.
Еще одна серия многофункциональных агентов, представляющих собой пиридоксин-ресвератрольные гибриды и полученные на их базе основания Манниха (схема 12 ), была синтезирована в работе Yang et al. [35].
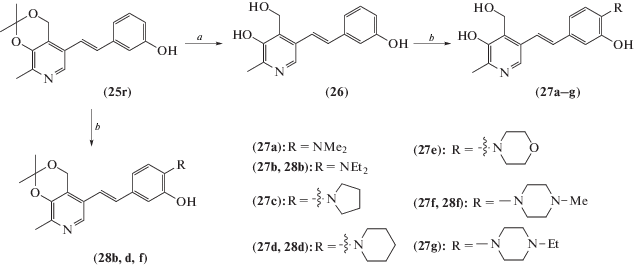
Схема 12 . Синтез соединений (26–28). Реагенты и условия реакций: a – 10% HCl, THF, 70°C, 3 ч; b – амин, параформ, EtOH, кипячение.
Результаты испытаний показали, что большинство из этих соединений могут избирательно ингибировать ацетилхолинэстеразу (АХЭ) и MAO-B. Среди них соединения (27d) и (28b) продемонстрировали наибольший потенциал ингибирования по отношению к АХЭ (IC50 = 2.11 и 1.56 мкМ соответственно), а соединение (27е) наиболее активно ингибировало МАО-В (IC50 = 2.68 мкМ). Кинетический анализ показал, что соединение (27d) является ингибитором смешанного типа, связываясь одновременно с каталитическим и периферическим анионным сайтом АХЭ. Кроме того, все исследованные соединения показали хорошие антиоксидантные и металлхелатообразующие свойства, что важно при создании многофункциональных агентов для лечения болезни Альцгеймера.
Так, в попытке создания эффективных средств для терапии болезни Альцгеймера для рассмотренных выше соединений (10i) и (11f), проявляющих одновременно выраженную антиоксидантную и металлхелатирующую активность, было оценено ингибирующее влияние на агрегацию Aβ. Было показано, что рассматриваемые соединения могут эффективно взаимодействовать с C‑концом Aβ1–42, ингибировать собственную или индуцированную металлом (железо, медь) агрегацию β-листов и образование фибрилл, показывая потенциал против агрегации Aβ1–42 больший, чем у ресвератрола.
В продолжение этой фармакофорно-комбинированной стратегии в исследовании Jiang et al. [36] была синтезирована серия производных 2-замещенных бензотиазолов (схема 13 ), в которых фрагмент деферипрона или мальтола были соединены с тиогавином T – флуоресценным индикатором, широко используемым in vitro для оценки агрегации Aβ.
Максимальную эффективность показали молекулы (29c) и (29i), проявляющие превосходную ингибирующую активность в отношении агрегации Aβ1–42, высокую радикалпоглощающую активность, выраженные хелатирующие свойства по отношению к биометаллам, а также дезагрегационную активность в отношении ранее образованных Aβ1–42 фибрилл.
В исследовании [37] были синтезированы соединения, нацеленные на терапию болезни Альцгеймера путем объединения фармакофоров ресвератрола и фрагмента известного хелатора металла – клиохинола (схема 14 ).

Схема 13 . Синтез соединений (29) и (30). Реагенты и условия реакций: a – PMB-Cl, K2CO3, DMF, 80°C; b – CH3NH2, H2O, кипячение; c – MnO2, CHCl3, кипячение; d – AcOH, Ac2O, MW; e – K2CO3, MeOH; f – NaH, THF; g – CF3COOH, CH2Cl2.

Схема 14 . Синтез соединений (31). Реагенты и условия реакций: a – 37% HCl, 37% HCHO; b – P(OEt)3, кипячение; c – MOMCl, (i-Pr)2NEt, CH2Cl2; d – ArCHO, CH3ONa, DMF, 0–80°C; e – 6 M HCl, CH3OH, кипячение; f – BBr3, CH2Cl2.
Гибриды показали значительную способность ингибировать автоиндуцированную агрегацию β‑амилоида (Aβ) и агрегацию Aβ, индуцированную медью(II), потенциальное антиоксидантное действие и хелатирующую способность. Среди этих соединений (31с) имело наиболее высокий потенциал ингибирования автоиндуцированной (IC50 = 8.50 мкМ) и индуцированной медью(II) агрегации Aβ, оно также способствовало разборке хорошо структурированных фибрилл Aβ, образованных путем самоиндукции агрегации и индуцированных медью(II) агрегаций Aβ. Кроме того, соединение (31c) было способно контролировать продукцию гидроксильных радикалов за счет блокирования окислительно-восстановительного цикла меди путем комплексообразования, что подтверждается экспериментами с использованием системы Cu(I)/Cu(II) с аскорбатом в качестве восстановителя. Важно отметить, что для соединения (31c) летальная доза у белых мышей была выше 2 г/кг, а также была продемонстрирована способность преодолевать гематоэнцефалический барьер.
В качестве другой важной мишени при терапии нейродегенеративных заболеваний рассматриваются резидентные макрофаги ЦНС – так называемая микроглия. Ее гиперактивация (при воздействии различных факторов) инициирует чрезмерную продукцию воспалительных медиаторов (оксид азота, цитокины, TNF-α), что оказывает повреждающее действе на нейроны и способствует патогенезу болезни Альцгеймера, Паркинсона и других нейродегенеративных заболеваний.
Основываясь на данных о способности ресвератрола подавлять активацию макрофагов и контролировать экспрессию iNOS, в ряде работ были синтезированы его гетероциклические аналоги (схема 15 ), и было изучено их ингибирующее влияние в экспериментальной модели активации микроглии липополисахаридами (LPS) [38, 39].
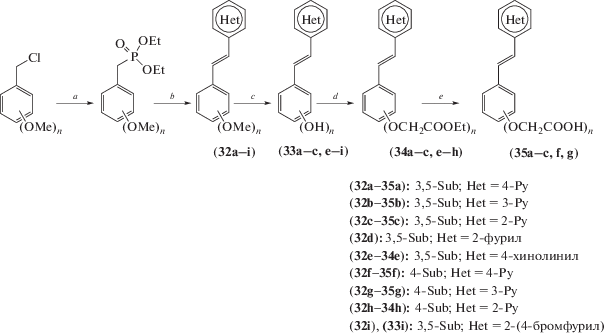
Схема 15 . Синтез соединений (32–35). Реагенты и условия реакций: a – P(OEt)3, кипячение, 3 ч; b – PyCHO, NaOEt, DMF; c – BBr3, СH2Cl2, –10°C, 2 ч; d – ClCH2COOEt, KOH, DMF; e – 1. NaOH, 2. HCl.
Было обнаружено, что соединения (32e), (33e) и (33i) значительно ингибировали продукцию NO и TNF-α микроглией, блокируя фосфорилирование и деградацию IκBα, но не проявляли способности взаимодействовать напрямую с NO в растворе нитропруссида натрия, а производное (33i), кроме того, эффективно блокировало образование ROS.
Сходные эффекты проявляло и ранее описанное производное (17g), для которого наряду со снижением экспрессии iNOS, TNF-α и уровня мРНК, была обнаружена способность снижать секрецию интерлейкина-6 (IL-6), экспрессию белка Toll-подобного рецептора 4 (TLR4), а также ингибировать сигнальные пути, ассоциированные с митоген-активируемой протеинкиназой (MAPK) и ядерным фактором транскрипции κB (NF-κB) [40]. Таким образом, мощные ингибирующие эффекты соединений на активацию микроглии представляют интерес для лечения нейродегенеративных заболеваний, сопровождаемых активацией микроглии.
Аналоги с противовоспалительной активностью
Было установлено, что ресвератрол способен проявлять противовоспалительный эффект не только путем подавления активации макрофагов и контроля экспрессии iNOS, но и за счет способности снижать экспрессию и активность ЦОГ-1 и ЦОГ-2 [41]. К сожалению, как и у большинства нестероидных противовоспалительных средств, у ресвератрола из-за недостаточной селективности ингибирования ЦОГ были показаны побочные эффекты со стороны желудочно-кишечного тракта, например, значимое негативное влияние на заживление индуцированной индометацином язвы желудка у мышей [42].
В попытке создания более эффективных и безопасных противовосполительных средств в работе Chen et al. [43] были синтезированы и исследованы ранее описанные производные (33–35), некоторые из которых были превращены в основания Манниха (36–38) (схема 16 ).

Схема 16 . Синтез соединений (36–38). Реагенты и условия реакций: a – HCHO, морфолин, EtOH, кипячение, 2 ч.
В испытаниях, проведенных на модели индуцированного ксилолом отека у мышей, наиболее активным соединением оказался ресвератрол, подавляющий индуцированный отек на 38.9%. Сравнимые результаты (37.0%) продемонстрировало соединение (35с). Другие производные (35f), (37b), (38) показали уменьшение отека в диапазоне от 30 до 35%.
Ранее описанная серия производных стирилхиназолина (17a–k) также исследовалась на наличие способности ингибировать ЦОГ-2 и снижать продукцию PGE2. Большая часть стирилхиназолинов (17a–k), за исключением 3,4-диацетилированных стирилхиназолинов (17d–f), показала сравнимую или лучшую ингибирующую активность, чем ресвератрол, при более низкой цитотоксичности, изученной на клеточной линии RAW264.7. При этом было установлено, что заместители в бензольном кольце стирилхиназолинов оказывали более сильное влияние на ингибирующую активность, чем заместители в хиназолиновом цикле.
В работе Kim et al. [44] реакцией Виттига–Хорнера между пиронилфосфонатом и различными ароматическими карбоксальдегидами (схема 17 ) были получены пиронилвинильные аналоги ресвератрола (39).
Оценка их способности ингибировать продукцию NO и PGE2 была исследована на LPS-активированных клетках макрофагов клеточной линии RAW264.7. Наилучшую ингибирующую активность показало производное (39b) с меньшей по сравнению с ресвератролом цитотоксичностью.

Схема 17 . Синтез соединений (39). Реагенты и условия реакций: a – PMB-Cl, K2CO3, DMF, 80°C; b – SOCl2, 2 ч; c – P(OMe)3; d – ArCHO, NaH, THF; e – BBr3, СH2Cl2.
Аналоги с иными физиологическими эффектами
Ранее было обнаружено, что ресвератрол обладает ингибирующим действием против вируса гриппа in vitro [45]. В исследовании Li et al. [46] с использованием оценки активности нейраминидазы была протестирована противогриппозная активность 50 производных ресвератрола. Среди веществ, проявлявших ингибирующее действие на активность нейраминидазы вируса гриппа (штамм A/PR/8/34 (H1N1)), оказался и описанный ранее аналог (33b) со значением IC50 = 186.1 мМ.
ЗАКЛЮЧЕНИЕ
Исследования по созданию аналогов ресвератрола в рамках обсуждаемой стратегии в значительной степени сосредоточены на получении гибридных структур, основным преимуществом которых выступает мультитаргетное воздействие, что особенно важно при лечении таких сложных с точки зрения механизмов патогенеза патологий, как онкологические и нейродегенеративные заболевания. При этом за основу берется сохранение и потенцирование известной для прототипа антирадикальной активности и ряд других ранее установленных мишень-ориентированных эффектов. Во многих случаях такой подход оказался весьма эффективным и обеспечил не только аддитивные эффекты отдельных фармакофорных фрагментов, но и их потенцирующее действие. Большинство исследований проводилось на культурах клеток, что сопряжено с рядом известных ограничений, ввиду чего для подтверждения фармакологических эффектов и установления истинных механизмов действия новых соединений необходимы испытания в рамках целостного организма.
Список литературы
Takaoka M. // J. Faculty Sci. Hokkaido Imper. Univ. 1940. V. 3. P. 1–16.
Perdue L. // The French Paradox and beyond / Sonoma, California: Renaissance, 1993.
Bonnefont-Rousselot D. // Nutrients. 2016. V. 8. P. 250–273. https://doi.org/10.3390/nu8050250
Khan O.S., Bhat A.A., Krishnankutty R., Mohammad R.M., Uddin S. // Nutr. Cancer. 2016. V. 68. P. 365–373. https://doi.org/10.1080/01635581.2016.1152386
Singh N., Agrawal M., Doré S. // ACS Chem. Neurosci. 2013. V. 4. P. 1151–1162. https://doi.org/10.1021/cn400094w
Poulsen M.M., Fjeldborg K., Ornstrup M.J., Kjær T.N., Nøhr M.K., Pedersen S.B. // Biochim. Biophys. Acta. 2015. V. 1852. P. 1124–1136. https://doi.org/10.1016/j.bbadis.2014.12.024
Markus M.A., Morris B.J. // Clin. Interv. Aging. 2008. V. 3. P. 331–339.
Westphal C.H., Dipp M.A., Guarente L. // Trends Biochem. Sci. 2007. V. 32. P. 555–560. https://doi.org/10.1016/j.tibs.2007.09.008
Gülçin I. // Innov. Food Sci. Emerg. Technol. 2010. V. 11. P. 210–218. https://doi.org/10.1016/j.ifset.2009.07.002
Sun X., Peng B., Yan W. // J. Chem. Thermodyn. 2008. V. 40. P. 735–738. https://doi.org/10.1016/j.jct.2007.10.006
Delmas D., Aires V., Limagne E., Dutartre P., Mazué F., Ghiringhelli F., Latruffe N. // Ann. N.Y. Acad. Sci. 2011. V. 1215. P. 48–59. https://doi.org/10.1111/j.1749-6632.2010.05871.x
Yang J., Liu G.-Y., Lu D.-L., Dai F., Qian Y.-P., Jin X.-L., Zhou B. // Chem. Eur. J. 2010. V. 16. P. 12 808–12 813. https://doi.org/10.1002/chem.201002020
Semenov A.V., Balakireva O.I., Tarasova I.V. Burtasov A.A., Semenova E.V., Petrov P.S., Minaeva O.V., Pyataev N.A. // Med. Chem. Res. 2018. V. 27. P. 1298–1308. https://doi.org/10.1007/s00044-018-2150-8
Semenov A.V., Balakireva O.I., Tarasova I.V., Semenova E.V., Zulfugarov P.K. // Med. Chem. Res. 2020. V. 29. P. 1590–1599. https://doi.org/10.1007/s00044-020-02585-6
Xu P., Zhang M., Sheng R., Ma Y. // Eur. J. Med. Chem. 2017. V. 127. P. 174–186. https://doi.org/10.1016/j.ejmech.2016.12.045
Kálai T., Borza E., Antus C., Radnai B., Gulyás-Fekete G., Fehér A., Sümegi B., Hideg K. // Bioorg. Med. Chem. 2011. V. 19. P. 7311–7317. https://doi.org/10.1016/j.bmc.2011.10.066
Sale S, Verschoyle R.D., Boocock D., Jones D.J.L., Wilsher N., Ruparelia K.C., Potter G.A., Farmer P.B., Steward W.P., Gescher A.J. // Br. J. Cancer. 2004. V. 90. P. 736–744. https://doi.org/10.1038/sj.bjc.6601568
Penthala N.R., Thakkar S., Crooks P.A. // Bioorg. Med. Chem. Let. 2015. V. 25. P. 2763–2767. https://doi.org/10.1016/j.bmcl.2015.05.019
Kamal A., Ashraf Md., Basha S.T., Hussaini S.M.A., Singh S., Vishnuvardhan M.V.P.S., Kiran B., Sridhar B. // Org. Biomol. Chem. 2016. V. 14. P. 1382–1394. https://doi.org/10.1039/C5OB02022K
Kim J.-Y., Choi H.-E., Lee H.-H., Shin J.-S., Shin D.-H., Choi J.-H., Lee Y. S., Lee K.-T. // Oncol. Reports. 2015. V. 33. P. 2639–2647. https://doi.org/10.3892/or.2015.3871
Park J.H., Min H.-Y., Kim S.S., Lee J.Y., Lee S.K., Lee Y.S. // Arch. Pharm. 2004. V. 337. P. 20–24. https://doi.org/10.1002/ardp.200300791
Park E.Y., Kim J.-I., Leem D.-G., Shin J.-S., Kim K.-T., Choi S.Y., Lee M.-H., Choi J.-H., Lee Y.S., Lee K.-T. // Oncol. Reports. 2016. V. 36. P. 3577–3587. https://doi.org/10.3892/or.2016.5168
Srivastava V., Lee H. // Bioorg. Med. Chem. 2015. V. 23. P. 7629–7640. https://doi.org/10.1016/j.bmc.2015.11.007
Trapp V., Parmakhtiar B., Papazian V., Willmott L., Fruehauf J.P. // Angiogenesis. 2010. V. 13. P. 305–315. https://doi.org/10.1007/s10456-010-9187-8
Lanzilli G., Fuggetta M.P., Tricarico M., Cottarelli A., Serafino A., Falchetti R., Ravagnan G., Turriziani M., Adamo R., Franzese O., Bonmassar E. // Int. J. Oncol. 2006. V. 28. P. 641–648. https://doi.org/10.3892/ijo.28.3.641
Martí-Centelles R., Murga J., Falomir E., Carda M., Alberto Marco J. // Med. Chem. Commun. 2015. V. 6. P. 1809–1815. https://doi.org/10.1039/c5md00197h
De Filippis B., De Lellis L., Florio R., Ammazzalorso A., Amoia P., Fantacuzzi M., Giampietro L., Maccallini C., Amoroso R., Veschi S., Cama A. // Med. Chem. Res. 2019. V. 28, P. 984–991. https://doi.org/10.1007/s00044-019-02351-3
Abdulla A., Zhao X., Yang F. // J. Biochem. Pharmacol. Res. 2013. V. 1. P. 56–63.
Duan Y.-C., Guan Y.-Y., Zhai X.-Y., Ding L.-N., Qin W.-P., Shen D.-D., Liu X.-Q., Sun X.-D., Zheng Y.-C., Liu H.-M. // Eur. J. Med. Chem. 2017. V. 126. P. 246–258. https://doi.org/10.1016/j.ejmech.2016.11.035
Duan Y., Qin W., Suo F., Zhai X., Guan Y., Wang X., Zheng Y., Liu H. // Bioorg. Med. Chem. 2018. V. 26. P. 6000–6014. https://doi.org/10.1016/j.bmc.2018.10.037
Bhat K.P.L., Lantvit D., Christov K., Mehta R.G., Moon R.C., Pezzuto J.M. // Cancer Res. 2001. V. 61. P. 7456–7463.
Pugachev M.V., Nguyen T.T.N., Bulatov T.M., Pavelyev R.S., Iksanova A.G., Bondar O.V., Balakin K.V., Shtyrlin Yu.G. // Journal of Chemistry. 2017. V. 2017. P. 1–7.https://doi.org/10.1155/2017/8281518
Pugachev M.V., Pavelyev R.S., Nguyen T.N.T., Iksanova A.G., Lodochnikova O.A., Shtyrlin Yu.G. // Russian Chem. Bull., Int. Ed. 2016. V. 65. P. 532–536. https://doi.org/10.1007/s11172-016-1333-z
Li W., Yang X., Song Q., Cao Z., Shi Y., Deng Y., Zhang L. // Bioorganic Chem. 2020. V. 97. P. 103 707. https://doi.org/10.1016/j.bioorg.2020.103707
Yang X., Qiang X., Li Y., Luo L., Xu R., Zheng Y., Cao Z., Tan Z., Deng Y. // Bioorganic Chem. 2017. V. 71. P. 305–314. https://doi.org/10.1016/j.bioorg.2017.02.016
Jiang L., Zhang M., Tang L., Weng Q., Shen Y., Hu Y., Sheng R. // RSC Adv. 2016. V. 6. P. 17 318–17 327. https://doi.org/10.1039/C5RA25788C
Mao F., Yan J., Li J., Jia X., Miao H., Sun Y., Huang L., Li X. // Org. Biomol. Chem. 2014. V. 12. P. 5936–5944. https://doi.org/10.1039/c4ob00998c
Meng X.-L., Yang J.-Y., Chen G.-L., Wang L.-H., Zhang L.-J., Wang S., Li J., Wu C.-F. // Chem.-Biol. Interact. 2008. V. 174. P. 51–59. https://doi.org/10.1016/j.cbi.2008.04.015
Meng X.L., Yang J.Y., Chen G.L., Zhang L.J., Wang L.H., Li J., Wang J.M., Wu C.F. // Int. Immunopharmacol. 2008. V. 8. P. 1074–1082. https://doi.org/10.1016/j.intimp.2008.03.011
Hou Y., Zhang Y., Mi Y., Wang J., Zhang H., Xu J., Yang Y., Liu J., Ding L., Yang J., Chen G., Wu C. // Mol. Nutr. Food Res. 2019. V. 63. P. 1 801 380. https://doi.org/10.1002/mnfr.201801380
Szewczuk L.M., Forti L., Stivala L.A., Penning T.M. // J. Biol. Chem. 2004. V. 279. P. 22727–22737. https://doi.org/10.1074/jbc.M314302200
Guha P., Dey A., Chatterjee A., Chattopadhyay S., Bandyopadhyay S.K. // Br. J. Pharmacol. 2010. V. 159. P. 726–734. https://doi.org/10.1111/j.1476-5381.2009.00572.x
Chen G., Shan W., Wu Y., Ren L., Dong J., Ji Z. // Chem. Pharm. Bull. 2005. V. 53. P. 1587–1590. https://doi.org/10.1248/cpb.53.1587
Kim M.H., Shin J.-S., Lee K.-T., Lee Y.S. // Bull. Korean Chem. Soc. 2011. V. 32. P. 299–302. https://doi.org/10.5012/bkcs.2011.32.1.299
Palamara A.T., Nencioni L., Aquilano K., De Chiara G., Hernandez L., Cozzolino F., Ciriolo M.R., Garaci E. // J. Infect. Dis. 2005. V. 191. P. 1719–1729. https://doi.org/10.1086/429694
Li C., Fang J.-S., Lian W.-W., Pang X.-C., Liu A.-L., Du G.-H. // Chem. Biol. Drug Des. 2015. V. 85. P. 427–438. https://doi.org/10.1111/cbdd.12425
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия