

Биоорганическая химия, 2021, T. 47, № 1, стр. 106-110
Селективное ацетилирование первичной гидроксильной группы в метил-D-гексопиранозидах смесью уксусного ангидрида и уксусной кислоты
Ю. Е. Цветков 1, Д. В. Яшунский 1, Н. Э. Нифантьев 1, *
1 Институт органической химии им. Н.Д. Зелинского РАН
119991 Москва, Ленинский просп., 47, Россия
* E-mail: nen@ioc.ac.ru
Поступила в редакцию 27.07.2020
После доработки 15.08.2020
Принята к публикации 16.08.2020
Аннотация
6-О-Ацетилированные производные D-гексопиранозидов являются ценными промежуточными продуктами в синтетической химии углеводов. Мы разработали новый простой метод избирательного ацетилирования первичной гидроксильной группы в метил-D-гексопиранозидах, заключающийся во взаимодействии со смесью уксусного ангидрида и уксусной кислоты при 50°С, в результате чего образуются соответствующие 6-ацетаты с выходами 40–50%.
ВВЕДЕНИЕ
Производные моно- и олигосахаридов, содержащие ацетильную группу при О6 и свободные гидроксильные группы в положениях 2, 3 и 4, широко используются в химии углеводов как исходные соединения для дальнейших манипуляций с защитными группами [1–6] или как гликозил-акцепторы в реакциях гликозилирования [7–10]. 6‑О-Ацетилированные производные моносахаридов, вторичные гидроксильные группы которых несут отличные от ацетатов защитные группы, находят применение в олигосахаридном синтезе, причем 6-О-ацетильные группы могут контролировать стереохимический результат реакций гликозилирования [11, 12].
Наиболее эффективным методом избирательного введения 6-О-ацетильной группы является энзиматическое ацетилирование в присутствии липаз из различных источников; в качестве донора ацетильной группы обычно используется винилацетат [7–10, 13, 14]. Выходы 6-О-ацетилированных производных в этих реакциях могут достигать 90%. Однако коммерческие ферментные препараты достаточно дороги, поэтому химические методы избирательного ацетилирования первичной гидроксильной группы в незащищенных гексопиранозидах также являлись предметом ряда исследований. Были предложены новые реагенты (например, N-ацетилимидазол в присутствии гидроксида тетраметиламмония [15]) и катализаторы селективного 6-О-ацетилирования (в частности, производные 4-диметиламинопиридина [16], соли лантанидов [17] и скандия [18]). В ряде случаев (см., например, работу Bianco et al. [17]), эффективность и селективность химических методов не уступала таковым для ферментативных реакций. Избирательное 6-О-ацетилирование с использованием ацетилхлорида в присутствии пространственно затрудненных аминов также продемонстрировало достаточно высокую эффективность [19].
Недавно мы показали, что уксусная кислота и уксусный ангидрид способны в отсутствие катализаторов избирательно ацетилировать первичную гидроксильную группу в метил-2,3-ди-О-бензоил-α-D-глюкопиранозиде, результатом чего является образование соответствующего 6-О-ацетильного производного с высоким выходом [20]. В настоящей статье мы описываем использование аналогичной реакции для моноацетилирования незащищенных метил-D-гексопиранозидов (I–V) с получением соответствующих 6-ацетатов (VI–X).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Свободные метил-D-гексопиранозиды практически нерастворимы в уксусном ангидриде, поэтому исходные соединения (I–V) сначала растворяли в уксусной кислоте, затем прибавляли уксусный ангидрид (объемное соотношение Ас2О–АсОН 1 : 2) и полученные растворы нагревали при 50 °С, контролируя протекание реакции методом ТСХ (схема 1 ). Реакцию останавливали, когда содержание исходного метилгликозида в реакционной смеси примерно равнялось содержанию продуктов ди- и триацетилирования.

Схема 1 . 6-О-Ацетилирование гексопиранозильных тетраолов (I–V).
Моноацетилированные производные выделяли колоночной хроматографией на силикагеле в системе хлороформ–метанол (4 : 1) с выходом 50–60% (табл. 1). По данным ЯМР, главными компонентами полученных хроматографически однородных продуктов являлись 6-ацетаты (VI–X), идентифицированные по характерному слабопольному положению сигналов протонов при С6 в спектрах 1Н-ЯМР. Минорными компонентами являлись моноацетилированные производные, несущие ацетильную группу на вторичных гидроксильных группах. Об этом свидетельствовало присутствие в спектрах 1Н-ЯМР минорных сигналов протонов Н1, а также сигналов в слабом поле (δ > 4.7 м.д.), отвечающих кольцевым протонам в положениях, несущих ацетильную группу. Количество вторичных ацетатов во фракции моноацетилированных производных варьировалось от 7 до 30% в зависимости от структуры исходного метилгликозида.
Таблица 1.
Ацетилирование метил-D-гексопиранозидов смесью Ас2О–АсОН
| Исходные метил-D-гексопиранозиды (I–V) |
Время реакции, ч | 6-Ацетаты (VI–X) | Суммарный выход фракции моноацетатов, % | Содержание моноацетатов по вторичным ОН-группам*, % | Выход кристаллического 6-ацетата, % |
|---|---|---|---|---|---|
 (I) |
10 | 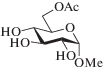 (VI) |
58 | 7 | –** |
 (II) |
10 | (VII) | 62 | 12 | 38 |
| (III) | 7 | 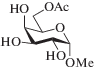 (VIII) |
51 | 30 | 28 |
 (IV) |
9 | (IX) | 57 | 14 | 38 |
 (V)
(V)
|
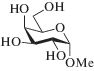
10 |  (X)
(X)
|
61 | 14 | –** |


Наиболее высокая региоселективность 6-О-ацетилирования наблюдалась в случае α-метилглюкозида (I), наименьшая – в случае α-метилгалактозида (III). β-Метилглюкозид (II), β-метилгалактозид (IV) и α-метилманнозид (V) занимали промежуточное положение (табл. 1). Поиск условий хроматографической очистки 6-О-ацетилированных производных (VI–X) от примесей вторичных ацетатов не привел к успеху. Соединения (VII–IX) были выделены в чистом виде кристаллизацией (табл. 1).
Таким образом, смесь уксусного ангидрида и уксусной кислоты избирательно ацетилирует первичную гидроксильную группу в незащищенных гексопиранозидах, в результате чего образуются соответствующие 6-О-ацетилированные производные с выходами 50–60%. Предлагаемый метод не уступает в ряде случаев известным методам избирательного ацетилирования первичных гидроксильных групп в производных углеводов [15, 16, 18]. Умеренные выходы целевых продуктов компенсируются простотой реакции и отсутствием необходимости использования малодоступных или дорогостоящих катализаторов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Тонкослойную хроматографию проводили на пластинках с силикагелем Kieselgel 60 F254 (Merck, Германия), вещества обнаруживали опрыскиванием раствором орцинола (180 мг орцинола в смеси 85 мл воды, 10 мл ортофосфорной кислоты и 5 мл этанола) с последующим нагреванием при ~150°С. Колоночную хроматографию проводили на силикагеле Silica gel 60 (40–63 мкм; Merck, Германия). Оптическое вращение измеряли на цифровом поляриметре P-2000 (JASСO, Япония) при комнатной температуре. Температуры плавления определяли на столике Кофлера. Спектры ЯМР регистрировали при 25°С на спектрометре AMX-400 (Bruker, Швейцария) в D2O. Отнесение сигналов проводили с использованием методик двумерной корреляционной спектроскопии COSY и HSQC. Обозначения 2-Ас, 3-Ас и 4-Ас в описании спектров 1Н-ЯМР соответствуют метил-D-гексопиранозидам, несущим ацетильную группу в положении 2, 3 или 4 соответственно.
Общая методика 6-О-ацетилирования. К раствору метил-D-гексопиранозида в АсОН (1–3 мл/100 мг) прибавили по каплям Ас2О (0.5 объема АсОН) при 50°С. Смесь перемешивали при этой температуре, контролируя протекание реакции ТСХ в системе CHCl3–MeOH (4 : 1). Когда визуально содержание исходного гексопиранозида (Rf 0.10–0.15) и суммы продуктов ди- (Rf 0.55–0.65) и триацетилирования (Rf 0.80–0.85) становилось равным (7–10 ч), смесь охлаждали, упаривали, затем остаточные АсОН и Ас2О удаляли соупариванием со смесью EtOH–толуол. Из остатка колоночной хроматографией (CHCl3–MeOH 95 : 5 → 9 : 1) выделяли хроматографически однородную фракцию продуктов моноацетилирования (Rf 0.40–0.45, CHCl3–MeOH (4 : 1)).
Метил-6-О-ацетил-α-D-глюкопиранозид (VI). После колоночной хроматографии получено соединение (VI) в виде бесцветного сиропа с общим выходом 58%, содержащее ~7% ацетатов по вторичным гидроксилам. 1Н-ЯМР (400 МГц, D2O, δ, м.д., J, Гц): 5.13 (т, 0.03Н, J 9.5, Н3 3-Ас), 4.97 (д, 0.025Н, J1,2 3.7, Н1 2-Ас), 4.86 (д, 0.05Н, J1,2 4.0, Н1 2-Ас и 3-Ас), 4.81 (д, 1Н, J1,2 3.8, Н1), 4.41 (дд, 1Н, J6a,5 2.4, J6a,6b 12.5, H6a), 4.30 (дд, 1Н, J6b,5 5.3, Н6b), 3.85 (ддд, 1Н, Н5), 3.68 (т, 1Н, J 9.4, Н3), 3.58 (дд, J2,3 9.8, H2), 3.46 (т, 1Н, J 9.5, Н4), 3.42 (с, 3Н, СН3О), 2.14 (с, 3Н, СН3СО). 13С-ЯМР (100 МГц, D2O, δ, м.д.): 175.0 (СО), 100.3 (С1), 73.9 (С3), 72.1 (С2), 70.5 (С4), 70.2 (С5), 64.3 (С6), 56.1 (СН3О), 21.1 (СН3СО). Данные спектров ЯМР соединения (VI) хорошо согласуются с данными, приведенными в работе Jansson et al. [21].
Метил-6-О-ацетил-β-D-глюкопиранозид (VII). После колоночной хроматографии получена фракция моноацетатов (выход 62%), содержащая 6‑ацетатат (VII) и ацетаты по вторичным гидроксилам (~12%). 1Н-ЯМР (400 МГц, D2O, δ, м.д., J, Гц): 4.98 (т, 0.06Н, J 9.1, Н3 3-Ас), 4.80 (т, 0.04Н, J 9.4, Н4 4-Ас), 4.69 (т, 0.04Н, J 9.2, Н2 2-Ас), 4.58 (д, 0.04Н, J1,2 8.1, Н1 2-Ас), 4.49 (д, 0.06Н, J1,2 8.1, Н1 3-Ас), 4.43 (дд, 1Н, J6a,5 2.2, J6a,6b 12.2, Н6a), 4.40 (д, 1Н, J1,2 7.9, Н1), 4.30 (дд, 1Н, J6b,5 5.1, H6b), 3.69–3.64 (м, 1Н, Н5), 3.56 (с, 3Н, СН3О), 3.53–3.44 (м, 2Н, Н3, Н4), 3.27 (т, 1 Н, J 8.8, Н2), 2.14 (с, 3Н, СН3СО). 13С-ЯМР (100 МГц, D2O, δ, м.д.): 175.0 (СО), 104.3 (С1), 76.5 (С3), 74.2 (С5), 74.0 (С2), 70.4 (С4), 64.1 (С6), 58.2 (СН3О), 21.1 (СН3СО). Данные спектров ЯМР соединения (VII) хорошо согласуются с данными, приведенными в работе Jansson et al. [21]. Кристаллизацией из смеси EtOH–Et2O выделено соединение (VII) (38%) в виде бесцветных кристаллов, т. пл. 129–131°С, [α]D –26° (c 1, EtOH). Литературные данные [22]: т. пл. 128–129°С, [α]D –34° (вода).
Метил-6-О-ацетил-α-D-галактопиранозид (VIII). После колоночной хроматографии получена фракция моноацетатов (выход 51%), содержащая 6-ацетат (VIII) и ацетаты по вторичным гидроксилам (~30%). 1Н-ЯМР (400 МГц, D2O, δ, м.д., J, Гц): 5.34 (д, 0.14Н, J3,4 3.3, Н4 4-Ас), 5.04–5.00 (м, 0.60Н, Н1 2-Ас, Н2 2-Ас, Н3 3-Ас), 4.91 (д, 0.34Н, J1,2 4.1, Н1 3-Ас, Н1 4-Ас), 4.85 (д, 1Н, J1,2 2.9, Н1), 4.32 (дд, 1Н, J6a,5 4.1, J6a,6b 11.6, H6a), 4.25 (дд, 1Н, J6b,5 8.1, Н6b), 4.13 (м, 1Н, Н5), 4.01 (уш. с, 1Н, Н4), 3.83 (м, 2Н, Н2, Н3), 3.41 (с, 3Н, СН3О), 2.13 (с, 3Н, СН3СО). 13С-ЯМР (100 МГц, D2O, δ, м.д.): 174.9 (СО), 100.3 (С1), 70.1 (С3), 70.0 (С4), 69.1 (С5), 68.9 (С2), 65.0 (С6), 55.9 (СН3О), 21.1 (СН3СО). Данные спектров ЯМР соединения (VIII) хорошо согласуются с данными, приведенными в работе Jansson et al. [21]. Кристаллизацией из смеси EtOH–Et2O получено соединение (VIII) (выход 28%) в виде бесцветных кристаллов, т. пл. 157–159°С, [α]D +147° (c 1, EtOH). Литературные данные [23]: т. пл. 154–156°С, [α]D +160° (EtOH).
Метил-6-О-ацетил-β-D-галактопиранозид (IX). После колоночной хроматографии получена фракция моноацетатов (выход 57%), содержащая 6-ацетат (IX) и ацетаты по вторичным гидроксилам (~14%). 1Н-ЯМР (400 МГц, D2O, δ, м.д., J, Гц): 5.30 (д, 0.05Н, J3,4 3.5, Н4 4-Ас), 4.92 (дд, 0.04Н, J1,2 8.1, J2,3 10.1, Н2 2-Ас), 4.87 (дд, 0.07Н, J2,3 10.1, J3,4 3.4, Н3 3-Ас), 4.52 (д, 0.04Н, J1,2 8.1, Н1 2-Ас), 4.44 (д, 0.07Н, J1,2 8.1, Н1 3-Ас), 4.41 (д, 0.05Н, J1,2 7.9, Н1 4-Ас), 4.33 (д, 1Н, J1,2 7.9, Н1), 4.29 (д, 2Н, J6,5 6.2, 2 Н6), 3.96 (д, 1Н, J3,4 3.5, Н4), 3.92 (т, 1Н, J5,6 6.2, Н5), 3.66 (дд, 1Н, J3,4 3.5, J2,3 10.0, Н3), 3.57 (с, 3Н, СН3О), 3.51 (дд, 1Н, J2,3 10.0, J1,2 7.9, Н2), 2.13 (с, 3Н, СН3СО). 13С-ЯМР (100 МГц, D2O, δ, м.д.): 174.9 (СО), 104.7 (С1), 73.5 (С3), 73.5 (С5), 71.4 (С2), 69.5 (С4), 64.7 (С6), 58.1 (СН3О), 21.1 (СН3СО). Данные спектров ЯМР соединения (IX) хорошо согласуются с данными, приведенными в работе Jansson et al. [21]. Кристаллизацией из смеси EtOH–Et2O получено соединение (IX) (выход 38%) в виде бесцветных кристаллов, т. пл. 146–148°С, [α]D –11° (c 1, EtOH). Литературные данные [24]: т. пл. 142–145°С, [α]D –14° (EtOH).
Метил-6-О-ацетил-β-D-маннопиранозид (X). После колоночной хроматографии получено соединение (X) в виде бесцветного сиропа с общим выходом 61%, содержащее ~14% ацетатов по вторичным гидроксилам. 1Н-ЯМР (400 МГц, D2O, δ, м.д., J, Гц): 5.03 (дд, 0.05Н, J1,2 1.6, J2,3 3.5, Н2 2-Ас), 4.96 (т, 0.04Н, J 9.5, Н4 4-Ас), 4.93 (дд, 0.07Н, J2,3 3.4, J3,4 9.8, Н3 3-Ас), 4.69 (уш. с, 1Н, Н1), 4.37 (дд, 1Н, J6а,5 2.2, J6a,6b 12.2, Н6а), 4.22 (дд, 1Н, J6b,5 5.6, Н6b), 3.88 (дд, 1Н, J1,2 1.8, J2,3 3.1, Н2), 3.70 (м, 1Н, Н5), 3.67 (дд, 1Н, J3,4 9.4, Н3), 3.64 (т, 1Н, J 9.7, Н4), 3.34 (с, 3Н, СН3О), 2.08 (с, 3Н, СН3СО). Данные спектра 1Н-ЯМР соединения (X) хорошо согласуются с данными, приведенными в работе Lassfolk et al. [25]. 13С-ЯМР (100 МГц, D2O, δ, м.д.): 175.2 (СО), 102.0 (С1), 71.4 (С3), 71.1 (С5), 70.8 (С2), 67.6 (С4), 64.6 (С6), 55.8 (СН3О), 21.2 (СН3СО).
Список литературы
Rana S.S., Matta K.L. // Carbohydr. Res. 1983. V. 113. P. C18–C21. https://doi.org/10.1016/0008-6215(83)88254-8
La Ferla B., Prosperi D., Lay L., Russo G., Panza L. // Carbohydr. Res. 2002. V. 337. P. 1333–1342. https://doi.org/10.1016/s0008-6215(02)00164-7
Kaji E., Komori T., Yokoyama M., Kato T., Nishino T., Shirahata T. // Tetrahedron. 2010. V. 66. P. 4089–4100. https://doi.org/10.1016/j.tet.2010.04.007
Muramatsu W., Kawabata T. // Tetrahedron Lett. 2007. V. 48. P. 5031–5033. https://doi.org/10.1016/j.tetlet.2007.05.098
Gening M.L., Titov D.V., Grachev A.A., Gerbst A.G., Y-udina O.N., Chizhov A.O., Tsvetkov Y.E., Nifantiev N.E. // Eur. J. Org. Chem. 2010. V. 13. P. 2465–2475. https://doi.org/10.1002/ejoc.200901275
Kochetkov N.K., Nifant’ev N.E., Backinowsky L.V. // Tetrahedron. 1987. V. 43. P. 3109–3121. https://doi.org/10.1016/S0040-4020(01)86852-6
Lawandi J., Rocheleau S., Moitessier N. // Tetrahedron. 2011. V. 67. P. 8411–8420. https://doi.org/10.1016/j.tet.2011.07.026
Legentil L., Cabezas Y., Tasseau O., Tellier C., Daligault F., Ferrières V. // J. Org. Chem. 2017. V. 82. P. 7114–7122. https://doi.org/10.1021/acs.joc.7b00565
Rencurosi A., Poletti L., Russo G., Lay L. // Eur. J. Org. Chem. 2003. P. 1672–1680. https://doi.org/10.1002/ejoc.200200425
Zhang X., Kamiya T., Otsubo N., Ishida H., Kiso M. // J. Carbohydr. Chem. 1999. V. 18. P. 225–239. https://doi.org/10.1080/07328309908543992
Komarova B.S., Orekhova M.V., Tsvetkov Y.E., Nifantiev N.E. // Carbohydr. Res. 2014. V. 384. P. 70–86. https://doi.org/10.1016/j.carres.2013.11.016
Komarova B.S., Tsvetkov Y.E., Nifantiev N.E. // Chem. Rec. 2016. V. 16. P. 488–506. https://doi.org/10.1002/tcr.201500245
Yasukochi T., Fukase K., Kusumoto S. // Tetrahedron Lett. 1999, V. 40. P. 6591–6593. https://doi.org/10.1016/S0040-4039(99)01280-0
La Ferla B., Lay L., Russo G., Panza L. // Tetrahedron Asymmetry. 2000. V. 11. P. 3647–3651. https://doi.org/10.1016/S0957-4166(00)00342-6
Lu Y., Wei P., Pei Y., Xu H., Xin X., Pei Z. // Green Chem. 2014. V. 16. P. 4510–4514. https://doi.org/10.1039/c4gc00770k
Kurahashi T., Mizutani T., Yoshida J.-i. // Tetrahedron. 2002. V. 58. P. 8669–8677. https://doi.org/10.1016/S0040-4020(02)01098-0
Bianco A., Brufani M., Melchioni C., Romagnoli P. // Tetrahedron Lett. 1997. V. 38. P. 651–652.
McClure M.S., Berry M.B., Caine D., Crawford C., Crump B.C., Glover B.N., Kedia S.B., Millar A., Mitchell M.B., Nichols C.J., Patterson D.E., Powers J. // Eur. J. Org. Chem. 2012. P. 3561–3565. https://doi.org/10.1002/ejoc.201200261
Ishihara K., Kurihara H., Yamamoto H. // J. Org. Chem. 1993. V. 58. P. 3791–3793. https://doi.org/10.1021/jo00067a005
Цветков Ю.Е., Генинг М.Л., Нифантьев Н.Э. // Изв. АН. Сер. хим. 2020. С. 2228–2230. [Tsvetkov Y.E., Gening M.L., Nifantiev N.E. // Russ. Chem. Bull. 2020. V. 69. P. 2228–2230.]
Jansson P.-E., Kenne L., Schweda E. // J. Chem. Soc. Perkin Trans. I. 1987. P. 377–383. https://doi.org/10.1039/P19870000377
Bouveng H.O. // Acta Chem. Scand. 1961. V. 15. P. 87–95. https://doi.org/10.3891/acta.chem.scand.15-0087
Borén H.B., Garegg P.G., Kenne L., Pilotti Å., Svensson S., Swahn C.-G. // Acta Chem. Scand. 1973. V. 27. P. 2740–2748.
Garegg P.G., Swahn C.-G. // Acta Chem. Scand. 1972. V. 26. P. 3895–3901. https://doi.org/10.3891/acta.chem.scand.26-3895
Lassfolk R., Rahkila J., Johansson M.P., Ekholm F.S., Wärnå J., Leino R. // J. Am. Chem. Soc. 2019. V. 141. P. 1646–1654. https://doi.org/10.1021/jacs.8b11563
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия