

Биоорганическая химия, 2021, T. 47, № 1, стр. 57-75
Защитные группы как стереоконтролирующий фактор в реакциях гликозилирования
А. И. Токатлы 1, Д. З. Винницкий 1, Н. Е. Устюжанина 1, Н. Э. Нифантьев 1, *
1 Институт органической химии им. Н.Д. Зелинского РАН
119991 Москва, Ленинский просп., 47, Россия
* E-mail: nen@ioc.ac.ru
Поступила в редакцию 27.07.2020
После доработки 11.08.2020
Принята к публикации 12.08.2020
Аннотация
Синтетические олигосахариды представляют интерес как модельные соединения для исследования биологической активности природных структур, а также как компоненты новых лекарственных препаратов, гликоконъюгатных вакцин, углеводных диагностикумов и других продуктов. Ключевым этапом синтеза олигосахаридов является реакция гликозилирования, приводящая к построению связи между углеводными остатками. Настоящий обзор посвящен актуальной проблеме современной гликохимии – методам стереоконтроля в реакции гликозилирования. В качестве стереоконтролирующих факторов рассмотрены защитные группы в структуре гликозил-доноров, способные влиять на механизм превращения тремя основными путями: 1) за счет соучастия или анхимерного содействия, 2) дезактивации аномерного центра, 3) предварительной пространственной координации гликозил-донора и гликозил-акцептора. Показан классический механизм соучастия защитной группы при О2 в синтезе 1,2-транс-гликозидов и его современные модификации, включающие активирующие простые эфирные и хиральные защитные группы, а также ахиральные бициклические гликозил-доноры. Подробно рассмотрены механизмы соучастия удаленных заместителей, расположенных при О3, О4 и О6 гликозил-донора. Помимо этого, представлены подходы к стереоконтролю с использованием дезактивирующей защиты: конформационно ограничивающих и электроноакцепторных групп. Наконец, рассмотрены синтетические подходы, основанные на предварительной координации гликозил-донора и гликозил-акцептора, включающие методы внутримолекулярного гликозилирования. Представлены как классические, так и новейшие защитные группы, применяемые для контроля стерического исхода гликозилирования, подробно рассмотрены механизмы, лежащие в основе представленных подходов, а также приведены примеры применения описанных стратегий в синтезе сложных олигосахаридных производных.
ВВЕДЕНИЕ
Производные углеводов, которые из-за наличия в них гликозидной связи часто называют гликосоединениями, образуют один из четырех важнейших классов природных веществ (наряду с нуклеиновыми кислотами, белками и липидами), критически важных для протекания процессов жизнедеятельности практически у всех живых организмов [1]. Все известные клеточные формы жизни содержат в своих структурах олиго- и полисахариды, а также их конъюгаты с биополимерами и различными неуглеводными органическими соединениями [2]. Углеводные цепи входят в состав секретируемых и мембранных гликопротеинов и протеогликанов, а также таких важных компонентов клеточных мембран и межклеточного матрикса, как гликолипиды [3]. Хотя взаимосвязь структуры и функций углеводов изучается достаточно интенсивно и успешно на протяжении последних десятилетий, высокая разнородность и сложность строения в сочетании с минимальной эволюционной и межвидовой консервативностью значительно усложняют исследования в этой области [4]. Из-за сложности выделения природных гликосоединений в значимых количествах, необходимых для проведения испытаний, химический синтез их аналогов является перспективной альтернативой для получения молекулярных проб, применимых к изучению строения и биологической активности природных гликосоединений.
Центральной проблемой углеводного синтеза является стереонаправленное построение гликозидной связи между аномерным центром гликозил-донора и гликозил-акцептором, причем последний может быть представлен как углеводным соединением, так и неуглеводным, например, аминокислотой, полифенолом или другим функциональным производным. Продуктом данной реакции часто может быть не одно вещество, а несколько изомеров, отличающихся как местом присоединения гликозильного остатка к акцептору, так и конфигурацией новообразованной гликозидной связи [5].
При биосинтезе эта проблема решается с помощью специальных ферментов – гликозилтрансфераз, однозначно определяющих место присоединения и конфигурацию образующейся гликозидной связи. Таким образом, в природе данная реакция обычно проходит регио- и стереоспецифично. В случае же синтетических соединений решение проблем селективности является одной из базовых задач. И если региоспецифичность достигается оптимальной расстановкой защитных групп на нежелательных в реакции гидроксильных, амино- или иных реакционных фрагментах, то решение проблемы стереоспецифичности, или хотя бы стереоселективности, является гораздо более нетривиальной задачей. Для понимания возможных путей решения проблемы стереоконтроля рассмотрим обобщенный механизм реакции гликозилирования.
Ее начальным этапом является активация гликозил-донора (I) за счет взаимодействия с кислотой Льюиса (A) или иными агентами (схема 1 ). В результате после отщепления легко уходящей группы LG образуется карбокатион (II), который затем стабилизируется за счет формирования дополнительной связи между аномерным углеродом и эндоциклическим кислородом, что приводит к образованию более устойчивого катиона оксокарбения (III). Обычно в реакциях гликозилирования применяются активаторы с ненуклеофильным противоионом (трифлат, перхлорат и др.), который не конкурирует с относительно слабонуклеофильным гидроксилом гликозил-акцептора. Образующийся в результате активации гликозил-донора катион оксокарбения (III) доступен для нуклеофильной атаки по механизму SN1 с двух сторон относительно плоскости пиранозного цикла с образованием двух возможных продуктов: α- и β-гликозидов [6].

Схема 1 . Общая схема протекания реакции гликозилирования. LG – уходящая группа, А — активатор, (I) – гликозил-донор, (II) – гликозил-катион, (III) – оксокарбениевый катион.
Реакция гликозилирования является сложным процессом, на протекание которого влияет множество факторов. Известно, что на стерический исход способны оказывать влияние растворитель, температура [7], концентрация реагентов [8], метод активации [9], а также химические, стерические и электронные свойства донора и акцептора. В настоящее время наиболее широко используются методы стереоконтроля, основанные на модификации гликозил-донора, в первую очередь за счет стратегического выбора защитных групп [10].
В рамках данного обзора мы ограничимся рассмотрением трех возможных путей, к которым можно отнести большинство методов стереоконтроля реакций гликозилирования посредством защитных групп: 1) соучастие защитных групп гликозил-донора (сопровождается блокированием нуклеофильной атаки с одной из сторон); 2) дезактивация аномерного центра (смещение механизма протекания реакции в сторону SN2); 3) координация акцептора (пространственная предорганизация гликозил-акцептора с определенной стороны).
СОУЧАСТИЕ ЗАЩИТНЫХ ГРУПП ГЛИКОЗИЛ-ДОНОРА
Под соучастием защитных групп в реакции гликозилирования понимается внутримолекулярная стабилизация оксокарбениевого катиона (III) нуклеофильным фрагментом защитной группы. В результате указанного анхимерного содействия образуется бициклическая система, в которой одна из сторон пиранозного цикла становится недоступной для атаки гликозил-акцептора.
В зависимости от расположения соучаствующие группы подразделяют на “соседние” (neighboring participating groups – NPG), когда заместитель находится при О2, и “удаленные” (remote participating groups – RPG), когда заместитель находится при О3, O4 или О6. Кроме того, выделяют контроль реакции гликозилирования с помощью соучаствующих хиральных защитных групп. Далее каждый из видов соучастия будет рассмотрен более подробно.
Построение 1,2-транс-гликозидов с использованием соучаствующей защиты при О2. Исторически первым и наиболее подробно изученным механизмом стереоконтроля реакции гликозилирования является соучастие ацильной защиты при О2. Так, еще в 1939 г. было отмечено, что взаимодействие полностью ацетилированного гликозилгалогенида с ацетатом серебра приводит к образованию исключительно 1,2-транс-гликозилацетата [11]. В качестве объяснения этого феномена в 1940 г. H.S. Isbell предложил механизм соучастия [12].
Отличительной особенностью данного механизма гликозилирования является нуклеофильная атака оксокарбениевого катиона (IV) карбонильным атомом кислорода ацильной защиты при О2 с образованием диоксолениевого (ацилоксониевого) иона (V) (схема 2 ). Последний намного более устойчив за счет наличия стабильного пятичленного цикла, и, в результате, равновесие практически полностью смещается в его сторону. Таким образом, для атаки нуклеофила более доступной становится транс-сторона, и, соответственно, предпочтительно образуется 1,2-транс-гликозид [6].
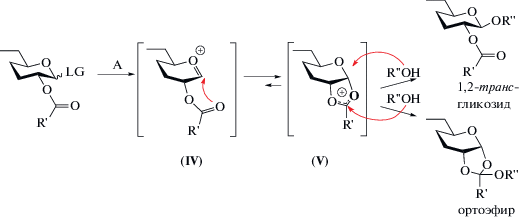
Схема 2 . Образование 1,2-транс-гликозида и ортоэфира при соучастии 2-О-ацильной группы. LG – уходящая группа, А — активатор, (IV) – оксокарбениевый катион, (V) – диоксолениевый (ацилоксониевый) ион.
Благодаря относительной стабильности, диоксолениевый катион был зарегистрирован различными методами, такими как РСА [13], ЯМР [14], низкотемпературная ИК-спектроскопия [15]. Кроме того, стабилизация гликозил-катиона заместителем при О2 была подтверждена результатами теоретических расчетов [6].
В связи с относительной простотой и однозначностью, данный метод стереоконтроля на сегодняшний день является доминирующим подходом к получению 1,2-транс-гликозидов. Однако, несмотря на широкое применение, использование ацильных защит при О2 имеет ряд известных недостатков [16]. Во-первых, нуклеофильная атака ацилоксониевого катиона (V) акцептором может привести к образованию ортоэфира (схема 2 ). Ортоэфиры неустойчивы в кислой среде и, как правило, могут быть перегруппированы в целевой гликозид. Однако перегруппировка требует дополнительных синтетических действий, а иногда оказывается и вовсе невозможной [17]. Во-вторых, ацильные группы обладают электронно-акцепторными свойствами и, соответственно, снижают реакционную способность аномерного центра в гликозил-доноре (эффект дезактивации подробнее рассмотрен ниже в разделе “Регулирование реакционной способности гликозил-доноров”), что, особенно в сочетании с малоактивным акцептором, может привести к ингибированию гликозилирования, вплоть до невозможности осуществления реакции между данными субстратами. Наконец, большинство ацильных защит удаляется в относительно жестких основных условиях, что в ряде случаев препятствует реализации стратегии ортогональных манипуляций с защитными группами [16].
Для предотвращения образования ортоэфиров был разработан ряд ацильных защитных групп более сложного строения. Так, в 2005 г. H.E. Ensley et al. была предложена 4-ацетокси-2,2-диметилбутаноильная (ADMB) защита. Считается, что данная группа препятствует образованию ортоэфира за счет стерических эффектов, возникающих вследствие дополнительной циклизации [17]. Аналогичным эффектом обладает и предложенная в 2019 г. 2,2-диметил-2-(о-нитрофенил)-ацетильная группа (DMNPA) [18].
Полностью избежать нежелательных эффектов ацильной защиты при О2 позволяют разработанные в последнее время соучаствующие простые эфирные группы. Впервые такой подход был изучен группой А.В. Демченко в 2005 г. с использованием пиколильной (Pic) защиты. В данном случае внутримолекулярная нуклеофильная атака оксокарбениевого катиона (VI) осуществляется атомом азота пиколильной группы с образованием стабилизированного шестичленного циклического интермедиата (VII) (схема 3 ) [19].
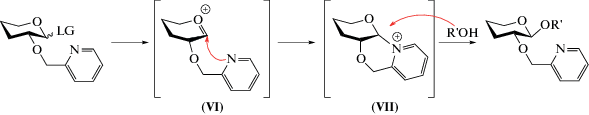
Схема 3 . Образование 1,2-транс-гликозида при соучастии пиколильной группы при О2. LG – уходящая группа, (VI) – оксокарбениевый катион, (VII) – стабилизированный циклический интермедиат.
Впоследствии был разработан ряд альтернативных простых эфирных защитных групп, способных к соучастию с образованием 1,2-транс-гликозидов: о-нитробензильная (NBn) [20] и о-цианобензильная (CBn) [21], а также алкоксиметильные производные – метоксиметил (МОМ), бензилоксиметил (BOM) и 2-нафтилметоксиметил (N-APOM) [22]. Все они не образуют ортоэфиров в качестве продуктов реакции гликозилирования и могут быть селективно удалены, когда это необходимо. Следует отметить, что рассмотренные в этом разделе защиты, альтернативные ацильным, не нашли заметного практического применения, и на сегодняшний день для построения 1,2-транс-гликозидов продолжают преимущественно использовать 2-О-ацилированные гликозил-доноры.
Стереоконтроль реакций гликозилирования с использованием хиральных защитных групп при О2 в гликозил-донорах. В то время как классический эффект соучастия 2-О-ацильных групп в гликозил-доноре в большинстве случаев решает проблему синтеза 1,2-транс-гликозидов, построение 1,2-цис-гликозидной связи является более сложной задачей. Исторически для этих целей использовались несоучаствующие группы при О2. Однако это приводило лишь к потере 1,2-транс-селективности, а не к 1,2-цис-стереонаправляющему эффекту. Достижение последнего осуществлялось посредством манипуляций с условиями реакции и попытками изменить электронные и стерические свойства гликозил-донора и гликозил-акцептора (см. следующий раздел), что, во-первых, являлось достаточно трудоемким процессом, зачастую значительно удлиняющим синтетическую схему, а во-вторых, требовало практически индивидуальной оптимизации условий в каждом отдельном случае.
Подход к использованию соучаствующей защиты при О2 для селективного получения 1,2-цис-гликозидов был впервые предложен в 2005 г. группой G.-J. Boons [23]. Предполагалось, что хиральная группа, несущая нуклеофильный фрагмент и объемный фенильный заместитель, в зависимости от конфигурации хирального центра, способна стабилизировать катион оксокарбения с селективным образованием транс- или цис-декалиновой системы (схема 4 ).

Схема 4 . Стереоконтроль реакции гликозилирования с использованием хиральных защитных групп.
Первой разработкой в этой области стала защитная группа, полученная из этилового эфира миндальной кислоты, в которой в качестве нуклеофильного фрагмента выступал карбонильный атом кислорода. S-Изомер глюкозилтрихлорацетимидатного донора (1) проявлял высокую активность и 1,2-цис-селективность в реакциях гликозилирования с серией акцепторов (выход до 95%, соотношение цис : транс до 20 : 1) (рис. 1). В то же время R-изомер донора (1) оказался намного менее эффективным: максимально достигнутое соотношение цис- и транс-продуктов составило только 1 : 8 [23]. Такая разница в стереонаправляющем эффекте была объяснена меньшей устойчивостью цис-декалиновой системы в силу дестабилизирующих стерических эффектов [8].

Результаты вышеописанной работы послужили основой для разработки хиральных защитных групп второго поколения. При этом наиболее эффективной оказалась 1-S-фенил-2-фенилтиоэтильная группа, в которой в качестве нуклеофила выступает более активный атом серы (см. пример гликозил-донора (2) на рис. 1). Как и предполагалось авторами, усиление нуклеофильных свойств заместителя привело к достижению стереоспецифичности 1,2-цис-гликозилирования глюкозильными и галактозильными донорами. Кроме того, образование транс-декалинового сульфониевого интермедиата было подтверждено низкотемпературным экспериментом ЯМР in situ [24].
Основываясь на методах стереоконтроля при помощи хиральных групп, были предприняты попытки создания ахиральной защиты, также способной образовывать транс-декалиновый интермедиат. Таким образом предполагалось упростить синтез, т.к. не было бы необходимости получать оптически чистые хиральные доноры. Однако предложенная (2-тиофенил)-метильная группа проявила относительно низкую селективность: наилучшее соотношение 1,2-цис- и транс-гликозидов составило только 9 : 1 [25].
Более эффективным подходом является использование гликозил-доноров, в которых часть уходящей группы в аномерном положении одновременно является частью защиты при О2. Благодаря этому при активации донора сразу же образуется декалиновая стабилизированная система, и таким образом нет необходимости ее дополнительно генерировать in situ (схема 5 ). В 2010 г. был впервые разработан синтез оксотианового донора, при помощи которого осуществили стереоспецифичное глюкозилирование ряда гликозил-акцепторов с выходами до 62% [26].

Схема 5 . Гликозилирование оксотиановым донором.
Опираясь на вышеописанную работу, в 2012 г. были предложены бициклические доноры, содержащие сульфоксидный фрагмент при аномерном центре [27]. В реакциях гликозилирования в присутствии триметоксибензола подобного рода доноры обладают высокой 1,2-цис-селективностью при условии наличия дезактивирующих заместителей. При этом в процессе гликозилирования образуется арилированная триметоксифенилом хиральная группа при О2 донора, которая селективно удаляется воздействием трифторуксусной кислоты. Эффективность гликозил-доноров такого типа была продемонстрирована при селективном построении α-глюкозидных связей в процессе сборки тетрасахарида (7) – фрагмента полисахарида, выделенного из грибов вида Pseudallescheria boydii (схема 6 ) [27]. Так, с использованием тиогликозида (3) сначала был получен бициклический продукт (4), который переводили в сульфоксид (5), после чего последний вводили в реакцию гликозилирования с акцептором (6). В результате был получен α-связанный тетрасахарид (7) с выходом 70%. Схожую стратегию использовали в синтезе гексасахаридного фрагмента гликозилфосфатидилинозитного якоря Trypanosoma brucei [28].

Схема 6 . Олигосахаридный синтез с применением декалиновых доноров для построения 1,2-цис-гликозидной связи. (i) – 1) TMS2O, TMSOTf, 0°C, 30 мин; 2) Et3SiH, 3 ч; 3) PhCH(OMe)2, CSA, пониж. давл., DMF, 50°C, 18 ч; 4) Ac2O, Py; 5) Et3SiH, PhBCl2, –78°С; 6) Ac2O, Py; (ii) метахлорнадбензойная кислота, CH2Cl2, –78°C; (iii) – 1) триметоксибензол, Tf2O, 2,6-дитретбутил-4-метилпиридин, –10°C; 2) (6). Здесь и далее в процентах указан выход реакций.
Стереоконтролирующее соучастие удаленных защитных групп. Исходя из классического механизма соучастия из соседнего положения, можно предположить, что удаленные нуклеофильные группы также способны взаимодействовать с оксокарбениевым катионом. При реализации такого механизма, как и в классическом случае соучастия, вероятно, образуется бициклический интермедиат, атака которого нуклеофилом предпочтительна лишь с одной стороны. Таким образом, образующаяся гликозидная связь находится в анти-положении относительно соучаствующей группы. Исходя из этого можно ожидать, что при отсутствии соучаствующей группы при О2 ацильные защиты при О3 и О6 глюкозил-доноров, а также при О3, О4 и О6 галактозил-доноров будут обладать α-направляющим эффектом (схема 7 ). Возможность осуществления такого процесса позволяет использовать соучастие удаленных ацильных групп при планировании стереонаправленного построения 1,2-цис-гликозидных связей [29].

Схема 7 . Стабилизация оксокарбениевого гликозил-катиона удаленными ацильными защитными группами. LG – уходящая группа, А – активатор.
Действительно, указанный подход показал отличные результаты при синтезе уже достаточно большого числа ранее труднодоступных олигосахаридов с 1,2-цис-гликозидными связями. В частности, 3,4-ди-О-ацилированные фукозил(6-дезоксигалактозил)-доноры были успешно использованы для стереоспецифичного построения α-связей в синтезе линейных [30–34] и разветвленных [35, 36] избирательно сульфатированных олигофукозидов, структурно родственных фукоиданам из бурых водорослей. Так, например, гликозилирование трихлорацетимидатом (8) гликозил-акцептора (9) приводило к образованию исключительно α-связанного тетрасахарида (10) (схема 8a ).

Схема 8 . Примеры использования соучастия удаленных О-защитных ацильных групп в олигосахаридном синтезе. (i) – 1) Me2S2, MeOTf, MS 4 Å, CH2Cl2; 2) 40% aq. HF, Py; 3) BzCl, Py, 0°C; (ii) – 1) NIS, TfOH, CH2Cl2; 2) UV.
Глюкозил-доноры, содержащие ацильные заместители при О3 и О6, показали эффективность для селективного построения α-гликозидной связи в синтезе трисахаридного [37] и двух пентасахаридных фрагментов внешней области кора липополисахарида бактерии Pseudomonas aeruginosa [38, 39], а также для получения пентасахаридного фрагмента клеточной стенки Aspergillus fumigatus [40, 41] и (1→6)-α-связанных глюканов, характерных для ряда патогенных микроорганизмов [42]. Пример получения трисахаридного блока (13) из дисахарида (12) и 3,6-ди-О-ацетилированного N-фенил-трифторацетимидата (11) показан на схеме 8б .
Соучастие бензоильной группы при О3 в сочетании со стерически ограничивающей 4,6-О-ди-(трет-бутилсилиленовой) защитой 2-дезокси-2-азидогалактозного донора (14) привело к успешному получению олиго-α-галактозаминов (16–20) [43, 44] (схема 8в), в то время как соучаствующая защита при О4 галактозил-донора (21) способствовала эффективному образованию α‑связанного олигосахарида (23) [45] (схема 8г ). Соучастие 3-О-ацетильной группы в ксилозил-доноре было успешно использовано в синтезе ксилоолигосахаридов [46], структурно родственных олигосахаридным цепям рецептора Notch. Полученные олигосахариды были использованы при изучении двух ксилозилтрансфераз [47, 48], участвующих в биосинтезе данного гликопротеина.
Однако, несмотря на отличные результаты, полученные в вышеупомянутых работах, удаленное соучастие не является столь же универсальным подходом, как соучастие соседней группы при О2 [29]. Наряду с удачным применением удаленного соучастия, существует значительное количество примеров, когда ожидаемый стереонаправляющий эффект удаленных защитных групп на практике оказывался не столь велик, сильно зависел от конкретного донора и положения соучаствующей группы, а иногда и вовсе отсутствовал. Так, при использовании соучаствующей диэтилкарбомоильной группы при О6 глюкозного донора возможно достижение α-селективности [49], однако стереонаправляющий эффект данной защиты не сохраняется в случае галактозы. Оказывается, что соучаствующая группа при О6 галактозы имеет минимальный α-направляющий эффект [50], а в некоторых случаях даже повышает β-селективность по сравнению с несоучаствующей защитой [45]. Это может быть связано с конкурирующими типами внутримолекулярных взаимодействий между защитными группами, включая их пространственное расталкивание, влияющее на предпочтительную конформацию пиранозного кольца в гликозил-доноре [51].
Одним из методов решения проблемы недостаточного стереоконтролирующего влияния удаленной соучаствующей защиты может быть увеличение числа соучаствующих защитных групп в одном моносахаридном остатке. В частности, было показано, что в случае гликозилирования N-фенил-трифторацетимидатным глюкозным донором сочетание ацетильной защиты при О3 и О6 приводит к наилучшему выходу и селективности реакции по сравнению с моноацетилированными донорами [37]. В то же время увеличение количества О-ацильных групп в соответствующих положениях далеко не всегда способствует повышению селективности. Авторы вышеупомянутой работы [37] провели систематическое исследование моно- и диацетилированных доноров по О3 и О6 с серией акцепторов и установили, что в то время как диацетилированный донор приводит к максимальной α-селективности в реакции с первичным гидроксилом акцептора, в случае гликозилирования вторичного гидроксила оптимальный результат достигается при использовании соучаствующей защиты только при О6 [52].
Как видно из приведенных примеров, эффект соучастия удаленных групп иногда может оказаться нереализуемым. Для успешного применения синтетической стратегии, основанной на удаленном соучастии, зачастую необходима комбинация стереоконтролирующих удаленных защитных групп с другими инструментами, способными оказывать влияние на стереохимический исход реакции, такими как растворитель или температура. Именно их грамотное сочетание способно дать необходимый результат.
Ввиду столь нерегулярных, а иногда и противоречивых свидетельств стереоконтроля с помощью удаленных соучаствующих групп, сама его возможность долгое время оставалась дискуссионным вопросом, а отсутствие надежно зафиксированного интермедиата – ацилоксониевого иона, а также конкурирующего продукта реакции – ортоэфира, ставших надежными доказательствами соучастия из соседнего положения, вызывало лишь дополнительные сомнения. Однако в последние десятилетия был осуществлен ряд серьезных исследований, направленных на подробное изучение и доказательство существования механизма стереоконтролирующего соучастия удаленных О-ацильных групп в реакциях гликозилирования.
Систематическое исследование механизма соучастия удаленных О-ацильных заместителей было выполнено D. Crich et al. в 2008 г. В рамках данной работы был осуществлен эксперимент, направленный на получение стабильных бициклических продуктов взаимодействия удаленной трет-бутоксикарбонильной (Boc) защиты с аномерным центром (схема 9 a). Таким образом предполагалось доказать способность удаленных карбонильных групп к соучастию. Как и ожидалось, бициклические продукты были получены для 2-О-Вос-защищенных доноров. Из удаленных положений были исследованы гликозил-доноры, содержащие ацильные заместители при О3 аллозы, О3 и О4 маннозы, О6 глюкозы и О4 галактозы, однако бициклический продукт (25) был получен лишь в первом случае [53]. Таким образом, была подтверждена способность к соучастию аксиально расположенной защиты при О3 для аллозы, однако остальные положения оставались под вопросом.

Схема 9 . Бициклические продукты анхимерного содействия защитной группы при О3.
В 2009 г. исследование влияния ацильных заместителей на стерический результат гликозилирования было осуществлено группой K.S. Kim с использованием в качестве соучаствующей нуклеофильной группы трихлорацетимидатоила при О3, О4 и О6 маннозного донора. Стабильный бициклический продукт (27) был получен в случае защиты при О3 (схема 9 б). Этот результат совпадает с экспериментально наблюдаемым α-направляющим влиянием O-ацильной защиты при О3 маннозы в реакции гликозилирования [54].
Наконец, в 2020 г. были опубликованы данные, подтверждающие способность к соучастию ацетильной группы при О4 галактозы. Эксперимент заключался в генерации гликозил-катионов физическими методами фрагментации и получении их низкотемпературных ИК-спектров. Полученные спектры подтверждают образование диоксолениевого катиона посредством взаимодействия карбонильного кислорода ацетильной группы при О4 с аномерным центром [55].
Возможность осуществления удаленного соучастия также подтверждается теоретическими работами, основанными как на полуэмпирических методах, так и на расчетах ab initio. Наиболее изученными в этом плане являются фукозильные доноры – бромиды и трихлорацетимидаты. В рамках серии работ были изучены моно- и диацилированные (бензоилированные, ацетилированные и хлорацетилированные) фукопиранозиды и даже несколько фукофуранозидов. Полученные в результате расчетов несколькими различными методами значения энергии стабилизации образующихся диоксолениевых ионов прекрасно коррелируют как между собой, так и с экспериментально полученными в реакциях гликозилирования соотношениями α- и β-изомерных продуктов [56, 57].
Еще одним примером комплексного исследования, сочетающего экспериментальные и теоретические методы, является вышедшая совсем недавно совместная работа групп J.D.C. Codée и T.J. Boltje [58]. В ней использованы ИК-ионная спектроскопия (IRIS), расчеты методом DFT и серия модельных реакций гликозилирования. На примере широкой серии 3-О-, 4-О- и 6-О-моно-ацетилированных производных глюкозы, галактозы и маннозы авторам удалось показать степень влияния удаленного соучастия для различных доноров. Например, показано, что удаленное соучастие может быть использовано в случае 3‑О-ацилированных маннозил-доноров и в несколько меньшей степени в случае 4-О-ацилированных галактозил-доноров. Показано также, что соучастие 3-О-ацильных групп в глюкозных и галактозных производных оказывает относительно слабое влияние на стереохимический результат гликозилирования, в то время как значимого влияния соучастия 6-О-ацильной группы зафиксировано не было. Предложенный авторами подход представляется перспективным как для более подробного изучения удаленного соучастия, так и для надежного предсказания стереохимического результата реакции. Однако полученные результаты находятся в противоречии с опубликованными и рассмотренными выше экспериментальными данными, например, о стереоконтролирующем эффекте 6-О-ацильных заместителей [29, 52].
Таким образом, в то время как стереонаправляющее соучастие ацильной защиты при О2 является подробно изученным синтетическим подходом с подтвержденным механизмом, соучастие удаленных О-ацильных групп (при О3, О4 и О6) пока только всесторонне исследуется. И хотя работы последних лет не оставляют сомнений в принципиальной возможности реализации данного механизма, на практике, в отличие от классического соучастия групп при О2, удаленное соучастие не является универсальным подходом. Тем не менее большое количество успешно проведенных синтезов показывает, что при должной проработке и применении сопутствующих инструментов стереоконтроля удаленное соучастие является основным, а в некоторых случаях и единственно возможным подходом при синтезе 1,2-цис-гликозидов, который есть в арсенале химии углеводов на сегодняшний день.
РЕГУЛИРОВАНИЕ РЕАКЦИОННОЙ СПОСОБНОСТИ ГЛИКОЗИЛ-ДОНОРОВ
Защитные группы способны оказывать значительное влияние на стерические и электронные свойства гликозил-донора и гликозил-акцептора, а также на интермедиаты и продукты реакции гликозилирования. При этом некоторые классы защитных групп затрудняют образование карбокатиона и, соответственно, снижают реакционную способность гликозил-донора. Впервые дезактивирующее действие защитных групп было установлено B. Fraser-Reid: экспериментально показано, что сложноэфирные группы снижают активность донора по сравнению с простыми эфирами [59]. Этот принцип нашел широкое применение в различных подходах к контролю реакций гликозилирования. С одной стороны, возможна селективная активация ряда гликозил-доноров и осуществление серии последовательных реакций гликозилирования без выделения промежуточных продуктов [60]. С другой стороны, затруднение диссоциации уходящей группы может послужить методом стереоконтроля реакции, приближая механизм замещения к бимолекулярному SN2 [61]. При этом, в зависимости от целевого стереохимического исхода реакции, решающим этапом в процессе гликозилирования может быть предактивация, которая проводится перед добавлением гликозил-акцептора в реакционную смесь. Для построения аксиально расположенной гликозидной связи, как правило, нет необходимости в предактивации, поскольку аномерный эффект и так способствует образованию продукта с аксиальным заместителем в аномерном положении. Но в случаях, когда формирование аксиального продукта нежелательно, предактивация позволяет изменить путь протекания реакции и достигнуть альтернативного стерического результата [9]. Некоторые работы в этой области рассмотрены ниже.
Использование конформационно-ограничивающих защитных групп для изменения реакционной способности гликозил-доноров. Впервые о возможности влияния на стереохимический результат реакции гликозилирования посредством конформационно-ограничивающих защитных групп стало известно в конце 90-х гг., когда метод гликозилирования с использованием сульфоксидных доноров, предложенный D. Kahne в 1989 г. [61], был применен группой D. Crich в работе с маннозными донорами [61]. В ходе исследований было обнаружено, что при изменении порядка добавления реагентов в реакции между соединением (28) и акцептором (29) происходит обращение стереоселективности с преимущественным образованием β-маннозида (30) в случае, когда активация донора трифторметансульфоновым ангидридом предваряет добавление акцептора (схема 10 ). Данное наблюдение быстро нашло применение для получения β-маннозидов, синтез которых до этого был затруднен.

Схема 10 . Обращение стереохимического результата реакции маннозилирования при изменении порядка добавления реагентов. DTBMP – 2,6-ди-трет-бутил-4-метилпиридин.
В результате дальнейших исследований данной реакции группой D. Crich было предложено объяснение наблюдаемого эффекта. При предактивации гликозил-донора образующийся оксокарбениевый катион атакуется трифлат-анионом с образованием устойчивого α-гликозил-трифлата (схема 11 ). При этом в случае моноциклической системы (на примере 4,6-ди-О-бензильного маннозида) данный ковалентно-связанный трифлат находится в равновесии с тесной ионной парой (ТИП), образующейся из него при разрыве связи между аномерным углеродом и трифлатом, и сольватно-разделенной ионной парой (СРИП), образующейся, в свою очередь, из ТИП. Добавляемый затем акцептор атакует в первую очередь наиболее реакционноспособную сольватно-разделенную пару, при этом атака возможна с обеих сторон. В результате такой реакции образуется в основном наиболее выгодный продукт с α-гликозидной связью, что полностью согласуется с образованием α-трифлата на первом этапе. В случае бициклической декалиновой системы (например, наличия 4,6-О-бензилиденовой защиты в структуре донора) равновесие между ковалентным трифлатом, ТИП и СРИП сильно смещается в сторону первого. Таким образом, атака акцептором происходит только со свободной стороны по SN2-механизму с образованием β-маннозида.

Схема 11 . Предложенный механизм образования α- и β-маннозидов.
В том случае, когда перед активацией в реакционной смеси присутствует акцептор, последний перехватывает образующийся на начальном этапе активации оксокарбениевый катион до того, как он будет атакован трифлат-анионом. Таким образом, ковалентный трифлат не образуется, и реакция протекает по SN1-механизму с образованием более выгодного α-маннозидного продукта.
Наличие ковалентно-связанного трифлата как ключевого интермедиата реакции в случае предварительной активации донора было подтверждено ЯМР-экспериментами in situ, в результате которых был описан ряд таких трифлатов, полученных как из исходных сульфоксидов, так и из соответствующих бромидов. Возможность существования в качестве основного интермедиата ТИП или СРИП была отвергнута по характерным химическим сдвигам сигналов аномерных углеродов.
Таким образом, воздействие, оказываемое бензилиденовой группой на стереохимический результат реакции, заключается в смещении равновесия между интермедиатами – ковалентно связанным трифлатом, ТИП и СРИП в сторону первого, что, в свою очередь, оказывает влияние на основной реализуемый в рамках реакции механизм – SN2 либо SN1. Суть данного эффекта связана с проявляемой бензилиденовой защитой торсионной дезактивацией – увеличением активационного барьера изменения конформации циклогексанового кольца из классического “кресла” за счет препятствования “уплощению” цикла, необходимого для образования оксокарбениевого иона. Впервые данный эффект был показан группой B. Fraser-Reid на примере гидролиза н-пентенил-глюкозидов [63], а впоследствии подтвержден H.H. Jensen при исследовании скорости диссоциации уходящей группы ряда маннозил-доноров. Было установлено, что 4,6-О-бензилиденовая защита способствует меньшей склонности к диссоциации по сравнению с нециклическими формами [64].
Недавние исследования привели к некоторому усложнению трактовки рассмотренного механизма [65]. Во-первых, было обнаружено, что в тех же условиях глюкозильный донор проявляет, наоборот, α-селективность, хотя продолжает наблюдаться образование α-трифлата; во-вторых, исследования кинетического изотопного эффекта в реакции маннозилирования противоречат осуществлению механизма SN2 в чистом виде.
Эти противоречия могут быть объяснены, если учесть, что свойства гликозил-донора способны смещать равновесие переходного состояния от СРИП к ТИП и ковалентному трифлату. Так, в настоящее время предполагается, что соединением, которое, собственно, подвергается атаке гликозил-акцептора, является ТИП, источником для генерации которой служит α-трифлат. Таким образом, трифлат-анион продолжает закрывать для атаки α-сторону аномерного центра, но реакция протекает по мономолекулярному механизму, что лучше согласуется с данными по исследованиям кинетического изотопного эффекта. В случае глюкозил-доноров предполагается, что гликозилирование акцептора осуществляется сольватированной ионной парой, и α-селективность объясняется аномерным эффектом [9]. Это различие аномерной селективности между глюкозильным и маннозильным донорами может быть объяснено конфигурацией хирального центра при C2. Так, изменение диэдрального угла (О2–С2)–(С3–О3) делает глюкозный катион оксокарбения более устойчивым за счет выгодного расположения диполя С2–О2 относительно оксокарбениевого фрагмента по сравнению с возможным маннозным катионом. Именно за счет этого и происходит смещение равновесия промежуточного состояния дальше от α-трифлата [65].
Впервые стереонаправляющий эффект 4,6-О-ацетальной защиты был обнаружен в реакции β‑маннозилирования α-сульфоксидным донором с бензилиденовой защитой при активации трифторметансульфоновым ангидридом, однако с тех пор круг субстратов, в которых возможны подобные эффекты, был расширен. Помимо введения в данную реакцию глюкозидов и галактозидов, хорошие результаты были получены и с использованием иных циклических защит, образующих шестичленный цикл с О4 и О6, например, фенилборонатной [66] и ди-трет-бутилсилиленовой [67].
Последующие исследования структурных факторов, влияющих на реакционную способность гликозил-доноров, показали, что помимо сульфоксидной уходящей группы возможно использование тиогликозидов, причем активация может осуществляться трифторметансульфокислотой в присутствии N-иодсукцинимида [68]. Также были успешно использованы трихлорацетимидатные [69], пентенилгликозидные [70] и гликозилфосфатные [71] доноры в сочетании с подходящими трифторметилсульфонатными активаторами. В то же время классическое β-маннозилирование бензилиден-защищенными сульфоксидными донорами, предложенное D. Crich, в настоящее время успешно применяется в олигосахаридном синтезе. Так, в 2016 г. была синтезирована серия олигосахаридов, родственных фрагментам β-маннанов клеточной стенки патогенного гриба Candida albicans, с использованием как сульфоксидных, так и тиогликозидных доноров [72]. Пример синтеза пентасахаридного производного (34) представлен на схеме 12 .
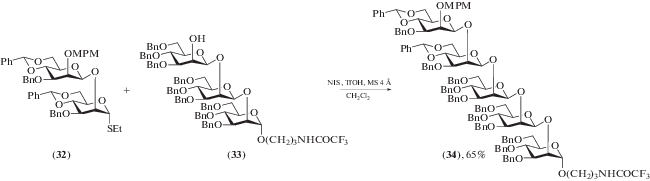
Схема 12 . Синтез β-маннозида (34) с использованием гликозил-донора (32), содержащего 4,6-О-бензилиденовую защиту.
Влияние электроноакцепторных заместителей на реакционную способность гликозил-доноров. В процессе исследования дезактивирующего и стереонаправляющего действия 4,6-О-бензилиденовой защиты в реакции маннозилирования группой D. Crich также было изучено действие электроноакцепторных заместителей на результат реакции. Авторы предполагали, что электроноакцепторные группы, аналогично конформационно-ограничивающим, будут способствовать повышению энергетического барьера образования катиона оксокарбения и, соответственно, образованию β‑маннозидов в условиях предактивации. Действительно, исследование реакции гликозилирования с серией 6-фторзамещенных рамнозных (6-дезоксиманнозных) доноров показало, что β‑селективность увеличивалась вместе с количеством атомов фтора [73].
В 2009 г. было проведено систематическое исследование влияния удаленных электроноакцепторных заместителей на стерический исход маннозилирования. В качестве контроля использовали полностью О-бензилированный маннозилтрихлорацетимидат (35), а также его аналоги с систематически замененными бензильными защитами при О3, О4, и О6 на серию электроноакцепторных сложноэфирных защит (табл. 1). Было установлено, что β-селективность возрастает с усилением электроноакцепторных свойств заместителя – самый сильный β-направляющий эффект проявила бензилсульфонильная (SO2Bn) группа (доноры (36–38)). При этом порядок возрастания селективности в ряду изменявшихся защит в целом сохранялся независимо от положения рассматриваемой группы в углеводном кольце. Это позволяло исключить стерический фактор как определяющий эффект стереохимического исхода реакции. Кроме этого, в ходе проведенного исследования были подтверждены образование ковалентных α-трифлатов и повышенная устойчивость трифлата при наличии более электроноакцепторной защиты. В связи с предполагаемой значимостью генерации α-трифлата в β‑маннозилировании, авторы также осуществили серию экспериментов с альтернативной активацией при помощи AgClO4. При этом наблюдалось предпочтительное образование α-продукта, что подтверждает значимость предактивации при использовании доноров подобного типа [54].
Таблица 1.
Влияние О-бензилсульфонильной защитной группы на стереохимический результат маннозилирования гликозил-донорами (35–38)
 |
||||
|---|---|---|---|---|
| Номер соединения | Варьируемые защитные группы | β : α | ||
| R' | R'' | R''' | ||
| (35) | Bn | Bn | Bn | 1 : 2.7 |
| (36) | SO2Bn | Bn | Bn | 15.9 : 1 |
| (37) | Bn | SO2Bn | Bn | 10.7 : 1 |
| (38) | Bn | Bn | SO2Bn | 13.8 : 1 |
Впоследствии авторы вышеописанного исследования расширили круг рассматриваемых гликозил-доноров и изучили влияние сложноэфирной защиты на глюкозилирование и галактозилирование. При этом было обнаружено, что ацильные заместители могут иметь как α-, так и β-направляющий эффект. α-Направляющими оказались ацильные группы при О3 глюкозил-донора, а также при О3 и О4 галактозил-донора. Это наблюдение в сочетании с тем, что неспособная к соучастию SO2Bn-группа имела исключительно β-направляющий эффект независимо от расположения, послужило подтверждением возможности удаленного соучастия О-ацильной защиты [74] (см. выше подраздел “Стереоконтролирующее соучастие удаленных защитных групп”).
К дезактивированным за счет электронного эффекта маннозил-донорам также можно отнести маннуронозил-доноры, в которых электроноакцепторной группой является карбоксильный фрагмент при С5. β-Селективность маннуронил-доноров была успешно использована группой G. van der Marel в синтезе соответствующего трисахарида [75]. Опираясь на эту работу, в 2015 г. с высоким выходом и селективностью (в среднем 75% на цикл) был осуществлен автоматизированный синтез β-(1→4)-связанного маннуроногексасахарида [76], причем полученный продукт был затем успешно восстановлен до соответствующего β-(1→4)-связанного манногексозида, получение которого с использованием маннозил-доноров представляется весьма сложной задачей.
ПРЕДВАРИТЕЛЬНАЯ КООРДИНАЦИЯ ГЛИКОЗИЛ-ДОНОРА И ГЛИКОЗИЛ-АКЦЕПТОРА ДЛЯ УЛУЧШЕНИЯ ИХ ВЗАИМОДЕЙСТВИЯ
В то время как наиболее распространенные подходы для стереоконтроля реакций гликозилирования можно упрощенно свести к блокированию подхода нуклеофила с определенной стороны гликозил-донора, существует противоположная стратегия. В ней проводится предварительное расположение гликозил-акцептора с требуемой стороны аномерного центра гликозил-донора при помощи ковалентного связывания посредством подходящего временного мостика.
Разработка внутримолекулярного гликозилирования в производных, содержащих соединенные гликозил-донор и гликозил-акцептор, может быть представлена как попытка воспроизвести химическими методами высокую эффективность и специфичность природных процессов ферментативного гликозилирования, обеспечиваемых за счет взаимного расположения донора и акцептора в активном центре фермента [77]. При этом методы внутримолекулярного гликозилирования условно можно разделить на три категории в зависимости от расположения связывающего их спейсера (схема 13 а) [78]. Первая является моделью “молекулярного зажима” (molecular clamp) или взаимного расположения реагентов (VIII). Во второй реализуется стратегия соединения гликозил-донорного и гликозил-акцепторного остатков через уходящую группу (IX), а в третьей осуществляется внутримолекулярная доставка агликона (X) при перегликозилировании атома кислорода, временно вовлеченного в межзвеньевой мостик.

Схема 13 . Внутримолекулярное гликозилирование. (i) – (39), (40), DCC, DMAP, THF : диоксан 1 : 5; (ii) – Ac2O, Py; (iii) – AcOH, 90°C; (iv) – Ac2O, Py, 0°C; (v) – RhCl(PPh3)3; (vi) – CaSO4, HgCl2, HgO, CH3CN, 60°C; (vii) – NaOMe (0.1 M).
Впервые возможность предварительного ковалентного связывания гликозил-донора и гликозил-акцептора была предложена в 1986 г. T. Shiba [79] на примере хитобиозного производного (43). Так, из предшественников (39) и (40) был получен их конъюгат (41), в котором моносахаридные звенья соединены сложноэфирной связью (схема 13 б). Замена О-защит, в том числе удаление аллильного агликона и формирование оксазолинового производного (42), а затем активация последнего, приводили к образованию целевого продукта (43). При этом наблюдалось стереоспецифичное образование исключительно β-гликозидной связи, присутствующей в продукте (43), а также весьма высокая эффективность гликозилирования: при попытке проведения реакции между соответствующими несоединенными моносахаридами образование продукта (43) не наблюдалось. Впоследствии рассмотренный подход был использован еще в некоторых случаях и стал известен как метод “молекулярного зажима”. При таком подходе межмоносахаридный мостик располагается вдали от реакционного центра и обеспечивает необходимую пространственную сближенность свободной ОН-группы гликозил-акцепторной части и аномерного центра гликозил-донорной части. Таким образом, в зависимости от структуры мостика и участвующих моносахаридных частей, возможно селективное построение как α-, так и β-гликозидов. При этом был предложен ряд типов мостика, в первую очередь симметричные бифункциональные производные фталевой кислоты, ксилолы и бисфенол А [78].
Метод внутримолекулярной доставки агликона основывается на получении алкилиденового или силилиденового мостика между гидроксильными группами донора и акцептора при помощи спейсера. Таким образом достигается стереоспецифичное образование гликозидной связи в син-положении относительно спейсера. При этом наиболее распространенные и успешные подходы заключаются в получении 1,2-цис-гликозидов за счет образования ацеталя при реакции между О2 донора и гидроксилом акцептора (схема 13 в). Примеры эффективных синтезов представлены в работах [80–82].
Последним подходом к внутримолекулярной доставке агликона является использование ариловых эфиров для получения временного мостика методом окисления в присутствии акцептора. Впервые этот метод был предложен в 1994 г. как очередной подход к получению β-маннозидов (схема 13 г). Поскольку вышеописанные методы внутримолекулярной доставки агликона предполагают осуществление манипуляций с малоустойчивыми интермедиатами [83], а кроме того, условия получения алкилацеталей при помощи реагента Теббе несовместимы с использованием сложноэфирных защитных групп [84], авторы стремились разработать более простой и универсальный подход к получению необходимого ацеталя.
ЗАКЛЮЧЕНИЕ
За время, прошедшее с первых попыток осуществления стереоконтроля в реакциях гликозилирования с помощью защитных групп, достигнут заметный прогресс. На основе соучастия защитной группы при О2 разработан и практически досконально изучен высокоэффективный механизм построения 1,2-транс-гликозидной связи. Стереонаправленное 1,2-цис-гликозилирование на сегодняшний день также не является неразрешимой проблемой в подавляющем большинстве случаев. Созданы подходы на основе содействия удаленных О-защит, а также концепции “активации–дезактивации”, позволяющей не только оказывать влияние на стереохимический результат реакций, но и осуществлять несколько последовательных реакций гликозилирования без промежуточного выделения продуктов. Для случаев же, когда данные подходы неприменимы или не работают, предложены методы, основанные на предварительной координации гликозил-донора и гликозил-акцептора. При этом в рамках каждого подхода существует возможность выбора конкретных защитных групп, различающихся по своим характеристикам. Помимо перечисленных подходов существует ряд многообещающих разработок, не получивших на настоящий момент широкого применения. Так, в последние годы были разработаны методы стереоконтроля, основанные на координировании донора и акцептора через водородную связь, образуемую акцепторным гидроксилом и защитной группой донора (схема 14 ) [85], а также на использовании объемных стерически ограничивающих групп при О6 для получения α-гликозидов [29, 86].

Схема 14 . Координирование донора и акцептора через водородную связь с пиколильной группой.
Тем не менее разработанные на сегодняшний день методы стереоконтроля гликозилирования пока не являются совершенными, а все возрастающая сложность синтезируемых углеводных структур, включая конъюгаты с олигосахаридными векторными лигандами [87] и конъюгированные углеводные вакцины [88–92], а также появление технологий автоматизированного олигосахаридного синтеза [93, 94] требуют использования стереоспецифичных методов на каждой стадии гликозилирования. Это наглядно демонстрирует необходимость развития обсуждаемой области гликохимии как посредством модернизации и универсализации уже известных методов, так и за счет разработки принципиально новых подходов, а также типов стереоконтролирующих защитных групп. Исследования последнего десятилетия подтверждают эту тенденцию и позволяют рассчитывать на дальнейшее совершенствование методов стереоспецифичного гликозилирования в ближайшем будущем.
Список литературы
Essentials of Glycobiology / Eds. Varki A., Cummings R.D., Esko J.D., Stanley P., Hart G.W., Aebi M., Darvill A.G., Kinoshita T., Packer N.H., Prestegard J.H., Schnaar R.L., Seeberger P.H. 3rd ed. Cold Spring Harbor (N.Y.): Cold Spring Harbor Laboratory Press, 2017. 823 p.
Varki A. // Cold Spring Harb Perspect Biol. 2011. V. 3. https://doi.org/10.1101/cshperspect.a005462
Moremen K.W., Tiemeyer M., Nairn A.V. // Nat. Rev. Mol. Cell Biol. 2012. V. 13. P. 448–462. https://doi.org/10.1038/nrm3383
Varki A. // Glycobiology. 2016. V. 27. P. 3–49. https://doi.org/10.1093/glycob/cww086
Бочков А.Ф., Афанасьев В.А., Заиков Г.Е. // Образование и расщепление гликозидных связей. Москва: Наука, 1978. 179 с.
Nukada T., Bereces A., Zgierski M.Z., Whitfield D. // J. Am. Chem. Soc. 1998. V. 120. P. 13291–13295. https://doi.org/10.1021/ja981041m
Chatterjee S., Moon S., Hentschel F., Gilmore K., Seeberger P.H. // J. Am. Chem. Soc. 2018. V. 140 I. 38. P. 11 942–11 953. https://doi.org/10.1021/jacs.8b04525
Kim J.-H., Yang H., Khot V., Whitfield D., Boons G.-J. // Eur. J. Org. Chem. 2006. P. 5007–5028. https://doi.org/10.1002/ejoc.200600440
Yang B., Yang W., Ramadan S., Huang X. // Eur. J. Org. Chem. 2018. V. 2018. P. 1075–1096. https://doi.org/10.1002/ejoc.201701579
Leng W.-L. Yao H., He J.-X., Liu X.-W. // Acc. Chem. Res. 2018. V. 51. P. 628–639. https://doi.org/10.1021/acs.accounts.7b00449
Tipson S. // J. Biol. Chem. 1939. V. 130. P. 55–59.
Isbell H.S. // Ann. Rev. Biochem. 1940. V. 9. P. 65–92.
Paulsen H., Herold C.-P. // Chem. Ber. 1970. V. 103. P. 2450–2462. https://doi.org/10.1002/cber.19701030817
Crich D., Dain Z., Gastaldi S. // J. Org. Chem. 1999. V. 64. P. 5224–5229. https://doi.org/10.1021/jo990424f
Mucha E., Marianski M., Xu F.-F., Thomas D.A., Meijer G., von Helden G., Seeberger P. H., Pagel K. // Nat. Commun. 2018. V. 9. 4174. https://doi.org/10.1038/s41467-018-06764-3
Boltje T.J., Liu L., Boons G.-J. // In: Glycochemical Synthesis / Eds. Hung S.-C., Zulueta M.M.L. Hoboken New Jersey: John Wiley & Sons Inc., 2016. P. 97–130. https://doi.org/10.1002/9781119006435.ch4
Yu H., Williams D.L., Ensley H.E. // Tetrahedron Lett. 2005. V. 46. P. 3417–3421. https://doi.org/10.1016/j.tetlet.2005.03.099
Liu H., Hansen T., Zhou S.-Y., Wen G.-E., Liu X.-X., Zhang Q.-J., Codée J.D.C., Schmidt R.R., Sun J.-S. // Org. Lett. 2019. V. 21. P. 8713–8717. https://doi.org/10.1021/acs.orglett.9b03321
Smoot J.T., Pornsuriyasak P., Demchenko A.V. // Angew. Chem. Int. Ed. 2005.V. 44. P. 7123–7126. https://doi.org/10.1002/anie.200502694
Buda S., Gołębiowska P. Mlynarski J. // Eur. J. Org. Chem. 2013. P. 3988–3991. https://doi.org/10.1002/ejoc.201300123
Le Mai Hoang K., Liu X.-W. // Nat. Commun. 2014. V. 5. 5051. https://doi.org/10.1038/ncomms6051
Karak M., Suenaga M. Oishi T., Torikai K. // Org. Lett. 2019. V. 21. I. 4. P. 1221–1225. https://doi.org/10.1021/acs.orglett.9b00220
Kim J.-H., Yang H., Boons G.-J. // Angew. Chem. Int. Ed. 2005. V. 44. P. 947–949. https://doi.org/10.1002/anie.200461745
Kim J.-H., Yang H., Park J., Boons G.-J. // J. Am. Chem. Soc. 2005. V. 127. P. 12 090–12 097. https://doi.org/10.1021/ja052548h
Cox D.A., Fairbanks A.J.// Tetrahedron: Asymmetry. 2009. V. 20. P. 773–780. https://doi.org/10.1016/j.tetasy.2009.02.018
Fascione, M.A., Turnbull W.B. // Beilstein J. Org. Chem. 2010. V. 6. https://doi.org/10.3762/bjoc.6.19
Fang T., Mo K.-F., Boons G.-J. // J. Am. Chem. Soc. 2012. V. 134 P. 7545−7552. https://doi.org/10.1021/ja3018187
Huang W., Gao Q., Boons G.-J. // Chem. Eur. J. 2015. V. 21. P. 12 920–12 926. https://doi.org/10.1002/chem.201501844
Komarova B.S., Tsvetkov Y.E., Nifantiev N.E. // Chem. Rec. 2016. V. 16. P. 488–506. https://doi.org/10.1002/tcr.201500245
Khatuntseva E.A., Ustuzhanina N.E., Zatonskii G.V., Shashkov A.S., Usov A.I., Nifant’ev N.E. // J. Carbohydr. Chem. 2000. V. 19. P. 1151–1173. https://doi.org/10.1080/07328300008544140
Gerbst A.G., Ustuzhanina N.E., Grachev A.A., Zlotina N.S., Khatuntseva E.A., Tsvetkov D.E., Shashkov A.S., Usov A.I., Nifantiev N.E. // J. Carbohydr. Chem. 2002. V. 21. P. 313–324. https://doi.org/10.1081/CAR-120013500
Gerbst A.G., Ustuzhanina N.E., Grachev A.A., Tsvetkov D.E., Khatuntseva E.A., Shashkov A.S., Usov A.I., Preobrazhenskaya M.E., Ushakova N.A., Nifantiev N.E. // J. Carbohydr. Chem. 2003. V. 22 P. 109–122. https://doi.org/10.1081/CAR-120020481
Ustyuzhanina N., Krylov V., Grachev A., Gerbst A., Nifantiev N. // Synthesis. 2006. V. 23. P. 4017–4031. https://doi.org/10.1055/s-2006-950333
Krylov V.B., Kaskova Z.M., Vinnitskiy D.Z., Ustyuzhanina N.E., Grachev A.A., Chizhov A.O., Nifantiev N.E. // Carbohydr. Res. 2011. V. 346. P. 540–550. https://doi.org/10.1016/j.carres.2011.01.005
Гербст А.Г., Грачев А.А., Устюжанина Н.Е., Хатунцева Е.А., Цветков Д.Е., Усов А.И., Шашков А.С., Преображенская М.Е., Ушакова Н.А., Нифантьев Н.Э. // Биоорг. химия. 2004. Т. 30. С. 1–12. [Gerbst A.G., Grachev A.A., Ustyuzhanina N.E., Khatuntseva E.A., Tsvetkov D.E., Usov A.I., Shashkov A.S., Preobrazhenskaya M.E., Ushakova N.A., Nifantiev N.E. // Russ. J. Bioorg. Chem. 2004. V. 30. P. 137–147.] https://doi.org/10.1023/B:RUBI.0000023099.48598.9a
Vinnitsky D.Z., Krylov V.B., Ustyuzhanina N.E., Dmitrenok A.S., Nifantiev N.E. // Org. Biomol. Chem. 2016. V. 14. P. 598–611. https://doi.org/10.1039/c5ob02040a
Komarova B.S., Tsvetkov Y.E., Knirel Y.A., Zähringer U., Pier G.B., Nifantiev N.E. // Tetrahedron Lett. 2006. V. 47. P. 3583–3587. https://doi.org/10.1016/j.tetlet.2006.03.045
Komarova B.S., Tsvetkov Y.E., Pier G.B., Nifantiev N.E. // J. Org. Chem. 2008. V. 73. P. 8411–8421. https://doi.org/10.1021/jo801561p
Komarova B. S., Tsvetkov Y.E., Pier G.B., Nifantiev N.E. // Carbohydr. Res. 2012. V. 360. P. 56–68. https://doi.org/10.1016/j.carres.2012.07.019
Komarova B.S., Orekhova M.V., Tsvetkov Y.E., Beau R., Aimanianda V., Latgé J.-P., Nifantiev N.E. // Chem. Eur. J. 2015. V. 20. P. 1029–1035. https://doi.org/10.1002/chem.201404770
Komarova B.S., Wong S.S., Orekhova M.V., Tsvetkov Y.E., Krylov V.B., Beauvais A., Bouchara J.-P., Kearney J., Aimanianda V., Latgé J.-P., Nifantiev N.E. // J. Org. Chem. 2018. V. 83. P. 12 965–12 976. https://doi.org/10.1021/acs.joc.8b01142
Komarova B.S., Dorokhova V.S., Tsvetkov Y.E., Nifantiev N.E. // Org. Chem. Front. 2018. V. 5. P. 909–928. https://doi.org/10.1039/c7qo01007a
Kazakova E.D., Yashunsky D.V., Krylov V.B., Bouchara J.P., Cornet M., Valsecchi I., Fontaine T., Latgé J.-P., Nifantiev N.E. // J. Am. Chem. Soc. 2020. V. 142. P. 1175–1179. https://doi.org/10.1021/jacs.9b11703
Kazakova E.D., Yashunsky D.V., Khatuntseva E.A., Nifantiev N.E. // Pure Appl. Chem. 2020. V. 92. P. 1047–1056. https://doi.org/10.1515/pac-2020-0105
Hahm H.S., Hurevich M., Seeberger P.H. // Nat. Commun. 2016. V. 7. 12482. https://doi.org/10.1038/ncomms12482
Krylov V., Ustyuzhanina N., Bakker H., Nifantiev N. // Synthesis. 2007. P. 3147–3154. https://doi.org/10.1055/s-2007-990784
Sethi M.K., Buettner F.F.R., Krylov V., Takeuchi H., Nifantiev N., Haltiwanger R.S., Gerardy-Schahn R. // J. Biol. Chem. 2010. V. 285. P. 1582–1586. https://doi.org/10.1074/jbc.C109.065409
Sethi M.K., Buettner F.F.R., Ashikov A., Krylov V.B., Takeuchi H., Nifantiev N.E., Haltiwanger R.S., Gerardy-Schahn R., Bakker H. // J. Biol. Chem. 2012. V. 287. P. 2739–2748. https://doi.org/10.1074/jbc.M111.302406
Mukaiyama T., Suenaga M., Chiba H., Jona H. // Chem. Lett. 2002. P. 56–57. https://doi.org/10.1246/cl.2002.56
Li Z., Zhu L., Kalikanda J. // Tetrahedron Lett. 2011. V. 52. P. 5629–5632. https://doi.org/10.1016/j.tetlet.2011.08.091
Komarova B.S., Gerbst AG., Finogenova A.M., Dmitrenok A.S., Tsvetkov Y.E., Nifantiev N.E. // J. Org. Chem.2017. V. 82. P. 8897–8908. https://doi.org/10.1021/acs.joc.7b01167
Komarova B.S., Orekhova M.V., Tsvetkov Y.E., Nifantiev N.E. // Carbohydr. Res. 2014. V. 384. P. 70–86. https://doi.org/10.1016/j.carres.2013.11.016
Crich D., Hu T., Cai F. // J. Org. Chem. 2008. V. 73. P. 8942–8953. https://doi.org/10.1021/jo801630m
Baek J.Y., Lee B.-Y., Jo M.G., Kim K.S. // J. Am. Chem. Soc. 2009. V. 131. P. 17 705–17 713. https://doi.org/10.1021/ja907252u
Marianski M., Mucha E., Greis K., Moon S., Pardo A., Kirschbaum K., Thomas D.A., Meijer G., von Helden G. Gilmore K., Seeberger P.H., Pagel K. // Angew. Chem. Int. Ed. 2020. V. 59. P. 6166–6171. https://doi.org/10.1002/anie.201916245
Gerbst A.G., Ustuzhanina N.E., Grachev A.A., Khatuntseva E.A., Tsvetkov D.E., Whitfield D.M., Bereces A., Nifantiev N.E. // J. Carbohydr. Chem. 2001. V. 20. P. 821–831. https://doi.org/10.1081/CAR-100108659
Gerbst A.G., Krylov V.B., Nifantiev N.E. // In: Carbohydrate Chemistry – a Specialist Periodical Reports / Eds.: Rauter A.P., Lindhorst T., Queneau Y. RSC. 2021. V. 44. P. 151–169. https://doi.org/10.1039/9781788013864-00151
Hansen T., Elferink H., van Hengst J.M.A., Houthuijs K.J., Remmerswaal W.A., Kromm A. Berden G., van der Vorm S., Rijs A.M., Overkleeft H.S., Filippov D.V., Rutjes F.PJ. van der Marel G., Martens J., Oomens J., Codée J.D.C., Boltje T.J. // Nat. Commun. 2020. V. 11. https://doi.org/10.1038/s41467-020-16362-x
Mootoo D.R., Konradsson P., Udodong U., Fraser-Reid B. // J. Am. Chem. Soc. 1988. V. 110. P. 5583–5584. https://doi.org/10.1021/ja00224a060
Yang B., Yoshida K., Huang X. // In: Glycochemical Synthesis / Eds. Hung S.-C., Zulueta M.M.L. Hoboken New Jersey: John Wiley & Sons Inc., 2016. P. 155–188. https://doi.org/10.1002/9781119006435.ch6
Crich D., Sun S. // J. Am. Chem. Soc. 1997. V. 119. P. 11 217–11 223. https://doi.org/10.1021/ja971239r
Kahne D., Walker S., Cheng Y., Van Engen D. // J. Am. Chem. Soc. 1989. V. 111. P. 6881–6882. https://doi.org/10.1021/ja00199a081
Fraser-Reid B., Wu Z., Andrews C.W., Skowronski E. // J. Am. Chem. Soc. 1991. V. 113. P. 1434–1435. https://doi.org/10.1021/ja00004a066
Jensen H.H., Nordstrom L.U., Bols M. // J. Am. Chem. Soc. 2004. V. 126. P. 9205–9213. https://doi.org/10.1021/ja047578j
Crich D. // Acc. Chem. Res. 2010. V. 43 I. 8. P. 1144–1153. https://doi.org/10.1021/ar100035r
Crich D., de la Mora M., Vinod A.U. // J. Org. Chem. 2003. V. 68. P. 8142–8148. https://doi.org/10.1021/jo0349882
Kumagai D., Miyazaki M., Nishimura S.-I. // Tetrahedron Lett. 2001. V. 42. P. 1953–1956. https://doi.org/10.1016/S0040-4039(01)00044-2
Yun M., Shin Y., Chun K.H., Jen S. // Bull. Korean Chem. Soc. 2000. V. 21. P. 562–566.
Weingart R., Schmidt R.R. // Tetrahedron Lett. 2000. V. 41. P. 8753–8758. https://doi.org/10.1016/S0040-4039(00)01497-0
Baek J.Y., Choi T.J., Jeon H.B., Kim K.S. // Angew. Chem. Int. Ed. 2006. V. 45. P. 7436–7440. https://doi.org/10.1002/anie.200602642
Tsuda T., Sato S., Nakamura S., Hashimoto S. // Heterocycles 2003. V. 59. P. 509–515. https://doi.org/10.3987/COM-02-S80
Karelin A.A., Tsvetkov Y.E., Paulovičová E., Paulovičová L, Nifantiev N.E. // Eur. J. Org. Chem. 2016. P. 1173–1181. https://doi.org/10.1002/ejoc.201501464
Crich D., Vinogradova O. // J. Am. Chem. Soc. 2007. V. 129. P. 11 756–11 765. https://doi.org/10.1021/ja0730258
Baek J.Y., Kwon H.-W., Myug S.J., Park J.J., Kim M.Y., Rathwell D.C.K., Jeon H.B., Seeberger P.H., Kim K.S. // Tetrahedron. 2015. V. 71. P. 5315–5320. https://doi.org/10.1016/j.tet.2015.06.014
van den Bos L.J., Dinkelaar J., Overkleeft H.S., van der Marel G.// J. Am. Chem. Soc. 2006. V. 128. P. 13 066–13 067. https://doi.org/10.1021/ja064787q
Tang S.-L., Pohl N.L.B. // Org. Lett. 2015. V. 17. P. 2642–2645. https://doi.org/10.1021/acs.orglett.5b01013
Behrendt M.E., Schmidt R.R. // Tetrahedron Lett. 1993. V. 34. P. 6733–6736. https://doi.org/10.1016/S0040-4039(00)61687-8
Xiao G.J., Demchenko A.V. // Beilstein J. Org. Chem. 2017. V. 13. P. 2028–2048. https://doi.org/10.3762/bjoc.13.201
Kusomoto S., Imoto M., Ogiku T., Shiba T. // Bull. Chem. Soc. Jpn. 1986. V. 59. P. 1419–1423. https://doi.org/10.1246/bcsj.59.1419
Barresi F., Hindsgau O. // J. Am. Chem. Soc. 1991. V. 113. P. 9376–9377. https://doi.org/10.1021/ja00024a057
Stork G., Kim G. // J. Am. Chem. Soc. 1992. V. 114. P. 1087–1088. https://doi.org/10.1021/ja00029a047
Bols M. // Tetrahedron. 1993. V. 49. P. 10 049–10 060. https://doi.org/10.1016/S0040-4020(01)80200-3
Ito Y., Ogawa T. // Angew. Chem. Int. Ed. Engl. 1994. V. 33 P. 1765–1767. https://doi.org/10.1002/anie.199417651
Ishiwata A., Ito Y. // In: Selective Glycosylations: Synthetic Methods and Catalysts / Ed. Bennet C.S. Weinheim: Wiley-VCH Verlag GmbH & Co. KgaA, 2017. P. 81–96.
Yasomanee J.P., Demchenko A.V. // J. Am. Chem. Soc. 2012. V. 134. P. 20 097–20 102. https://doi.org/10.1021/ja307355n
Zhang Y., Zhou S., Wang X., Zhan H., Guo Z., Gao J. // Org. Chem. Front., 2019. V. 6. P. 762–772. https://doi.org/10.1039/C8QO01177J
Щегравина Е.С., Сачкова А.А., Усова С.Д., Нючев А.В., Грачева Ю.А., Федоров А.Ю. // Биоорг. химия. 2021. Т. 47. С. 76–105. [Scchegravina E.S., Sachkova A.A., Usova S.D., Nyuchev A.V., Gracheva Yu.A., Fedorov A.Yu. // Russ. J. Bioorg. Chem. 2021. V. 47. P. 69– 96.] https://doi.org/10.31857/S013234232101022X
Генинг М.Л., Курбатова Е.А., Нифантьев Н.Э. // Биоорг. химия. 2021. Т. 47. С. 3–28. [Gening M.L., Kurbatova E. A., Nifantiev N.E. // Russ. J. Bioorg. Chem. 2021. V. 47. P. 1–24.] https://doi.org/10.31857/S0132342321010073
Хатунцева Е.А., Нифантьев Н.Э. // Биоорг. химия. 2021. Т. 47. С. 29–56. [Khatuntseva E.A., Nifantiev N.E. // Russ. J. Bioorg. Chem. 2021. V. 47. P. 25–51.] https://doi.org/10.31857/S0132342321010103
Adamo R., Nilo A., Castagner B., Boutureira O., Berti F., Bernardes G.J.L. // Chem. Sci. 2013. V. 4. P. 2995–3008. https://doi.org/10.1039/C3SC50862E
Hu Q.-Y., Berti F., Adamo R. // Chem. Soc. Rev. 2016. V. 45. P. 1691–1719. https://doi.org/10.1039/c4cs00388h
MacCalman T.E., Phillips-Jones M.K., Harding S.E. // Biotechnol. Genet. Eng. Rev. 2019. V. 35. P. 93–125. https://doi.org/10.1080/02648725.2019.1703614
Seeberger P.H. // Acc. Chem. Res. 2015. V. 48. P. 1450−1463. https://doi.org/10.1021/ar5004362
Pardo-Vargas A., Delbianco M., Seeberger P.H. // Curr. Opin. Chem. Biol. 2018. V. 46. P. 48–55. https://doi.org/10.1016/j.cbpa.2018.04.007
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия