

Биоорганическая химия, 2021, T. 47, № 2, стр. 184-194
Получение новых представителей класса фосфорилгуанидиновых олигонуклеотидов
С. А. Жуков 1, Д. В. Пышный 1, 2, М. С. Купрюшкин 1, *, **
1 ФГБУН “Институт химической биологии и фундаментальной медицины” СО РАН
630090 Новосибирск, просп. акад. Лаврентьева, 8, Россия
2 ФГБОУ ВПО ”Новосибирский национальный исследовательский государственный университет”
630090 Новосибирск, ул. Пирогова, 2, Россия
* E-mail: kuprummax@gmail.com
** E-mail: pyshnyi@niboch.nsc.ru
Поступила в редакцию 24.09.2020
После доработки 30.09.2020
Принята к публикации 10.10.2020
Аннотация
В данной работе были получены новые представители класса фосфорилгуанидиновых олигонуклеотидных производных. Предложена и осуществлена синтетическая схема, позволяющая получать из различных вторичных аминов широкий набор диаминокарбенийазидов для последующего введения тетразамещенных гуанидиновых остатков в состав олигонуклеотидов по реакции Штаудингера. Был выявлен ряд факторов, влияющих на выход фосфорилгуанидинового производного, таких как размер алкильных заместителей в составе используемого азида и его чистота, а также проведение процедуры элиминирования защитной цианэтильной группы, предшествующей финальному деблокированию олигонуклеотида.
ВВЕДЕНИЕ
Синтетические олигонуклеотиды в настоящее время нашли широкое применение в различных областях молекулярной биологии, биотехнологии и медицины. На данный момент более 10 олигонуклеотидных препаратов уже одобрены организацией FDA (Food and Drug Administration), а более 40 препаратов находятся на различных стадиях клинических испытаний [1–3]. Для терапевтического применения олигонуклеотидов особое значение имеют такие свойства, как химическая и энзиматическая устойчивость, эффективность проникновения в клетки и биораспределение, которые достигаются введением различных модификаций в состав создаваемых НК-конструкций [4].
Существует множество путей введения химических модификаций в структуру олигонуклеотидов [5, 6], при этом реализация большинства из них зачастую является отдельной синтетической задачей. Разработка подходов, позволяющих унифицировать процедуру введения различных модификаций и их комбинаций в состав олигонуклеотида, является актуальным технологическим этапом, а применение способов, совместимых с протоколами твердофазного амидофосфитного синтеза, позволит сделать создание различных олигонуклеотидных конструкций рутинной процедурой.
В Институте химической биологии и фундаментальной медицины СО РАН был разработан новый класс НК-производных – фосфорилгуанидиновые олигонуклеотиды [7]. Для их получения использовали окисление фосфиттриэфирной группы (промежуточного продукта олигонуклеотидного синтеза) тетразмещенным диаминокарбенийазидом по реакции Штаудингера. В настоящее время эффективность данного подхода продемонстрирована на примере коммерчески доступного диаминокарбенийазида – гексафторфосфата 2-азидо-1,3-диметилимидазолидиния (ADMP) [8]. Новый класс соединений и метод их получения в настоящий момент запатентованы в России и Японии и находится на стадии патентования в национальных ведомствах других стран [9].
Данный подход позволяет вводить замещенные гуанидиновые остатки по межнуклеотидному фосфату олигонуклеотидной цепи с высокой эффективностью, даже в случае получения олигонуклеотидов с полностью модифицированным остовом. Реакция не требует применения высококонцентрированных растворов и нагревания, что позволило адаптировать метод введения данной модификации к автоматическому синтезу олигонуклеотидов на ДНК/РНК-синтезаторах.
Данная статья посвящена разработке схемы синтеза различных тетразамещенных диаминокарбенийазидов и получения новых представителей класса фосфорилгуандиновых олигонуклеотидов по реакции Штаудингера.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Получение тетразамещенных диаминокарбенийазидов. В настоящее время единственным коммерчески доступным диаминокарбенийазидом является ADMP (рис. 1а), используемый для введения 1,3-диметилимидазолидиновой группы в состав олигонуклеотидного остова (рис. 1б). Для получения фосфорилгуанидиновых производных с другими заместителями (рис. 1в) необходимо разработать схему синтеза соответствующих диаминокаребнийазидов.
Рис. 1.
(а) – Гексафторфосфат 2-азидо-1,3-диметилимидазолидиния (ADMP) – единственный коммерчески доступный диаминокарбенийазид; (б) – фосфорилгуанидиновое производное олигонуклеотида c 1,3-диметилимидазолидиновой группой; (в) – фосфорилгуанидиновое производное олигонуклеотида с различными заместителями R1–R4.

Для синтеза набора азидов-модификаторов, несущих различные заместители, была выбрана схема, включающая следующие этапы: получение замещенной мочевины, конвертацию мочевины в соответствующий диаминокарбенийхлорид и замещение атома хлора на азидогруппу.
Учитывая трудоемкость получения несимметричных замещенных мочевин, было решено использовать различные вторичные амины как исходные соединения, содержащие различные функциональные остатки. В качестве карбонилирующего агента был выбран тиофосген (рис. 2, стадия (1)). Далее взаимодействием с оксалилхлоридом тиомочевины конвертировали в диамиокарбенийхлориды (рис. 2. стадия (2)), так называемые соли Вильсмайера [10]. Для получения диаминокарбенийазидов действием азида натрия замещали атом хлора на азидогруппу (рис. 2, стадия (3)) [11].
Рис. 2.
Схема получения диаминокарбенийазидов: (1) – получение тиомочевины; (2) – получение соли Вильсмайера; (3) – замещение атома хлора на азидогруппу.

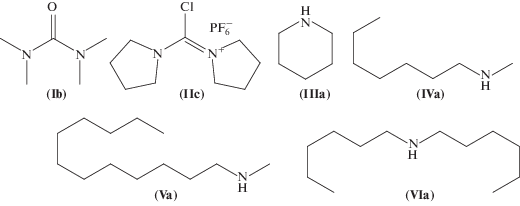
В качестве исходных соединений для получения целевых азидов были выбраны вторичные амины с алкильными заместителями различной длины (рис. 3, (IIIa–VIa)), а также тетраметилмочевина (рис. 3, (Ib)) и коммерчески доступная соль Вильсмайера (рис. 3, (IIc)) для синтеза азидов по сокращенной схеме. На первой стадии синтеза взаимодействием тиофосгена с избытком вторичного амина получали симметричные тетразамещенные тиомочевины (IIIb–VIb). Для связывания образующегося HCl в реакционную смесь добавляли N,N-диизопропилэтиламин (DIPEA) либо, если образующаяся соль используемого амина (Va) и (VIa) выпадала в осадок, вносили дополнительное количество такого амина. Получаемые тиомочевины отделяли от избытка амина и побочных продуктов методом колоночной хроматографии. Полноту протекания реакций и чистоту образующихся продуктов контролировали при помощи анализа методами ТСХ и 1H-ЯМР.
Рис. 3.
Соединения-предшественники для получения диаминокарбенийазидов: тетраметилмочевина (Ib); гексафторфосфат хлородипирролидинокарбения (IIc); амины: пиперидин (IIIa), N-гептилметиламин (IVa), N-додецилметиламин (Va), дигексиламин (VIa).
На следующей стадии взаимодействием тиомочевин с оксалилхлоридом получали диамиокарбенийхлориды, так называемые соли Вильсмайера. В ходе реакции образуются газообразные CO и CSO, что позволяет избежать необходимости очистки целевого продукта от продуктов деградации используемого реагента. Кроме того, оксалилхлорид обладает достаточно низкой температурой кипения (61°C), что позволяет отделять его избыток от реакционной смеси при упаривании. В данной реакции образуется соль Вильсмайера с анионом Cl–, но для повышения растворимости конечного продукта в органических растворителях противоион замещали на гексафторфосфат в реакции обмена с солью KPF6 [12], после чего отделяли реакционную смесь от осадка хлорида калия фильтрованием.
Поскольку соли Вильсмайера обладают крайне высокой реакционной способностью, данные соединения не выделяли, и после фильтрования и упаривания получаемую реакционную смесь вводили в реакцию с азидом натрия для замещения атома хлора на азидогруппу. Азид натрия мало растворим в органических растворителях, поэтому для проведения реакции использовали суспензию азида натрия в ацетонитриле. Анализ 1H-ЯМР-спектров полученных азидов показал, что сигналы, соответствующие примесям непрореагировавших веществ и возможным продуктам их деградации, составляют не более 10% от суммарной интенсивности. Таким образом, реакции получения соли Вильсмайера и замещения атома хлора на азидогруппу протекают с высокой эффективностью.
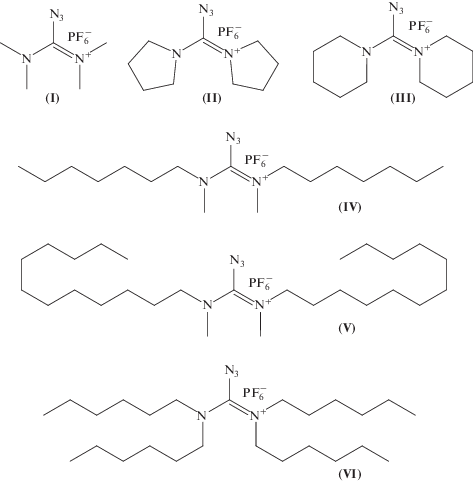
C использованием разработанной синтетической схемы был получен набор из шести азидов-модификаторов (рис. 4).
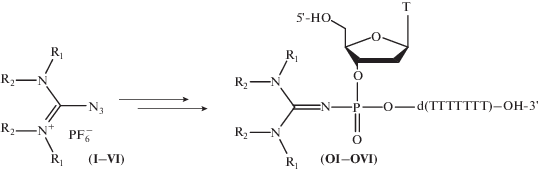
Использование синтезированных азидов для введения модификаций в состав олигонуклеотидов. Для проверки эффективности протекания реакции Штаудингера с использованием полученных азидов-модификаторов были синтезированы модельные октатимидилаты 5'-T*TTTTTTT-3' (рис. 5). После этапа присоединения мономера стадию окисления не проводили, автоматический синтез останавливали, реактор вынимали из синтезатора, полимерный носитель (CPG) с иммобилизованным фосфиттриэфиром переносили из реактора в пластиковую пробирку и проводили реакцию Штаудингера с раствором диамиокарбенийазида. Затем переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали обработки в рамках автоматизированного твердофазного синтеза.
Рис. 5.
Схема введения модификации в состав модельного октатимидилата 5'-T*TTTTTTT-3' по реакции Штаудингера. (I) R1 = R2 = –CH3; (II) R1 = R2 = –(CH2)4–; (III) R1 = R2 = –(CH2)4–; (IV) R1 = –C7H15, R2 = –CH3; (V) R1 = –C12H25, R2 = –CH3; (VI) R1 = R2 = –C6H13.
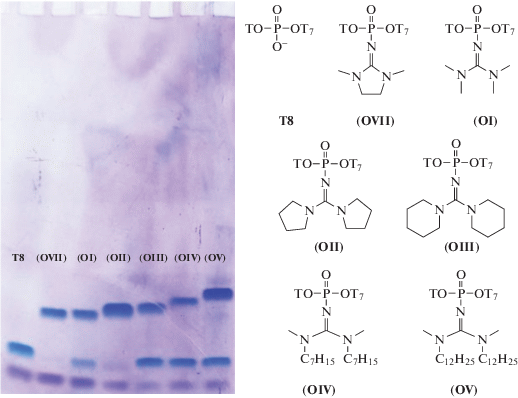
Эффективность введения модификации (конверсию) оценивали по профилю офВЭЖХ как отношение площади пика, соответствующего модифицированному олигонуклеотиду, к суммарной площади пиков на хроматограмме. Для обеспечения введения модификации с эффективностью не менее 50% при проведении реакции Штаудингера в большинстве случаев использовали концентрированные 1 М растворы азидов-модификаторов в ацетонитриле при умеренном нагревании (40°С). Условия, значения конверсии введения модификаций, а также результаты масс-спектрометрии полученных представителей класса фосфорилгуанидинов представлены в табл. 1. Кроме того, в табл. 1 приведено значение конверсии при использовании коммерчески доступного азида ADMP, получаемое при введении модификации в ручном режиме по описанному выше протоколу. Стоит отметить, что при проведении реакции с ADMP в автоматическом режиме синтеза значения конверсии превышают 99% [8].
Таблица 1.
Эффективность введения различных модификаций олигонуклеотида 5'-T*TTTTTTT-3' (* – место введения модификации по межнуклеотидному фосфату) и результаты масс-спектрометрического анализа модифицированных олигонуклеотидов
| Шифр азида | Шифр олигонуклеотида | Условия | Конверсия, % | Mr (теор), г/моль | Mr (эксп), г/моль |
|---|---|---|---|---|---|
| (I) | (OI) | 1 М, 40°С, 1 ч | ~80 | 2467.5 | 2467.5 |
| (II) | (OII) | 0.25 М, 40°С, 1 ч | ~90 | 2519.5 | 2519.4 |
| (III) | (OIII) | 1 М, 40°С, 1 ч | ~70 | 2547.6 | 2547.6 |
| (IV) | (OIV) | 1 М, 40°С, 1 ч | ~60 | 2635.8 | 2635.7 |
| (V) | (OV) | 1 М, 50°С, 6 ч | ~50 | 2775.9 | 2775.8 |
| (VI) | (OVI) | 1 М, 40°С, 1 ч | 0 | – | – |
| ADMP | (OVII) | 0.1 М, 40°С, 1 ч | ~90 | 2465.4 | 2465.5 |
| – | T8 | – | – | 2370.4 | 2370.4 |
Анализируя значения конверсии, можно выявить закономерность: эффективность введения модификации падает с нарастанием размера алкильных заместителей, что можно объяснить стерическими затруднениями при взаимодействии азида с фосфиттриэфирной группой в составе растущей олигонуклеотидной цепи. В случае азида (VI), содержащего четыре гексильных заместителя, реакция Штаудингера не протекает вовсе (конверсия 0%). Для азида (V), содержащего две метильные и две объемные додецильные группы, при проведении реакции в течение 1 ч при 40°С наблюдалось низкое значение конверсии (10%), и только при повышении температуры до 50°С и существенном увеличении времени (6 ч) эффективность введения модификации увеличилась до 50%. В то же время для азида (II), содержащего компактные циклические заместители, высокое значение конверсии (~90%) было достигнуто при 40°С и более низкой концентрации азидопроизводного (0.25 М).
На рис. 6а представлены профили офВЭЖХ реакционных смесей полученных модифицированных олигонуклеотидов. Пик со временем удерживания 6.8 мин соответствует немодифицированному олигонуклеотиду (градиент ацетонитрила 0–50% за 15 мин). Более гидрофобные фосфорил-гуанидиновые олигонуклеотиды имеют большее время удерживания, увеличивающееся с нарастанием размера алкильных заместителей. Для анализа наиболее гидрофобного производного (OV), содержащего додецильные остатки, был использован градиент ацетонитрила 0–90% (рис. 6б).
Рис. 6.
(а) – Профили офВЭЖХ олигонуклеотидов (OI–OIV). Градиент ацетонитрила 0–50% за 15 мин; (б) – профиль офВЭЖХ олигонуклеотида (OV). Градиент ацетонитрила 0–90% за 15 мин. T8 – контрольный октатимидилат.

Несимметричность (OI, OIV, OV) или раздвоенность (OII, OIII) пиков, соответствующих целевым продуктам синтеза, обусловлена существованием двух диастереомерных форм мономодифицированных олигонуклеотидов, имеющих различное время удерживания.
Кроме того, был проведен эксперимент по исследованию электрофоретической подвижности синтезированных олигонуклеотидов в условиях денатурирующего ПААГ-электрофореза (рис. 7). Меньшая электрофоретическая подвижность модифицированных олигонуклеотидов в сравнении с нативным олигонуклеотидом равной длины объясняется наличием в них незаряженной фосфорилгуанидиновой группы. Снижение суммарного заряда олигонуклеотида при сохранении его длины приводит к уменьшению электрофоретической подвижности. С нарастанием размеров алкильных заместителей подвижность уменьшается незначительно.
Рис. 7.
Подвижность модифицированных олигонуклеотидов (OI–OV) и (OVII), а также контрольного немодифицированного олигонуклеотида (T8) в условиях денатурирующего электрофореза в 15%-ном ПААГ.
Влияние предварительного элиминирования цианэтильной группы на эффективность конверсии реакции модификации олигонуклеотидов. В первоначально проведенных экспериментах по введению модификации на примере азида (II) даже в сравнительно жестких условиях (1 М, 40°С, 1 ч) значения конверсии не превышали 50%, а в качестве единственного побочного продукта присутствовал соответствующий немодифицированный олигонуклеотид (рис. 8, кривая 1).
Рис. 8.
Профили офВЭЖХ олигонуклеотидов: 1 – введение модификации с использованием азида (II) без предварительного элиминирования (5'-T*TTTTTTT-3'); 2 – контрольный эксперимент, последний мономер присоединяли без стадии окисления (5'-T*TTTTTTT-3'). T8 – контрольный октатимидилат. Градиент ацетонитрила 0–50% за 15 мин.

При этом в случае использования азида (VI), когда обнаружить целевой продукт не удавалось вовсе, на хроматографическом профиле реакционной смеси можно отчетливо наблюдать два пика, соответствующих продуктам деградации неокисленного фосфиттриэфирного производного. Такой же профиль хроматографии наблюдался в случае контрольного эксперимента, в котором при синтезе октатимидилата последнее звено вводили без проведения стадии окисления (рис. 8, кривая 2). Данное наблюдение позволило предположить, что низкие значения конверсии реакции модификации азидом (II) объясняются не низкой реакционной способностью используемого азида, а щелочным гидролизом получающегося фосфазенового производного (промежуточного продукта реакции Штаудингера) при финальном деблокировании синтезируемого олигонуклеотида водным раствором аммиака или метиламина (рис. 9, (1)).
Рис. 9.
Возможные пути превращения промежуточного фосфазенового продукта: (1) – побочная реакция в ходе постсинтетической обработки, приводящая к образованию нативного олигонуклеотида; (2) – элиминирование цианэтильной группы действием DIPEA.

Для предотвращения протекания данной побочной реакции в протокол синтеза модифицированной олигонуклеотидной цепи после проведения реакции Штаудингера был введен этап обработки 10%-ным раствором DIPEA в ацетонитриле в течение 20 мин при 40°С для элиминирования защитной цианэтильной группы, что приводило к превращению фосфазенового остатка в стабильный фосфорилгуанидин еще до проведения финального деблокирования олигонуклеотда [13] (рис. 9, (2)).
Предварительное элиминирование цианэтильной группы привело к увеличению конверсии до значений >90%. В дальнейшем при введении всех модификаций использовали обработку 10%-ным раствором DIPEA в ацетонитриле. Значения конверсии, представленные в табл. 1, соответствуют экспериментам, проведенным с предварительным элиминированием цианэтильной группы до финального деблокирования олигонуклеотидного производного.
Еще одним наблюдением является увеличение выхода целевого продукта при синтезе олигонуклеотида с модификацией межнуклеотидного звена, отличного от 5'-терминальной позиции, даже без этапа обработки раствором DIPEA. Так, для олигонуклеотида 5'-TT*TTTTTTT-3' конверсия составила 70%. Данное наблюдение однозначно указывает на возможность протекания процесса элиминирования цианэтильной группы в составе фосфазенового остатка при прохождении полного синтетического цикла по стандартному амидофосфитному протоколу.
При синтезе модельного олигонуклеотида 5'-T*TTTTTTT-3' по протоколу с двукратно увеличенным этапом кэпирования, следующим сразу после этапа реакции Штаудингера, конверсия возросла до 90%. Вероятно, раствор N-метилимидазола в пиридине, обработка которым является частью протокола кэпирования, способен обеспечивать элиминирование цианэтильной группы при атоме фосфора в составе фосфазенового производного, чего не наблюдается в случае менее реакционноспособного “немодифицированного” фосфиттриэфирного звена растущей цепи.
Таким образом, при синтезе фосфорилгуанидиновых олигонуклеотидов возможно проводить необходимый этап элиминирования защитной группы в автоматическом режиме, используя растворы и регенты, применяемые в стандартном амидофосфитном протоколе олигонуклеотидного синтеза.
Интересно отметить, что для различных азидов степень влияния предварительного этапа β-элиминирования на значение конверсии различна. Так, при использовании азида ADMP значения конверсии синтеза олигонуклеотида 5'-T*TTTTTTT-3' с этапом β-элиминирования и при его отсутствии составляют 90 и 60% соответственно. Вероятно, алкильные заместители при гуанидиновом остатке в составе получаемого фосфазенового производного способны влиять на распеределение между продуктами щелочного гидролиза при финальном деблокировании синтезируемого олигонуклеотида.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и растворители. В работе были использованы следующие реагенты и растворители: гексафторфосфат калия, дигексиламин, N-гептилметиламин, N-додецилметиламин, пиперидин, гексафторфосфат хлородипирролидинокарбения (Sigma Aldrich, США); ацетонитрил, азид натрия, триэтиламин, диизопропилэтиламин (Panreac, Испания); гексафторфосфат 2-азидо-1,3-диметил-имидазолидиния, оксалилхлорид (TCI, Япония); дихлорметан, водный раствор аммиака, этиловый спирт, толуол (Реахим, Россия); тиофосген (Fluka, Швейцария); тетраметилмочевина (Merck, Германия).
Для ТСХ использовали пластинки DC-Alufolien Kieselgel 60 F254 (Merck, Германия), для колоночной адсорбционной хроматографии – колонку объемом 200 мл, заполненную рассчитанным количеством сорбента Kieselgel 60 (размер частиц 0.060–0.200 мм, размер пор 60 Å; Merck, Германия).
Для упаривания водных растворов использовали вакуумный концентратор SpeedVac (ThermoFisher, США), для упаривания органических растворов – Rotavapor R200 (Buchi, Швейцария). Получаемые соединения высушивали в эксикаторе до постоянной массы.
Хроматографический анализ. Полученные олигонуклеотиды анализировали методом офВЭЖХ на хроматографе Милихром A02 с использованием колонки ProntoSIL-120-5-C18, 2 × 75 мм (Эконова, Россия). Анализ проводили в системе “0.02 М ацетат триэтиламмония – 90%-ный ацетонитрил”, градиент ацетонитрила 0–90% за 30 мин, поток 200 мкл/мин, температура термостата 35°С. Детекцию осуществляли при длине волны 260 нм.
Электрофорез проводили в 15%-ном ПААГ в денатурирующих условиях (акриламид : N,N '-метиленбисакриламид (30 : 1), 8 М мочевина, 89 мМ Трис-борат, pH 8.3, 2 мM Na2-EDTA) в буферном растворе ТВЕ (89 мМ Трис-борат, pH 8.3, 2 мM Na2-EDTA) при напряжении 50 В/см. Олигонуклеотидные образцы наносили на гель в растворе 8 М мочевины, содержавшем 0.05% ксиленцианола FF и 0.05% бромфенолового синего. Результаты электрофоретического разделения визуализировали при помощи красителя Stains-all.
Спектры 1Н-ЯМР регистрировали на спектрометре Spinsolve 80 (Magritek, Германия; 80 МГц).
Масс-спектрометрия. Молекулярные массы олигонуклеотидов и олигонуклеотидных аналогов определяли с помощью масс-спектрометрии (ионизация методом электрораспыления) на приборе G6410A LCMS/MS (Agilent, США). Образцы олигонуклеотидов готовили растворением в 20 мМ триэтиламмоний ацетате в 60%-ном водном ацетонитриле до концентрации 0.1 мМ и объема 10 мкл. Для анализа использовали 80%-ный водный ацетонитрил в качестве элюента при скорости потока 0.1 мл/мин в режиме регистрации отрицательно заряженных ионов.
Все реакции проводили без доступа воздуха в атмосфере аргона. Абсолютирование органических растворителей проводили стандартными методами с последующим выдерживанием над молекулярными ситами или гидридом кальция.
Синтез олигонуклеотидов осуществляли на автоматическом ДНК-синтезаторе ASM-800 (Biosset, Россия) согласно стандартному протоколу твердофазного амидофосфитного синтеза, используя коммерческие дезоксирибонуклеозидные мономеры и соответствующие пористые стекла (Glen Research, США).
Для получения олигонуклеотидов, содержащих модифицированные звенья, присоединение соответствующего мономера проводили без стадий кэпирования, окисления и деблокирования. Затем реактор вынимали из синтезатора, полимерный носитель (CPG) с иммобилизованным промежуточным продуктом переносили из реактора в пластиковую пробирку и обрабатывали раствором азида-модификатора, после чего обрабатывали промежуточный продукт 10%-ным раствором N,N-диизопропилэтиламина в ацетонитриле в течение 20 мин при 40°С. Далее переносили CPG в реактор, и все последующие обработки проводили в условиях автоматического твердофазного синтеза.
Органический синтез. Бис(пентаметилен)тиомочевина (IIIb). К раствору пиперидина (1.3 мл, 12.7 ммоль) в 6 мл ацетонитрила при перемешивании добавляли тиофосген (250 мкл, 3.1 ммоль). Реакцию проводили при комнатной температуре. Через 3 ч отделяли образовавшийся осадок фильтрованием на бумажном фильтре, фильтрат упаривали на ротационном испарителе, 2 раза соупаривали с 6 мл толуола при 45°С, затем упаривали реакционную смесь в течение 1 ч при 60°С для удаления избытка пиперидина. После высушивания в эксикаторе было получено 158 мг продукта в виде желто-оранжевого масла (выход 24%).
1Н-ЯМР (80 МГц, (CD3)2SO, δ, м.д.): 1.54 (m, 12H, N–CH2–CH2–CH2–), 3.40 (m, 8H, N–CH2–).
N,N '-дигептил-N,N '-диметилтиомочевина (IVb). К раствору N-гептилбутиламина (550 мкл, 3.2 ммоль) в 6 мл ацетонитрила при перемешивании добавляли N-диизопропилэтиламин (550 мкл, 3.2 ммоль) и тиофосген (125 мкл, 1.5 ммоль). Реакцию проводили при комнатной температуре. Через 4 ч реакционную смесь упаривали на ротационном испарителе. Очистку проводили методом колоночной хроматографии в системе 20%-ный этанол в толуоле с добавлением 1% триэтиламина. Объединенные фракции упаривали на ротационном испарителе. После высушивания в эксикаторе было получено 180 мг продукта в виде желтого масла (выход 55%).
N,N '-дидодецил-N,N '-диметилтиомочевина (Vb). К раствору N-додецилметиламина (1630 мкл, 3.2 ммоль) в смеси 4 мл гексана и 2 мл ацетонитрила при перемешивании добавляли тиофосген (125 мкл, 1.5 ммоль). Реакцию проводили при температуре 60°С. Через 20 ч реакционную смесь упаривали на ротационном испарителе. Очистку проводили методом колоночной хроматографии в дихлорметане с добавлением 1% триэтиламина. Объединенные фракции упаривали на ротационном испарителе. После высушивания в эксикаторе было получено 333 мг продукта в виде желтых кристаллов (выход 49%).
1Н-ЯМР (80 МГц, CDCl3, δ, м.д.): 0.87 (t, 6H, –N–(CH2)11–CH3), 1.25 (m, 40H, –N–CH2–(CH2)10–CH3), 2.99 (s, 6H, N–CH3), 3,51 (t, 4H, –N–CH2–(CH2)10–CH3).
Тетрагексилтиомочевина (VIb). К раствору тетрагексиламина (3 мл, 12.7 ммоль) в смеси 4 мл гексана и 2 мл ацетонитрила при перемешивании добавляли тиофосген (250 мкл, 3.1 ммоль). Реакцию проводили при температуре 60°С. Через 20 ч реакционную смесь упаривали на ротационном испарителе. Очистку проводили методом колоночной хроматографии в системе 2%-ный этилацетат в гексане с добавлением 1% триэтиламина. Объединенные фракции упаривали на ротационном испарителе. После высушивания в эксикаторе было получено 637 мг продукта в виде желтых кристаллов (выход 50%).
1Н-ЯМР (80 МГц, CDCl3, δ, м.д.): 0.87 (t, 12H, –N–(CH2)5–CH3), 1.26 (m, 32H, –N–CH2–(CH2)4–CH3), 3.45 (t, 8H, –N–CH2–(CH2)4–CH3).
Общая методика получения солей Вильсмайера и диаминокарбенийазидов (I, III–VI). К раствору соотвествующей тиомочевины или мочевины в 6 мл ацетонитрила (или дихлорметана для тиомочевин (Vb, VIb)) при перемешивании по каплям добавляли оксалихлорид (3 экв.). Реакцию проводили при комнатной температуре. Через 15 мин после завершения выделения газа добавляли твердый гексафторфосфат калия (1.1 экв.). Через 4 ч фильтровали реакционную смесь на бумажном фильтре, фильтрат упаривали на ротационном испарителе для удаления избытка оксалилхлорида, растворяли в 6 мл ацетонитрила и при перемешивании добавляли твердый азид натрия (5 экв.). Через 20 ч реакционную смесь фильтровали на бумажном фильтре и упаривали на ротационном испарителе. После высушивания в эксикаторе получали соответствующий азид в виде желтого или коричневого масла.
Гексафторфосфат азидо-N,N,N ',N '-тетраметилформамидиния (I). Получено 172 мг продукта (выход 30%). 1Н-ЯМР (80 МГц, CDCl3, δ, м.д.): 2.80 (s, 12H, –CH3).
Гексафторфосфат азидодипиперидинокарбения (III). Получено 73 мг продукта (выход 48%). 1Н-ЯМР (80 МГц, (CD3)2SO, δ, м.д.): 1.58 (m, 12H, N–CH2–CH2–CH2–), 3.25 (m, 8H, N–CH2–).
Гексафторфосфат азидо-N,N '-дигептил-N,N '-диметилформамидиния (IV). Получено 65 мг продукта (выход 24%). 1Н-ЯМР (80 МГц, (CD3)2SO, δ, м.д.): 0.85 (t, 6H, N–(CH2)6–CH3), 1.24 (m, 20H, N–CH2–(CH2)5), 2.85 (s, 6H, N–CH3), 3.07 (t, 4H, N–CH2–).
Гексафторфосфат азидо-N,N '-дидодецил-N,N '-диметилформамидиния (V). Получено 162 мг продукта (выход 36%). 1Н-ЯМР (80 МГц, CDCl3, δ, м.д.): 0.87 (t, 6H, –N–(CH2)11–CH3), 1.25 (m, 40H, –N–CH2–(CH2)10–CH3), 2.85 (s, 6H, N–CH3), 3.45 (t, 4H, –N–CH2–(CH2)10–CH3).
Гексафторфосфат азидо-N,N,N ',N '-тетрагексилформамидиния (VI). Получено 337 мг продукта (выход 38%). 1Н-ЯМР (80 МГц, CDCl3, δ, м.д.): 0.87 (t, 12H, –N–(CH2)5–CH3), 1.26 (m, 32H, –N–CH2–(CH2)4–CH3), 3.35 (t, 8H, –N–CH2–(CH2)4–CH3).
Гексафторфосфат азидодипирролидинокарбения (II). К раствору гексафторфосфата хлородипирролидинокарбения (350 мг, 1 ммоль) в 6 мл сухого ацетонитрила добавляли твердый азид натрия (260 мг, 5 ммоль). Реакцию проводили при комнатной температуре. Через 2 ч реакционную смесь фильтровали на бумажном фильтре, фильтрат упаривали на ротационном испарителе. После высушивания в эксикаторе было получено 280 мг продукта в виде белых кристаллов (выход 82%).
1Н-ЯМР (80 МГц, (CD3)2SO, δ, м.д.): 1.91 (m, 8H, N–CH2–CH2–), 3.67 (m, 8H, N–CH2–).
ЗАКЛЮЧЕНИЕ
В данной работе была предложена синтетическая схема, позволяющая получать широкий набор тетразамещенных диаминокарбенийазидов из вторичных аминов. В случае использования коммерчески доступных мочевин или солей Вильсмайера схема синтеза может быть сокращена, однако в каталогах можно найти лишь ограниченное количество таких реагентов, тогда как широкий набор доступных вторичных аминов позволяет получать большое количество различных азидов.
С использованием данной схемы был синтезирован набор азидов-модификаторов, содержащих алкильные заместители различной длины, которые были использованы для введения модификаций в состав олигонуклеотидов по реакции Штаудингера. Было показано, что эффективность введения модификации падает с увеличением размера алкильных заместителей. Высокие значения конверсии в случае коммерческого азида ADMP, а также азида (II), полученного в одну стадию из соли Вильсмайера, свидетельствуют о существенном влиянии даже незначительного количества примесей в используемом азиде на эффективность протекания реакции.
Предварительное элиминирование цианэтильной группы в безводных условиях позволяет существенно увеличить эффективность введения модификации за счет превращения фосфазенового остатка в стабильный фосфорилгуанидин до финального деблокирования олигонуклеотида водным раствором аммиака.
Список литературы
Yin W., Rogge M. // Cts-Clin. Transl. Sci. 2019. V. 12. P. 98–112. https://doi.org/10.1111/cts.12624
Heo Y.A. // Drugs. 2020. V. 80. P. 329–333. https://doi.org/10.1007/s40265-020-01267-2
Balwani M., Sardh E., Ventura P., Peiro P.A., Rees D.C., Stolzel U., Bissell D.M., Bonkovsky H.L., Windyga J., Anderso, K.E., Parker C., Silver S.M., Keel S.B., Wang J.D., Stein P.E., Harper P., Vassiliou D., Wang B., Phillips J., Ivanova A., Langendonk J.G., Kauppinen R., Minder E., Horie Y., Penz C., Chen J.H., Liu S.B., Ko J.J., Sweetser M.T., Garg P., Vaishnaw A., Kim J.B., Simon A.R., Gouya L., Investigators E. // New Engl. J. Med. 2020. V. 382. P. 2289–2301. https://doi.org/10.1056/NEJMoa1913147
Rinaldi C., Wood M.J.A. // Nat. Rev. Neurol. 2018. V. 14. P. 9–21. https://doi.org/10.1038/nrneurol.2017.148
Deleavey G.F., Damha M.J. // Chem. Biol. 2012. V. 19. P. 937–954. https://doi.org/10.1016/j.chembiol.2012.07.011
Roy S., Caruthers M. // Molecules. 2013. V. 18. P. 14 268–14 284. https://doi.org/10.3390/molecules181114268
Kupryushkin M.S., Pyshnyi D.V., Stetsenko D.A. // Acta Naturae. 2014. V. 6. P. 116–118. https://doi.org/10.32607/20758251-2014-6-4-116-118
Dyudeeva E.S., Kupryushkin M.S., Lomzov A.A., Pyshnaya I.A., Pyshnyi D.V. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 709–718. https://doi.org/10.1134/S1068162019060153
Stetsenko D., Kupryshkin M., Pyshnyi D. // Patent WO2016028187A1, 2016.
Wieland G., Simchen G. // Liebigs Ann. Chem. 1985. V. 1985. P. 2178–2193. https://doi.org/10.1002/jlac.198519851108
Kitamura M., Murakami K. // Org. Synth. 2015. V. 92. P. 171. https://doi.org/10.15227/orgsyn.092.0171
El-Faham A. // Org Prep. Proced. Int. 1998. V. 30. P. 477–481. https://doi.org/10.1080/00304949809355316
Bazhenov M.A., Shernyukov A.V., Kupryushkin M.S., Pyshnyi D.V. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 699–708. https://doi.org/10.1134/S1068162019060074
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия