

Биоорганическая химия, 2021, T. 47, № 4, стр. 464-471
Фосфолипазы с бактерий рода Bacillus: биологическая роль, свойства и области применения
Ю. А. Меркульева 1, 2, Д. Н. Щербаков 1, 2, Е. А. Шарлаева 1, В. Ю. Чиркова 1, *
1 Алтайский государственный университет
656049 Барнаул, просп. Ленина, 61, Россия
2 Государственный научный центр вирусологии и биотехнологии “Вектор”
630559 Кольцово, Новосибирская обл., Россия
* E-mail: varvara.chirkova@gmail.com
Поступила в редакцию 19.07.2020
После доработки 20.08.2020
Принята к публикации 26.08.2020
Аннотация
Фосфолипазы – ферменты, катализирующие гидролитическое расщепление связей в фосфолипидах, обнаружены почти у всех организмов. Наибольший интерес вызывают фосфолипазы микробного происхождения. Популярность бактериальных ферментов обусловлена их огромным разнообразием и технологическими свойствами: высокой удельной активностью, термостабильностью, широкой субстратной специфичностью. Получение рекомбинантных бактериальных фосфолипаз и их совершенствование остаются актуальными задачами, для решения которых необходимы углубление и систематизация знаний о ферментах данной группы. В настоящем обзоре описаны свойства, структура и механизм действия бактериальных фосфолипаз C, которые получили широкое применение в различных областях практической деятельности человека: научных исследованиях, медицине, пищевой, химической промышленности и др.
ВВЕДЕНИЕ
Фосфолипазы (PL) (КФ 3.1.1, 3.1.4) – ферменты, осуществляющие гидролиз различных связей в фосфолипидах (рис. 1) и которые наряду с гликолипидами и холестерином относятся к основным компонентам биологических мембран. Действуя на поверхности раздела “липид–вода”, фосфолипазы выступают биокатализаторами межфазного катализа [1, 2]. Данные ферменты широко распространены в природе и выполняют разнообразные функции: участвуют в поддержании липидного состава мембран, играют существенную роль в развитии воспалительного процесса, запуская синтез медиаторов воспаления, принимают участие в работе инозитолфосфатной системы, обеспечивающей трансмембранную передачу гормональных сигналов, выступают активными компонентами змеиного яда гемолитического действия и др. [3, 4].
Рис. 1.
Общее строение фосфолипидов: заместители R1 и R2 – остатки жирных кислот, X – головная группа: холин, этаноламин, глицерол, инозитол или серин.

В зависимости от сайта расщепления связи в молекуле фосфолипида выделяют четыре основных семейства фосфолипаз: A, B, C и D (рис. 2) [5]. Фосфолипазы типов А и В – ацилгидролазы, типов C и D – фосфодиэстеразы. Фосфолипазы A1 (PLA1) и A2 (PLA2) гидролизуют SN-1 или SN-2 ацильную цепь с высвобождением свободной жирной кислоты и 1- или 2-ацил-лизофосфолипида соответственно. В случае отщепления обеих жирных кислот говорят о фосфолипазе типа B (PLB).
Рис. 2.
Реакции, катализируемые фосфолипазами: ЖК – жирная кислота, ФК – фосфатидная кислота, ДАГ – диацилглицерол, X – головная группа, P-X – фосфорилированная головная группа [5].
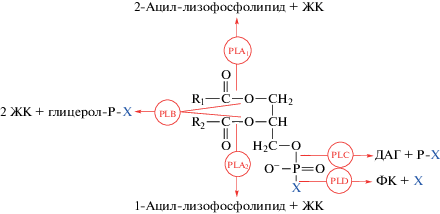
Фосфолипаза C (PLC) расщепляет глицерофосфатную связь с образованием диацилглицерола и фосфат-содержащей полярной группы, а фосфолипаза D (PLD) гидролизует связь между фосфатной и спиртовой группами, при этом высвобождаются фосфатидная кислота и спирт.
В зависимости от молекулярной массы, клеточной локализации, способа регуляции и субстратной специфичности выделяют несколько изоформ для каждого типа фосфолипаз [3, 4, 6]. Например, в клетках человека идентифицированы 10 изоформ фосфолипазы C, а у вирусов, бактерий, дрожжей, слизевиков, растений и млекопитающих обнаружены различные изоформы фосфолипазы D [7].
БАКТЕРИАЛЬНЫЕ ФОСФОЛИПАЗЫ
Большое разнообразие фосфолипаз продуцируется патогенными и непатогенными бактериями. Некоторые из бактериальных фосфолипаз проявляют свойства токсинов, другие, не будучи токсинами, играют важную роль в патогенезе заболеваний. Токсический эффект фосфолипаз проявляется как непосредственно в лизисе, так и в изменении метаболизма клеток хозяина. Накапливающиеся продукты их катализа – лизофосфолипиды – обладают сильными поверхностно-активными свойствами, что приводит к разрушению липопротеиновых структур клетки и активации гидролаз, ответственных за автолитический распад клеточных полимеров [8, 9].
У микроорганизмов обнаружены все типы фосфолипаз, различающиеся по положению гидролизуемой связи. Фосфолипаза A1 (КФ 3.1.1.32), отщепляющая SN-1 ацильную цепь, обладает широкой субстратной специфичностью, кофактором выступает Ca2+ [10]. PLA1 обнаружена у бактерий Serratia spp., Yersinia enterocolitica, Streptomyces alboflavus, Escherichia coli, Bacillus subtilis, B. megaterium, Mycobacterium phlei [11, 12]. Фосфолипаза A1 грамотрицательных бактерий выступает одним из факторов вирулентности, что вызывает повышенный интерес к изучению этой фосфолипазы. Она усиливает гемолитические свойства клеток бактерий и повышает их инвазивность [13].
Фосфолипаза A2 (КФ 3.1.1.4), отщепляющая SN-2 ацильную цепь, действует на фосфатидилэтаноламин, холинплазмалоген и фосфатиды. Кофактором также выступает Ca2+ [14]. PLA2 обнаружена у бактерий E. coli, Streptomyces coelicolor, St. violaceoruber, Helicobacter pylori [15], Yersinia enterocolitica [16]. У клеток E. coli фосфолипаза A2 увеличивает уровень лизофосфолипидов и жирных кислот в мембране, повышая ее проницаемость и участвуя таким образом в выбросе токсина бактериоцина из клетки.
Фосфолипазу B (L) (КФ 3.1.1.5) также называют лизофосфолипаза. Фермент действует на лизолецитин (лизофосфатидилхолин), образующийся в результате действия фосфолипазы А1 на лецитин (фосфатидилхолин) [17]. PLB обладает активностями фосфолипаз А1 и А2 – отщепляет обе SN-1 и SN-2 ацильные цепи фосфолипида. Фосфолипаза B не имеет кофактора. Ингибиторами для некоторых PLB служат диизопропилфторфосфат и п-хлормеркурбензойная кислота, для всех без исключения – поверхностно-активные вещества. Фосфолипаза B обнаружена у Pseudomonas fluorescens, Bacillus subtilis, Streptomyces sp., Mycoplasma laidlawii, M. phlei, Serratia plymuthica, Dictyostelium discoideum [18–20].
Фосфолипаза C (КФ 3.1.4.3) гидролизует глицерофосфатную связь, что приводит к образованию диацилглицерола и фосфат-содержащей полярной группы. PLC – ключевой фермент метаболизма фосфатидилинозитола и липидных сигнальных путей. Она гидролизует фосфатидилинозитол на два вторичных медиатора – инозитолтрифосфат и диацилглицерол, которые, вовлекаясь в сигнальные пути, активируют кальциевые каналы эндоплазматического ретикулума и протеинкиназу С соответственно. Кофактором данного фермента выступает Zn2+ [21]. Фосфолипаза С обнаружена у Listeria monocytogenes, Clostridium perfringens, Bacillus cereus, B. mycoides, B. аnthracis, Pseudomonas aeruginosa, P. cepacia, P. fluorescens, Burkholderia pseudomallei, Legionella pneumophila, Acinetobacter calcoaceticus, Staphylococcus aureus [22, 23]. Активность PLC зависит не только от состава фосфолипидов клеточных мембран, но и от компонентов среды, например, снижение активности фосфолипазы С микроорганизмов Clostridium perfringens и Bacillus cereus наблюдается под действием фосфат- и глицерол-содержащих соединений, возможно, за счет их конкуренции с субстратом за соответствующие центры связывания с ферментом [2, 24–26].
Фосфолипаза D (КФ 3.1.4.4) гидролизует связь между фосфатной и спиртовой группами фосфатидилхолина, при этом высвобождаются фосфатидная кислота и растворимый холин [27]. Активаторы PLD – анионные поверхностно-активные вещества, а ингибиторы – катионные. Фосфолипаза D обнаружена у Acinetobacter baumanii, E. coli, Neisseria gonorrhoeae, Yersinia pestis, Chlamydia trachomatis, Pseudomonas aeruginosa, Streptomyces sp. PMF, Rickettsiа conorii, R. рrowazekii [28].
ФОСФОЛИПАЗА С БАКТЕРИЙ РОДА Bacillus: СТРУКТУРА, СВОЙСТВА, МЕХАНИЗМЫ ДЕЙСТВИЯ
Фосфолипаза С, катализирующая стереоспецифический гидролиз фосфолипидов, обнаружена у широкого спектра грамположительных и некоторых грамотрицательных бактерий. Бактериальные фосфолипазы С, представляющие собой мономерные белки с типичными сигнальными последовательностями в структуре и секретирующиеся во внеклеточное пространство [6], по схожести структур принято разделять на несколько групп (табл. 1).
Таблица 1.
Группы бактериальных фосфолипаз по схожести структур
| Источник фосфолипаз | Группа | Микроорганизмы |
|---|---|---|
| Грамположительные бактерии | Zn-зависимые металлофосфолипазы C (α-токсин, BC-PLC) |
Clostridium perfringens Bacillus cereus |
| Сфингомиелиназы | Bacillus cereus Staphylococcus aureus |
|
| Фосфолипазы С, гидролизующие фосфатидилинозитол (PLC-A) |
Bacillus cereus Bacillus thuringiensis Listeria monocytogenes |
|
| Грамотрицательные бактерии | Фосфолипазы С Pseudomonas sp. (PLC-H и PLC-N) |
Pseudomonas aeruginosa |
| Фосфолипазы С Legionella sp. | Legionella sp. |
Гены, кодирующие α-токсин Clostridium perfringens, PLC Bacillus cereus, PLC из Clostridium bifermentans и Listeria monocytogenes, были секвенированы. Для них показана высокая степень гомологии – общие ~250 первых аминокислот. У α‑токсина Clostridium perfringens, в отличие от других фосфолипаз грамположительных бактерий, имеется 120 дополнительных аминокислотных остатков на C-конце. PLC со специфичностью к фосфатидилхолину (PC-PLC) B. thuringiensis и B. cereus, входящие в одну группу, имеют высокую степень гомологии.
Наиболее изучены структура и свойства PC-PLC Bacillus cereus. В результате обширных исследований этот белок получил статус прототипа фосфолипазы С [29]. Фермент – небольшой белок (28 кДа), довольно устойчивый к воздействию денатурирующих агентов: мочевине, додецилсульфату натрия и температуре (в присутствии 1 мМ Zn2+) [30]. Благодаря стабилизирующим ионам цинка, белок обладает способностью выдерживать нагрев до 100°C в течение коротких периодов времени [31]. Оптимум действия фермента наблюдается при температуре 50°С и рН 8–10. Фосфолипаза C обладает высокой стабильностью, а также толерантностью к замене ионов Zn2+ на другие двухвалентные катионы с сохранением (Co2+, Ni2+, Mg2+, Ca2+, Ba2+) либо увеличением (Mn2+) активности фермента [31, 32].
Фермент представляет собой единственную полипептидную цепь, свернутую в виде семи спиралей, образующих скрученную структуру (рис. 3а). PLC Bacillus spp. синтезируется с сигнальной последовательностью на N-конце (24 а.о.), секретируется во внеклеточное пространство в форме пропептида. Активная форма фермента (245 а.о.) образуется при отщеплении 14 N-концевых аминокислотных остатков клеточными протеазами (рис. 3б) [33–35].

В активном центре расположены три иона Zn2+, один из которых слабо связан. Ионы Zn2+ поддерживают конформационную стабильность фермента и участвуют в связывании субстрата независимо от его количества [36]. Определены 9 а.о., участвующих в связывании ионов: 5 His, 2 Asp, 1 Glu и 1 Trp. Считается, что первая аминокислота (Trp) в зрелом пептиде – незаменима для ферментативной активности, поскольку она способствует координации с основными ионами цинка. Ионы связываются с аминокислотными остатками из разных спиралей и, следовательно, стабилизируют конформацию молекулы.
PLC бактерий рода Bacillus обладает широкой субстратной специфичностью – распознает разные фосфолипидные субстраты, которые отличаются только структурой головной группы: фосфатидилхолин, фосфатидилэтаноламин, фосфатидилинозитол, фосфатидилглицерол, фосфатидилсерин (рис. 4) [37–39]. Фосфатидилхолин с шестью атомами углерода в каждой из ацильных боковых цепей подвергается воздействию фермента с большей каталитической эффективностью, чем субстрат, содержащий С2–С4 жирные кислоты [40, 41]. Пространственная ориентация боковых цепей глицерола на фосфатидилхолине, по-видимому, – важный фактор, способствующий связыванию и катализу [42].
Рис. 4.
Спектр субстратов для фосфолипазы C B. cereus: (a) – общее строение фосфолипида; (б) – головная группа [37].

Эукариотические и бактериальные фосфолипазы имеют общий механизм реакции – “пинг-понг” с промежуточным звеном, в ходе которого субстратная фосфатная группа ковалентно связывается с нуклеофильным аминокислотным остатком из активного центра [23]. Остатки аминокислот, составляющие активный сайт (Glu4, Tyr56 и Phe66), образуют каркас для связывания с основанием субстрата [43]. Карбоксильная группа Glu4 взаимодействует с атомом азота в головной группе холина с помощью полярной или ионной связи, а Phe66 – через катион-π-взаимодействие [44, 45]. Предполагается, что Tyr56 может стабилизировать положительный заряд на ингибиторе или субстрате и, по-видимому, определяет специфичность фермента [46].
В отношении молекулярного механизма действия PLC B. cereus существуют две точки зрения. Согласно первой, Asp55 играет роль основания, которое атакует нуклеофильную молекулу воды посредством депротонирования; вода, в свою очередь, запускает атаку на фосфор фосфолипида (рис. 5a) [38, 41].

Согласно второй точке зрения, Zn1 и Zn3 инициируют нуклеофильную атаку на фосфор, в то время как Zn2 активирует молекулу воды для протонирования уходящей группы (рис. 5б) [47, 48].
ПОЛУЧЕНИЕ БАКТЕРИАЛЬНЫХ ФОСФОЛИПАЗ С
В качестве источников получения ферментов могут выступать растения, животные и микроорганизмы. Но выделять ферменты из растительных или животных источников экономически и технологически менее выгодно, чем получать их с помощью микроорганизмов. Использование E. coli позволяет нарабатывать большое количество неактивной формы данного фермента в тельцах включения. Для получения активного фермента дополнительно осуществляют стадии разрушения клеток, очистки, рефолдинга и активации белка, что повышает стоимость конечного продукта [49].
Первая попытка внеклеточного синтеза PLC была проведена с использованием системы Pichia pastoris. N-Конец фермента объединяли с сигнальным пептидом α-фактора из Saccharomyces cerevisiae для секреции и His6-тегом, с дальнейшей очисткой после отщепления сигнального пептида [50]. Однако культивирование этого продуцента требует присутствия метанола в качестве индуктора и дорогих сред, что делает его использование невыгодным. Ферментативная активность PLC зависит от цинка, однако активность роста P. pastoris ингибируется избытком Zn2+ (5 мМ). Учитывая это, а также длительность культивирования P. pastoris (несколько суток), для получения PLC в промышленных масштабах предпринимаются попытки использовать экспрессионную систему B. subtilis. Как правило, для секреции применяется сигнальная последовательность α-амилазы amyE B. subtilis [51].
ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ ФОСФОЛИПАЗ С
Бактериальные фосфолипазы С используются для исследования механизмов активации арахидоновой кислоты и протеинкиназы С в клетках млекопитающих. Кроме того, они могут выступать в качестве реагентов для изучения структуры клеточных мембран, например, мембран эритроцитов [52, 53]. В отличие от многих бактериальных токсинов, для PLC не требуется интернализация белка, что привлекло внимание ученых к исследованию возможности применения фермента для доставки лекарственных средств. PLC, связанная с подходящим антителом, может составлять основу активного цитотоксического агента. На сегодняшний день фосфолипаза С считается одним из ключевых ферментов системы доставки лекарств по пути эндоцитоза, поскольку может катализировать мембранное слияние между клеточными мембранами и фосфолипидами средств доставки – липосом [54].
PLC имеет терапевтический потенциал при тромбогеморрагическом синдроме, т.к. способна инактивировать тромбопластин, запускающий процесс свертывания крови [31]. Гемолитическая фосфолипаза С (α-токсин) Clostridium perfringens играет основную роль в патогенезе газовой гангрены, в связи с чем выступает перспективным компонентом вакцины против данного заболевания [23].
В пищевой промышленности фосфолипаза C используется для рафинирования масел: соевого, пальмового, подсолнечного, рапсового и др. Неочищенное растительное масло состоит преимущественно из триглицеридов, но может содержать до 3% фосфолипидов, которые вызывают потемнение масла и изменение вкуса при хранении [55]. Для удаления фосфолипидов при рафинировании используют фосфолипазы. Участие PLС в процессе рафинирования не приводит к образованию свободных жирных кислот, которые необходимо удалять. Это позволяет сократить потери сырья, поэтому использование PLC предпочтительнее, чем PLА. В настоящее время для производства рафинированного растительного масла предложен препарат PLC из B. anthracis (Purifine™, Verenium) [56]. PLC из B. cereus – оптимальный фермент для использования в промышленном рафинировании растительных масел. Он обладает высокой удельной активностью, наиболее подходящим спектром субстратной специфичности и термостабильностью [5].
Фосфолипазу C применяют также для предварительной обработки масла при получении биодизельного топлива. При одновременном использовании PLC и лизофосфолипаз наблюдается эффект синергии, который делает ферментативный процесс производства биодизельного топлива более экономичным и экологичным. Сочетание рафинирования и трансэтерификации высвободившихся жирных кислот в этом процессе возможно осуществлять с одновременным использованием фосфолипаз и жидкой липазы Callera Trans L. [57].
При формировании структурно-механических свойств молока, а также в процессах созревания сыров важную роль играют фосфолипиды, в связи с чем фосфолипазы находят широкое применение в сыроделии. В настоящее время используют коммерческий препарат PLA1 из A. oryzae (YieldMAX® PL, Novozymes), PLC B. cereus и PLD St. сhromofuscus. При производстве сыра большая часть фосфолипидов удаляется вместе с сывороткой. При использовании фосфолипаз в результате гидролиза фосфолипидов удаляется только полярная группа, что позволяет увеличить выход продукта [58].
Еще одна перспективная область использования фосфолипазы С – получение диацилглицеридов с определенной энантиомерной структурой, которые находят применение в синтезе стереоспецифичных соединений, а также выступают клеточными медиаторами [59]. Последовательное действие двух классов фосфолипаз С и D может быть использовано, например, для синтеза дигидроксиацетонфосфата (DHAP) – интермедиата тонкого химического синтеза, или такого важного для медицины и фармацевтики биоактивного соединения, как сфингозин-1-фосфат (S1P), – главного регулятора сосудистой и иммунной системы [60, 61].
Повышение эффективности PLC может быть достигнуто путем иммобилизации фермента, т.к. катализаторы, полученные путем ковалентного присоединения, имеют более высокую удельную активность и пригодны для многократного использования [62].
ЗАКЛЮЧЕНИЕ
Бактериальные фосфолипазы не только играют важную роль в биологических процессах, но и находят широкое применение в научных исследованиях, медицине, пищевой, химической и других отраслях промышленности. Полученные в настоящее время рекомбинантные ферменты отличаются улучшенными технологическими свойствами (высокой удельной активностью, термостабильностью, широкой субстратной специфичностью), что открывает дополнительные возможности для их использования, поэтому создание эффективных продуцентов фосфолипаз на основе непатогенных бактерий остается важной биотехнологической задачей.
Список литературы
Aloulou A., Ali Y.B., Bezzine S., Gargouri Y., Gelb M.H. // Methods in Molecular Biology. 2012. V. 861. P. 63–85. https://doi.org/10.1007/978-1-61779-600-5_4
Литвинко Н.М. // Известия Национальной академии наук Беларуси. Серия химических наук. 2015. № 4. С. 109–121.
Ulbrich-Hofmann R. // ChemBioChem. 2012. V. 13. P. 2148–2149. https://doi.org/10.1002/cbic.201200542
Филькин C.Ю., Липкин А.В., Федоров А.Н. // Усп. биол. химии. 2020. № 60. С. 369–410.
Borrelli G.M., Trono D. // Int. J. Mol. Sci. 2015. V. 16. P. 20774–20840. https://doi.org/10.3390/ijms160920774
Titball R.W. // Symp. Ser. Soc. Appl. Microbiol. 1998. V. 27. P. 127S–137S.
Ramenskaia G.V., Melnik E.V., Petukhov A.E. // Biomed. Khim. 2018. V. 64. P. 84–93. https://doi.org/10.18097/PBMC20186401084
Titball R.W. // Microbiol. Rev. 1993. V. 57. P. 347–366.
Songer J.G. // Trends Microbiol. 1997. V. 5. P. 156–161.
Scandella C.J., Kornberg A. // Biochemistry. 1971. V. 10. P. 4447–4456.
Richmond G.S., Smith T.K. // Int. J. Mol. Sci. 2011. V. 12. P. 588–612. https://doi.org/10.3390/ijms12010588
Murayama K., Kano K., Matsumoto Y., Sugimori D. // J. Struct. Biol. 2013. V. 182. P. 192–196. https://doi.org/10.1016/j.jsb.2013.02.003
Bakholdina S.I., Tischenko N.M., Sidorin E.V., Isaeva M.P., Likhatskaya G.N., Dmitrenok P.S., Kim N.Yu., Chernikov O.V., Solov’eva T.F. // Biochemistry. 2016. V. 81. P. 47–57. https://doi.org/10.1134/s0006297916010053
Hanahan D.J., Brockerhoff H., Barron E.J. // J. Biol. Chem. 1960. V. 235. P. 1917–1923.
Ishiwata S. // Dainihon Sanshi Kaiho. 1901. V. 114. P. 1–5.
Köhler G.A., Brenot A., Haas-Stapleton E., Agabian N., Deva R., Nigam S. // Biochim. Biophys. Acta. 2006. V. 1761. P. 1391–1399. https://doi.org/10.1016/j.bbalip.2006.09.011
Saito K., Sugatani J., Okumura T. // Methods in Enzymology. 1991. V. 197. P. 446–456. https://doi.org/10.1016/0076-6879(91)97170-4
Jiang F., Huang S., Imadad K., Li C. // Bioresour. Technol. 2012. V. 104. P. 518–522. https://doi.org/10.1016/j.biortech.2011.09.112
Matsumoto Y., Mineta S., Murayama K., Sugimori D. // FEBS J. 2013. V. 280. P. 3780–3796. https://doi.org/10.1111/febs.12366
Masayama A., Kato S., Terashima T., Mølgaard A., Hhemmi H., Yoshimura T., Moriyama R. // Biosci. Biotechnol. Biochem. 2010. V. 74. P. 24–30. https://doi.org/10.1271/bbb.90391
Taguchi R., Ikezawa H. // Arch. Biochem. Biophys. 1978. V. 186. P. 196–201.
Djordjevic J.T. // Front. Microbiol. 2010. V. 1. P. 1–13. https://doi.org/10.3389/fmicb.2010.00125
Pokotylo I., Pejchar P., Potocký M., Kocourková D., Krčková Z., Ruelland E., Kravets V., Martinec J. // Prog. Lipid Res. 2013. V. 52. P. 62–79. https://doi.org/10.1016/j.plipres.2012.09.001
Ивинскене В.Л. // Энтомопатогенные бактерии и их роль в защите растений: сб. науч. тр. ВАСХНИЛ. Сиб. отд-ние. Новосибирск, 1987. С. 57–75.
Ikezawa H., Nakabayashi T., Suzuki K., Nakajima M., Taguchi T., Taguchi R. // J. Biochem. 1983. V. 93. P. 1717–1719. https://doi.org/10.1093/oxfordjournals.jbchem.a134315
Volwerk J.J., Koke J.A., Wetherwax P.B., Griffith O.H. // FEMS Microbiol. Lett. 1989. V. 61. P. 237–241. https://doi.org/10.1111/j.1574-6968.1989.tb03629.x
Jenkins M.G., Frohman M.A. // Cell. Mol. Life Sci. 2005. V. 62. P. 2305–2316. https://doi.org/10.1007/s00018-005-5195-z
Selvy P.E., Lavieri R.R., Lindsley C.W., Brown H.A. // Chem. Rev. 2011. V. 111. P. 6064–6119. https://doi.org/10.1021/cr200296t
Sakurai J., Nagahama M., Oda M. // J. Biochem. 2004. V. 136. P. 569–574. https://doi.org/10.1093/jb/mvh161
González-Bulnes P., González-Roura A., Canals D., Delgado A., Casas J., Llebaria A. // Bioorg. Med. Chem. 2010. V. 18. P. 8549–8555. https://doi.org/10.1016/j.bmc.2010.10.031
Otnaess A.-B., Little C., Sletten K., Wallin R., Johnsen S., Flengsrud R., Prydz, H. // Eur. J. Biochem. 1977. V. 79. P. 459–468. https://doi.org/10.1111/j.1432-1033.1977.tb11828.x
Elleboudy N.S., Aboulwafa M.M., Hassouna N.A. // Asian Pacific J. Trop. Med. 2014. V. 7. P. 860–866. https://doi.org/10.1016/s1995-7645(14)60150-4
Hough E., Hansen L.K., Birknes B., Jynge K., Hansen S., Hordvik A., Little C., Dodson E., Derewenda Z. // Nature. 1989. V. 338. P. 357–360. https://doi.org/10.1038/338357a0
Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlić A., Rose P.W. // Proceedings of the 21st International Conference on Web3D Technology – Web3D’16. 2016. P. 185–186. https://doi.org/10.1145/2945292.2945324
Rose A.S., Hildebrand P.W. // Nucleic. Acid Res. 2015. V. 43. P. W576–W579. https://doi.org/10.1093/nar/gkv402
Beecher D.J., Wong A.C.L. // Microbiology. 2000. V. 146. P. 3033–3039. https://doi.org/10.1099/00221287-146-12-3033
Lyu Y., Ye L., Xu J., Yang X., Chen W., Yu H. // Biotechnol. Lett. 2016. V. 38. P. 23–31. https://doi.org/10.1007/s10529-015-1962-6
Antikainen N.M., Hergenrother P.J., Harris M.M., Corbett W., Martin S.F. // Biochem. 2003. V. 42. P. 1603–1610. https://doi.org/10.1021/bi0267285
Shinitzky M., Friedman P., Haimovitz R. // J. Biol. Chem. 1993. V. 268. P. 14109–14115.
El-Sayed M.Y., DeBose C.D., Coury L.A., Roberts M.F. // Biochim. Biophys. Acta. 1985. V. 837. P. 325–335. https://doi.org/10.1016/0005-2760(85)90056-6
Martin S.F., Follows B.C., Hergenrother P.J., Trotter B.K. // Biochemistry. 2000. V. 39. P. 3410–3415. https://doi.org/10.1021/bi9919798
Snyder W.R. // Biochim. Biophys. Acta. 1987. V. 920. P. 155–160.
Benfield A.P., Goodey N.M., Phillips L.T., Martin S.F. // Arch. Biochem. Biophys. 2007. V. 460. P. 41–47.
Burley S.K., Petsko G.A. // Advances in Protein Chemistry. 1988. V. 39. P. 125–189. https://doi.org/10.1016/s0065-3233(08)60376-9
Dougherty D.A. // Science. 1996. V. 271. P. 163–168. https://doi.org/10.1126/science.271.5246.163
Celandroni F., Salvetti S., Senesi S., Ghelardi E. // FEMS Microbiol. Lett. 2014. V. 361. P. 95–103. https://doi.org/10.1111/1574-6968.12615
Sundell S., Hansen S., Hough E. // Protein Eng. 1994. V. 7. P. 571–577. https://doi.org/10.1093/protein/7.4.571
Liao R.Z., Yu J.G., Himo F. // Phys. Chem. B. 2010. V. 114. P. 2533–2540. https://doi.org/10.1021/jp910992f
Martin S.F., Spaller M.R., Hergenrother P.J. // Biochem. 1996. V. 35. P. 12970–12977. https://doi.org/10.1021/bi961316
Seo K.H., Rhee J.I. // Biotechnol. Lett. 2004. V. 26. P. 1475–1479. https://doi.org/10.1023/b:bile.0000044447.15205.90
Durban M.A., Silbersack J., Schweder T., Schauer F., Bornscheuer U.T. // Appl. Microbiol. Biotechnol. 2007. V. 74. P. 634–639. https://doi.org/10.1007/s00253-006-0712-z
Kent C., Evers A., Haun S.S.L. // Arch. Biochem. Biophys. 1986. V. 250. P. 519–525.
Parkinson E.K. // Carcinogenesis. 1987. V. 8. P. 857–860. https://doi.org/10.1093/carcin/8.6.857
Shimanouchi T., Kawasaki H., Fuse M., Umakoshi H., Kuboi R. // Colloids Surf. B Biointerfaces. 2013. V. 103. P. 75–83.
Mounts T.L., Nash A.M. // J. Am. Oil Chem. Soc. 1990. V. 67. P. 757–760. https://doi.org/10.1007/bf02540486
De Maria L., Vind J., Oxenboll K.M., Svendsen A., Patkar S. // Appl. Microbiol. Biotechnol. 2007. V. 74. P. 290–300. https://doi.org/10.1007/s00253-006-0775-x
Cesarini S., Haller R.F., Diaz P., Nielsen P.M. // Biotechnol. Biofuels. 2014. V. 7. P. 1–12. https://doi.org/10.1186/1754-6834-7-29
Casado V., Martín D., Torres C., Reglero G. // Lipases and Phospholipases: Methods and Protocols. New York: Springer, 2012. V. 861. P. 495–523.
Arrigo P.D., Servi S. // Trends in Biotechnology. 1997. V. 15. P. 90–96. https://doi.org/10.1016/s0167-7799(97)01012-3
Schümperli M., Pellaux R., Panke S. // Appl. Microbiol. Biotechnol. 2007. V. 75. P. 33–45. https://doi.org/10.1007/s00253-007-0882-3
Morigaki E., Miura Y., Takahata K., Tada M., Nakajima S., Baba N. // J. Chem. Res. 1998. V. 12. P. 774–775. https://doi.org/10.1039/a804770g
Anthonsen T., D’Arrigo P., Pedrocchi-Fantoni G., Secundo F., Servi S., Sundby E. // J. Mol. Catal. B Enzym. 1999. V. 6. P. 125–132. https://doi.org/10.1016/s1381-1177(98)00141-6
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия