

Биоорганическая химия, 2021, T. 47, № 5, стр. 610-621
Генно-инженерный синтез и свойства гиспидин-3-гидроксилазы – ключевого фермента биосинтеза люциферина грибов
А. С. Герасимов 1, 2, *, С. О. Рогожкин 2, Е. С. Шахова 1, Т. В. Чепурных 1, А. Ю. Гороховатский 1, Н. М. Мышкина 1, А. В. Балакирева 1, И. В. Ямпольский 1
1 ФГБУН “Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова” РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
2 Вятский государственный университет
610000 Киров, ул. Московская, 36, Россия
* E-mail: asgerasimoff@mail.ru
Поступила в редакцию 28.10.2020
После доработки 11.11.2020
Принята к публикации 16.11.2020
Аннотация
Биолюминесценция – это способность живых организмов испускать свет. В ее основе лежит биохимическая реакция окисления субстрата – люциферина, катализируемая ферментом люциферазой. Гриб Neonothopanus nambi – первый эукариотический организм, для которого была расшифрована биолюминесцентная система: были установлены структура люциферина, ген люциферазы, а также описаны интермедиаты и ферменты биосинтеза люциферина. Гиспидин-3-гидроксилаза (nnH3H) представляет собой один из ключевых ферментов данного каскада, катализирующий превращение гиспидина (предшественника люциферина) в 3-гидроксигиспидин – люциферин грибов. Для полного понимания механизма и субстратной специфичности nnH3H необходимо провести структурные исследования молекулы белка. Для этого требуется разработать протокол получения высокоочищенного препарата функционально активной nnH3H в достаточных количествах. Мы показали, что коэкспрессия гена гибридного белка SUMO-nnH3H с геном шаперонина GroEL/ES в клетках бактерии Escherichia coli позволяет получить фермент в растворимой и активной форме с выходом 20 мг со 100 мл бактериальной культуры. Также мы впервые продемонстрировали, что FAD – кофактор гиспидин-3-гидроксилазы грибов.
ВВЕДЕНИЕ
Биолюминесценция – это способность живых организмов испускать свет. В основе биолюминесценции лежит биохимическая реакция окисления субстрата – люциферина, катализируемая ферментом люциферазой. На сегодняшний день известны более сорока различных биолюминесцентных систем, однако только для некоторых из них были установлены структуры люцифераз и люциферинов [1, 2]. Для еще меньшего числа были охарактеризованы пути их биосинтеза и участвующие в них гены. Гриб Neonothopanus nambi – первый эукариотический организм с полностью расшифрованной биолюминесцентной системой [3]. Были установлены основные интермедиаты пути биосинтеза люциферина, а также участвующие в нем ферменты. Основной компонент системы – люциферин – образуется в ходе ключевой реакции гидроксилирования гиспидина, катализируемой ферментом гиспидин-3-гидроксилазой (nnH3H) (рис. 1).
Рис. 1.
Путь биосинтеза и утилизации люциферина грибов Neonothopanus nambi. Гиспидинсинтаза (nnHispS) катализирует превращение кофейной кислоты в гиспидин, затем гиспидин-3-гидроксилаза (nnH3H) катализирует гидроксилирование гиспидина с образованием люциферина грибов. В присутствии молекулярного кислорода люциферин окисляется люциферазой (nnLuz) до высокоэнергетического промежуточного соединения – эндопероксида, который испускает свет, распадаясь до оксилюциферина (каффеоилпирувата). В итоге оксилюциферин разлагается до кофейной кислоты в присутствии каффеоилпируватгидролазы (nnCPH).

На данный момент гиспидин-3-гидроксилазы высших грибов практически не охарактеризованы как на биохимическом, так и на структурном уровне. Гиспидин-3-гидроксилаза из N. nambi состоит из 422 а.о. и обладает молекулярной массой ~46 кДа. С помощью биоинформатических методов нами было предсказано, что nnH3H имеет домен с укладкой Россмана, который содержит β-складчатые структуры [4]. Это позволило предположить, что nnH3H относится к семейству растворимых NAD(P)Н/FAD-зависимых монооксигеназ. В литературе нет данных о разрешении пространственной структуры гиспидин-3-гидроксилаз высших грибов. К ближайшим гомологам данного белка (60–67% аминокислотной идентичности) можно отнести гидроксилазы других биолюминесцентных грибов, в частности родов Armillaria и Mycena [3], однако их структура также не изучена. Ближайшие гомологи с известной пространственной структурой обладают сходством с nnH3H по аминокислотной последовательности не более чем на 28%, что исключает возможность проведения на их основе структурного моделирования и предсказания эффектов аминокислотных замен. Также мало изучены биохимические особенности гидроксилаз данного семейства, включая их субстратную специфичность.
На сегодняшний день существует недостаточно экспериментальных исследований, посвященных изучению гиспидин-3-гидроксилаз грибов. Для понимания механизма действия и субстратной специфичности nnH3H следует провести структурные исследования молекулы белка. Для этого необходимо разработать протокол получения высокоочищенного препарата функционально активной nnH3H в достаточных для проведения анализа количествах. Поэтому целью данной работы стало получение рекомбинантной, высокоочищенной и ферментативно активной гиспидин-3-гидроксилазы из гриба N. nambi путем гетерологичной экспрессии в клетках бактерии Escherichia coli.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Экспрессия гена гибридного белка SUMO-nnH3H в клетках E. coli. Для наработки nnH3H мы выбрали бактериальную экспрессионную систему. Бактерии E. coli способны производить рекомбинантные белки с выходом >1 г/л. Более того, способность роста этих бактерий на минеральных средах делает E. coli подходящим организмом для продукции меченых белков для изучения при помощи ЯМР-спектроскопии. Однако известно, что клетки E. coli не способны проводить ряд посттрансляционных модификаций, в частности образование S–S-связей. Это приводит к формированию так называемых телец включения. Для получения функционально активного фермента требуется проведение ренатурации in vitro, разработка и осуществление которой зачастую трудоемки и малоэффективны.
Также активно применяются технологии гибридной экспрессии генов: целевой продукт получают в виде гибрида с белком-партнером, который улучшает физико-химические показатели молекулы белка, такие как растворимость, и его биологические свойства, например, активность, а также снижает токсическое воздействие на клетку. Использование соответствующих белков-партнеров, а также подбор оптимальных экспрессионных векторов, условий культивирования и индукции, коэкспрессия целевых генов с генами шаперонов – все это сделало возможным применение E. coli для получения сложных белков с правильной структурой [5].
Сначала мы использовали стратегию прямой экспрессии гена, используя систему на основе промотора РНК-полимеразы фага Т7. Уровень биосинтеза nnH3H согласно результатам дот-иммуноблоттинга составил ~100 мг/л. Подбор условий культивирования показал, что снижение температуры культивирования вплоть до 15°C, а также варьирование состава среды и концентрации индуктора не оказали влияния на растворимость целевого продукта. Таким образом, было принято решение перейти к использованию гибридной экспрессии гена. В качестве белков-партнеров были выбраны варианты, представленные в табл. 1. Согласно данным литературы, перечисленные белки-партнеры были успешно применены для повышения выхода и растворимости многих рекомбинантных белков [6–8].
Таблица 1.
Характеристика белков-партнеров, использованных для получения гиспидин-3-гидроксилазы гриба Neonothopanus nambi
| Название белка-партнера | Суммарный размер белка-партнера, кДа | Назначение белка-партнера | Ссылка |
|---|---|---|---|
| His6-TrxA | 13.67 | Способен накапливаться в больших количествах в цитоплазме и в растворимой форме. Как N-, так и C-конец белка находятся на поверхности молекулы и достаточно подвижны, что делает его “удобным” партнером | [6] |
| DsbA-His6 | 4.08 | Лидерный пептид периплазматического белка E. coli DsbA, необходимый для транслокации целевого белка в периплазму | [7] |
| NSP4-His6 | 4.07 | Синтетическая лидерная последовательность для транслокации целевого белка в периплазму, полученная путем комбинации лидерного пептида периплазматического белка E. coli DsbA и пектиназы PelB E. carovotora | [7] |
| His6-SUMO | 12.07 | При синтезе целевого белка, слитого с SUMO, наблюдается заметное увеличение экспрессии целевого полипептида и его растворимости. Внешняя гидрофильная оболочка и внутренний гидрофобный кор могут оказывать тот же эффект, что и детергенты при солюбилизации | [8] |
Для экспрессии генов гибридных белков в клетках бактерий были созданы генетические конструкции на основе экспрессионного вектора pET39b(+), который имеет в своем составе ген репрессора lac-оперона laci, предназначенного для дополнительного контроля транскрипции целевого гена. Мы установили, что гибридная экспрессия целевого гена значительно повышает выход продуцируемого белка (рис. 2). В случае использования конструкций, в которых целевой белок синтезируется в виде гибрида с SUMO или TrxA, выход составил >1 г/л, но только в виде телец включения. Изменение концентрации индуктора и температуры культивирования c 37 до 15°С не привели к увеличению растворимости целевого белка. При использовании лидерных последовательностей DsbA и NSP4 белок также накапливался в виде телец включения и отсутствовал во фракции периплазматических белков.
Рис. 2.
Анализ уровня экспрессии гена nnh3h и его вариантов в клетках E. coli BL21(DE3) при помощи дот-иммуноблоттинга. Указаны названия белков-партнеров и концентрация индуктора ИПТГ. К+ – положительный контроль – препарат белка, содержащего гистидиновую метку, с известной концентрацией; Auto – культивирование клеток на автоиндукционной среде TBP-5052 [12].

Максимальный уровень биосинтеза целевого продукта был достигнут при 30°С при использовании белка-партнера SUMO и составил ~2 г/л. С целью получения nnH3H в растворимой форме были созданы генетические конструкции, кодирующие гиспидин-3-гидроксилазу с различными партнерами, на основе экспрессионного вектора pCOLDIII [9]. При использовании вектора pCOLDIII происходит селективная индукция синтеза целевого белка при низкой температуре (15–20°С), что подавляет экспрессию других клеточных белков и снижает активность протеаз. С целью получения белка в растворимой форме нами были исследованы следующие условия: температура культивирования (20, 15, 13, 10°C), концентрация индуктора экспрессии – изопропил-β-D-1-тиогалактопиранозида (ИПТГ), концентрация посевного материала и специальных шоковых добавок, таких как этанол (1–3%, V/V), сорбитол (0.1–0.5 М), аргинин (0.1–0.3 М), KCl (0.05–0.2 М). Однако данные модификации условий культивирования не оказали положительное влияние на растворимость целевого белка.
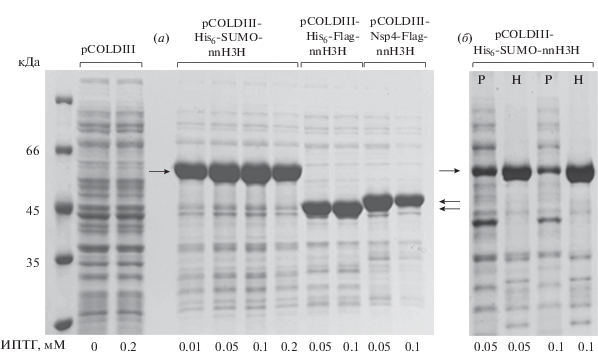
На рис. 3а представлен общий уровень экспрессии nnh3h при оптимальных условиях культивирования и индукции с использованием разных конструкций в клетках E. coli BL21(DE3). Оптимальная концентрация ИПТГ составила 0.05 мM для гибридной конструкции SUMO-nnH3H. По экспериментальным данным, выход целевого фермента составил ~1 г/л. В дальнейшем результаты фракционирования внутриклеточного белка показали, что в случае SUMO-nnH3H наблюдалось появление полосы с заданной молекулярной массой в области, соответствующей размеру целевого продукта, во фракции растворимых белков (рис. 3б). Как и ожидалось, растворимость целевого продукта была выше при меньшей концентрации ИПТГ. Также в ходе экспериментов по подбору оптимальных условий культивирования продуцента SUMO-nnH3H мы выяснили, что добавление в среду 60–180 мM KCl значительно повышало растворимость рекомбинантного фермента. Лизаты демонстрировали специфическую активность nnH3H – способность конвертировать гиспидин в люциферин, однако в нерастворимой форме содержание фермента было значительно больше.
Рис. 3.
(а) – Общий уровень экспрессии гена nnh3h в составе разных конструкций в E. coli BL21(DE3) при подборе среды для индукции; (б) – фракционирование белков в клетках E. coli BL21(DE3), продуцирующих SUMO-nnH3H. Р – фракция растворимого клеточного белка, H – фракция нерастворимого белка. Стрелками отмечено положение целевого белка.
Мы заключили, что SUMO-nnH3H может быть наработан в клетках E. coli в функционально активном состоянии. При оптимизации условий культивирования (температура 15°С, время – 48 ч, среда TBP + 60 мM KCl + 0.05 мM ИПТГ) удалось значительно увеличить долю растворимого белка. Однако большая доля целевого фермента (~90%) агрегировала в тельца включения. Стоит также отметить то, что SUMO-nnH3H растворимой фракции склонен к выпадению в осадок. Агрегация белка может происходить из-за того, что во время синтеза он не успевает приобрести правильную пространственную структуру, поэтому было решено коэкспрессировать целевой ген в присутствии шаперонина GroEL/ES.
GroEL/ES представляет собой сложную гетеромерную молекулу цилиндрической формы размером 145 Å в высоту и 135 Å в диаметре. Неправильно свернутый белок попадает в пространство цилиндра GroEL/ES – центральный канал диаметром ~45 Å, в котором создаются оптимальные условия для сворачивания белковой молекулы. Для экспериментов по коэкспрессии был использован экспрессионный вектор pACYC-TF-GroEL/ES [10]. Уровень биосинтеза SUMO-nnH3H в условиях коэкспрессии снизился до 1 г/л, однако ~30% целевого продукта находилось в растворимой форме.
Выделение SUMO-nnH3H из клеток E. coli. Для предотвращения агрегации целевого белка клеточную биомассу лизировали в буферном растворе, содержащем 300 мМ аргинина или 2 М мочевины или 1 М гидрохлорида гуанидина. Как видно из электрофореграммы (рис. 4а), добавление хаотропных агентов по-разному влияло на эффективность выделения и очистки. Показано значительное различие в поведении молекулы белка при адсорбции на сорбенте Ni-sepharose HP. Использование аргинина не только способствовало повышению динамической емкости сорбента, но также препятствовало неспецифическому связыванию. Для предотвращения неспецифического связывания с сорбентом хроматографию проводили в условиях перегрузки колонки. В результате за одну стадию хроматографической очистки удалось получить препарат целевого фермента с чистотой >95%, которая удовлетворяет требованиям к чистоте препаратов для структурных исследований. Стоит отметить, что лизаты, полученные на буферных растворах с добавлением гидрохлорида гуанидина или мочевины, имели активность, соответственно, в 20 и 10 раз меньше по сравнению с лизатом, содержащим аргинин.
Рис. 4.
(а) – Выделение SUMO-nnH3H из клеток E. coli BL21(DE3), трансформированных векторами pCOLDIII-His6-SUMO-nnH3H и pACYC-TF-GroEL/ES, в присутствии 300 мМ аргинина, 2 М мочевины и 1 М гидрохлорида гуанидина. Общий б. – общий растворимый белок; Несвяз. б. – фракция белков, не связавшихся с сорбентом; Элюция — элюированный белок; (б) – расщепление очищенного SUMO-nnH3H (в 50 мМ Трис-HCl, pH 7.5, 150 мМ NaCl, 50 мМ аргинин, 20 мМ имидазол). М – маркер; 1 – очищенный SUMO-nnH3H; 2–9 – образцы после расщепления (1 ч, комнатная температура) в условиях добавления 0.5 мМ DТТ (2), 2 мМ DТТ (3), 0.2% IGEPAL (4), 0.4% IGEPAL (5), 0.5 M аргинина (6), 1 М аргинина (7), 5% глицерина (8) и 10% глицерина (9).

Следующим шагом стало отщепление SUMO от nnH3H при помощи протеазы Ulp1 [11]. В ходе экспериментов по подбору условий гидролиза SUMO были проанализированы различные соотношения фермента и субстрата (от 1 : 1 до 1 : 10 000), разное время реакции (0.5–48 ч), температура (4–37°С) и pH (7.0–8.5). Также было проанализировано влияние различных компонентов на эффективность расщепления: аргинина (0.05–1 М), мочевины (0.1–2 М), тритона Х100 (0.01–0.5%), CHAPS (5–50 мМ), дитиотреитола (0.1–5 мМ), триметиламиноксида (0.1–0.5 М), KCl (50–200 мМ) и гиспидина (0.5–10 мкМ). Тем не менее получить гиспидин-3-гидроксилазу грибов без белка-партнера SUMO не удалось. Объяснений данному явлению может быть несколько. Во-первых, nnH3H оказался склонным к агрегации, и после добавления протеазы Ulp1 часть фермента выпадала в осадок (данные не представлены). Также отщепление SUMO от гибридного белка может быть затруднено стерической недоступностью сайта. Чтобы проверить эту гипотезу, была создана генетическая конструкция, в которой между белком-партнером и ферментом был вставлен гибкий линкер GSG4SG4. Также ген nnh3h в этой конструкции был укорочен с N- и С-концов, т.к. по данным молекулярного моделирования структуры гиспидин-3-гидроксилазы первые и последние 5 а.о. не влияют на структурные особенности целевой молекулы. Введение дополнительного линкера не повлияло на схему наработки, выделения и очистки фермента. Негативного влияния на активность не наблюдалось, однако эффективность расщепления также не изменилась. Расщепление очищенного SUMO-nnH3H было возможно лишь при значительном разведении целевого белка до концентрации 0.1–0.2 мг/мл и использовании относительно высокого количества протеазы Ulp1 (1 : 5 – 1 : 10). Расщепление протекало в течение 1 ч. Любая попытка масштабировать протокол заканчивались выпадением весомой части целевого белка в осадок (рис. 4б). Активность протеазы Ulp1 была подтверждена расщеплением модельного гибрида SUMO-DTX (данные не представлены).
Полученный рекомбинантный фермент nnH3H в виде гибрида с белком-партнером SUMO – функционально активный и пригодный для проведения структурных исследований. Имеющиеся в литературе данные позволяют предположить, что разница в молекулярных массах и наличие SUMO не будут препятствовать проведению таких исследований [8]. В результате выделения и очистки, которая включала в себя металло-хелатную аффинную хроматографию, мы получили очищенный фермент с выходом ~20 мг со 100 мл бактериальной культуры.
Оценка функциональной активности очищенного рекомбинантного фермента SUMO-nnH3H. Помимо гетерологичной экспрессии гена nnh3h, важной задачей стала оценка функциональной активности полученного фермента. Нами был разработан протокол, основанный на люциферин-люциферазной реакции грибов. При добавлении фермента к раствору гиспидина (предшественника люциферина, рис. 1) и NAD(P)Н протекает реакция гидроксилирования субстрата с образованием люциферина. Последний, в свою очередь, можно идентифицировать при помощи люциферазы N. nambi по эмиссии квантов света. Разработанный биолюминесцентный метод – чувствительный и позволяет быстро осуществить оценку активности фермента nnH3H.
Оказалось, что при выделении и очистке гибридного белка SUMO-nnH3H активность фермента существенно снижается. Мы предположили, что это происходит из-за диссоциации определенных кофакторов из молекулы активного фермента. Данными кофакторами, вероятнее всего, не выступают катионы двухвалентных металлов, т.к. фермент сохраняет аналогичную активность в буферном растворе, содержащем 10 мМ ЭДТА. При сравнении аминокислотных последовательностей с гомологами можно отметить, что nnH3H с большей долей вероятности имеет сайты связывания с FAD. Сам фермент относится к семейству NAD(P)Н-зависимых монооксигеназ, катализирующих реакцию: NAD(P)Н + + R-H + O2 → NAD(P)+ + R-OH + H2O, а также он, вероятнее всего, относится к семейству PHBH-подобных гидроксилаз, таких как р-гидроксибензоатгидроксилаза Pseudomonas fluorescens (UniProt: P00438), монооксигеназа Pseudomonas aeruginosa (UniProt: Q9HWG9) и дигидропиридингидроксилаза Paenarthrobacter nicotinovorans (UniProt: Q93NG3). С помощью биоинформатического анализа последовательности nnH3H были предсказаны предположительные сайты связывания FAD, помимо сайтов связывания с NAD(P)Н. Действительно, оказалось, что добавление FAD на несколько порядков повышает активность фермента, в отличие от его флавиновых предшественников (рис. 5).
Рис. 5.
Определение влияния кофактора FAD и его флавиновых предшественников на активность гиспидин-3-гидроксилазы N. nambi путем измерения биолюминесценции сопряженной реакции в присутствии люциферазы N. nambi, гиспидина и NADН.

Чтобы оценить специфичность взаимодействия FAD и SUMO-nnH3H, мы определили константу диссоциации (Kd) этого комплекса. Исследование проводили по методу, предложенному Loomans et al. (табл. 2) [12]. По нашим оценкам, Kd для FAD составляет ~0.78 мкМ. Таким образом, мы впервые показали, что молекула FAD не только связывается с молекулой фермента, но и важна для его активности.
Таблица 2.
Результаты измерения интенсивности биолюминесценции, использованные для расчета константы диссоциации комплекса FAD–SUMO-nnH3H
| Интенсивность свечения, ед. | Концентрация FAD, мкМ | ||||||
|---|---|---|---|---|---|---|---|
| 0 | 0.0333 | 0.3333 | 3.333 | 16.667 | 33.333 | 83.333 | |
| Без разведения SUMO-nnH3H (n = 1 для оценки Kd) | 101 | 2954 | 13 212 | 38 494 | 118 121 | 129 877 | 133 289 |
| 118 | 3120 | 12 876 | 41 198 | 115 736 | 132 816 | 134 783 | |
| 143 | 3016 | 13 561 | 40 182 | 115 967 | 129 765 | 134 536 | |
| 121 ± 21 | 3030 ± 84 | 13 216.3 ± 343 | 39 958 ± 1366 | 116 608 ± 1315 | 130 819 ± 1730 | 134 203 ± 801 | |
| При 4-кратном разведении (n = 4 для оценки Kd) | 123 | 1923 | 6441 | 14 573 | 68 820 | 76 465 | 78 746 |
| 129 | 1753 | 6262 | 15 001 | 67 121 | 75 983 | 77 391 | |
| 156 | 1801 | 6495 | 14 323 | 68 191 | 76 010 | 78 425 | |
| 136 ± 18 | 1826 ± 88 | 6399 ± 122 | 14 632 ± 343 | 68 044 ± 859 | 76 153 ± 271 | 78 187 ± 708 | |
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Штаммы E. coli. Для создания генетических конструкций использовали штамм E. coli XL1-Blue (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F'proAB lacI qZΔM15 Tn10 (Tetr)] (Stratagene, США), для бактериальной экспрессии гена nnh3h – штамм BL21(DE3) [F–ompT hsdSB(rB– mB–) gal dcm (DE3)] (Novagen, США).
Конструирование экспрессионных векторов для получения белка nnH3H. Приготовление компетентных клеток и трансформацию E. coli, выделение плазмидной ДНК, гидролиз ДНК эндонуклеазами рестрикции, лигирование и другие генно-инженерные манипуляции проводили согласно стандартным методикам [13]. Химический синтез небольших фрагментов дцДНК, олигонуклеотидов, а также контрольное секвенирование полученных конструкций проведены ЗАО “Евроген” (Россия). Создание генетической конструкции с последовательностью гена nnh3h было описано в предыдущей работе [4]. Для получения экспрессионных векторов с целью продукции целевого фермента в бактериях использовали плазмиды pET39b(+) (Novagen, США) и pCOLDIII (Takara Bio, Япония). Для создания генетических конструкций His6-Flag-nnH3H, HELIX-His6-Flag-nnH3H, DsbA-His6-Flag-nnH3H и NSP4-His6-Flag-nnH3H амплификацию гена nnh3h производили с использованием 3–4 олигонуклеотидов (табл. 3) методом step-out ПЦР. Для создания конструкции His6-Flag-nnH3H были использованы олигонуклеотиды, последовательности которых приведены в табл. 3.
Таблица 3.
Последовательности олигонуклеотидов, используемых при создании экспрессионных векторов для получения гиспидин-3-гидроксилазы
| Название конструкции | Название олигонуклеотида | Последовательность олигонуклеотида (5'–3') |
|---|---|---|
| pCOLDIII_His6-Flag-nnH3H, pET39b_His6-Flag-nnH3H | His-Flag-H3H dir1 | GGTTCCGACTACAAAGACGATGACGATAAAGCATCGTTTGAGAATTCTCTAAG |
| His-Flag-H3H XbaI dir2 | ATGCTCTAGAGAAGGAGATATACATATGGGCCACCATCATCACCACCATGGTT-CCGACTACAAAGACGAT | |
| pCOLDIII_DsbA-His6-Flag-nnH3H, pET39b_DsbA-His6-Flag-nnH3H | His-flag-H3H dir1 | CACCACCATGGTTCCGACTACAAAGA-CGATGACGATAAAGCATCGTTTGAGA-ATTCTCTAAG |
| DsbA-His-flag-H3H dir2 | TGGCTGGTTTAGTTTTAGCGTTTAGCGCATCGGCGGCGGGCCACCATCATCA-CCACCATGGTTCCGACTA | |
| DsbA-His-flag-H3H XbaI dir3 | ATGCTCTAGAGAAGGAGATATACATATGAAAAAGATTTGGCTGGCGCTGGCTGGTTTAGTTTTAGCGTT | |
| pCOLDIII_NSP4-His6-Flag-nnH3H, pET39b_NSP4-His6-Flag-nnH3H | His-flag-H3H dir1 | CACCACCATGGTTCCGACTACAAAGA-CGATGACGATAAAGCATCGTTTGAGA-ATTCTCTAAG |
| Nsp4-His-flag-H3H dir2 | GTCTGCTGCTCCTCGCTGCCCAGCC-GGCGATGGCGGCGGGCCACCATCATCACCACCATGGTTCCGACTA | |
| Nsp4-His-flag-H3H XbaI dir3 | ATGCTCTAGAGAAGGAGATATACATATGAAAAAGATTACCGCTGCGGCGGGT-CTGCTGCTCCTCGCTGC | |
| pCOLDIII_His6-SUMO-nnH3H, pET39b_His6-SUMO-nnH3H | His-Sumo XbaI dir | ATGCTCTAGAGAAGGAGATATACATATGGGCCACCATCATCACCACCATGGTT-CCGATTCTGAAGTGAACCAG |
| Sumo-partH3H rev | ACGATGCGCCACCAATCTGCTCACGG | |
| H3H-partSumo dir | ATTGGTGGCGCATCGTTTGAGAATTC-TCTAAG | |
| pCOLDIII_His6-TrxA-Flag-nnH3H, pET39b_His6-TrxA-Flag-nnH3H | His-Trx XbaI dir | ATGCTCTAGAGAAGGAGATATACATATGGGTCACCATCACCATCACCACGGTA-GCGATAAAATTATTCACCTGACT |
| Trx-flag rev | TCATCGTCTTTGTAGTCGGAACCGGCCAGGTTAGCGTCGAGG | |
| flag-H3H dir | CGACTACAAAGACGATGACGATAAAGCATCGTTTGAGAATTCTCTAAG | |
| Для всех конструкций | H3H HindIII rev | GCATAAGCTTATTAGGCAGAATTAGA-GCTTCTTAGGAGCGTCTCGAGAAC |
Полученные фрагменты ДНК клонировали в вектор pET39b(+), pCOLDIII по двум сайтам рестрикции (XbaI и HindIII). Для создания конструкций His6-SUMO-nnH3H и His6-TrxA-Flag-nnH3H проводили отжиг частично комплементарных ПЦР-продуктов, их амплификацию и вставку в вектор по двум сайтам рестрикции (XbaI и HindIII). Для создания конструкций His6-SUMO-nnH3H использовали олигонуклеотиды His-Sumo XbaI, Sumo-partH3H rev, H3H-partSumo dir, для конструкции His6-TrxA-Flag-nnH3H – His-Trx XbaI dir, Trx-flag rev, flag-H3H dir (табл. 3). Для создания всех конструкций использовали олигонуклеотид H3H HindIII rev (табл. 3).
Оценка уровня экспрессии гена nnh3h в клетках E. coli. Трансформацию бактериальных клеток E. coli для синтеза белка проводили согласно стандартному протоколу [14], затем добавляли раствор ИПТГ до конечных концентраций 0, 0.01, 0.05, 0.25 и 1 мМ. Клетки выращивали при температуре 10–37°С в течение 1–3 сут. Содержание целевого белка анализировали методами электрофореза по Лэммли [15] и дот-иммуноблоттинга [16], используя моноклональные антитела мыши против полигистидинового тэга, конъюгированные с пероксидазой хрена (A7058, Sigma, США). Анализируемые образцы наносили на нитроцеллюлозную мембрану Hybond-C (Cytiva, Швеция). Мембрану высушивали на воздухе и блокировали 5%-ным раствором сухого обезжиренного молока в ТBS-Tween 80 в течение 1 ч. Мембрану инкубировали с моноклональными антителами к гистидиновому тэгу, конъюгированными с пероксидазой хрена, в разведении 1 : 1000 (A7058, Sigma, США). Визуализацию проводили с помощью готового хромогенного субстрата для мембран (T0565, Sigma, США). Денситометрический анализ мембраны осуществляли при помощи программы ImageJ (www.imagej.net).
Фракционирование белков из клеток E. coli. Биомассу E. coli, собранную с 1 мл культуры, размораживали и суспендировали в 500 мкл лизирующего буфера (50 мМ Трис-HCl, рН 8.0, 250 мМ NaCl, 1 мМ ЭДТА, 0.5% Тритона Х-100). Суспензию переносили в пробирки (2 мл), добавляли 5 мкл раствора лизоцима (10 мг/мл) и инкубировали при комнатной температуре в течение 15 мин. Затем добавляли PMSF до концентрации 0.5 мМ. Образец трижды обрабатывали ультразвуком в течение 3–5 с, перерыв между циклами 2–3 мин. Во время всего процесса пробирки находились во льду. Обработанный образец затем центрифугировали в течение 10 мин при 12 000 g и 4°С. Супернатант (растворимый белок) отбирали в отдельную пробирку. Для получения раствора белка из телец включения осадок растворяли в 500 мкл TES-буфера (100 мМ Трис-HCl, pH 6.8, 1% SDS, 10 мМ ЭДТА). Фракцию периплазматических белков получали согласно методике Goffin et al. [17]. Локализацию целевого белка оценивали методом денатурирующего гель-электрофореза по методу Лэммли (12%-ный разделяющий и 8%-ный формирующий гели) [15].
Выделение целевого фермента из клеток E. coli. Среду TBP объемом 100 мл, содержащую ампициллин (200 мкг/мл) и хлорамфеникол (140 мкг/мл), инокулировали 50 колониями штамма E. coli BL21(DE3), трансформированного векторами pCOLDIII-His6-SUMO-nnH3H и pACYC-TF-GroEL/ES. Культуру выращивали при 37°С, 300 об/мин в течение 2–4 ч до достижения ОD550 1.0–1.5. Затем добавляли ИПТГ до конечной концентрации 0.05 мМ и снижали температуру культивирования до 15°С. Индукцию проводили в течение 24 ч. Биомассу отделяли с помощью центрифугирования при 8000 g в течение 5 мин, после чего осадок суспендировали в 25 мл лизирующего буфера (100 мМ KPi, pH 7.5, 300 мM NaCl, 20 мМ имидазол, 300 мМ аргинин, 0.5 мМ 2-меркаптоэтанол) и разрушали при помощи ультразвука (5 циклов по 30 с, перерывы по 2 мин). После разрушения в полученный лизат добавляли 0.1% (V/V) полиэтиленимина для удаления ДНК и центрифугировали в течение 25 мин при 12 000 g. Супернатант разводили в 2 раза водой Milli-Q, пропускали через фильтр с диаметром пор 0.22 мкм и наносили на колонку HiTrap Ni-sepharose HP (Cytiva, Швеция) объемом 5 мл, предварительно уравновешенную буфером А (50 мМ KPi, pH 7.5, 200 мM NaCl, 10 мМ имидазол, 150 мМ аргинин). Затем осуществляли промывку сначала буфером А (не менее трех объемов сорбента), а затем буфером В: 50 мМ KPi, pH 7.5, 200 мM NaCl, 40 мМ имидазол (не менее трех объемов сорбента). Элюцию проводили буфером С (50 мМ KPi, pH 7.5, 200 мM NaCl, 200 мМ имидазол). Элюат переводили в буферный раствор (50 мМ KPi, pH 7.5, 200 мM NaCl) на колонке PD10 (Cytiva, Швеция), содержащей сорбент Сефадекс G25 (Cytiva, Швеция), и использовали для проведения тестов на активность.
Определение активности nnH3H. Все стоковые растворы компонентов для измерения активности были приготовлены непосредственно перед экспериментом и находились во льду: 0.2 мМ гиспидин, 0.5 мМ FAD и 10 мМ NADPН в 10 мМ калий-фосфатном буфере (рН 7.5), 2%-ный раствор додецилмальтозида (DDM). Раствор nnH3H стандартизировали по концентрации белка методом Бредфорда до концентрации 0.1 мг/мл. Реакционная смесь для гидроксилирования гиспидина содержала 1 мкл раствора фермента, 10 мкМ гиспидин, 500 мкМ NADPН, 25 мкМ FAD. Объем реакции доводили до 10 мкл при помощи 10 мМ калий-фосфатного буфера, рН 7.5. Реакцию проводили при комнатной температуре в течение 10 мин. Концентрацию образовавшегося 3-гидроксигиспидина оценивали по интенсивности свечения, используя рекомбинантную люциферазу N. nambi. Для данных целей 10 мкл мембранной фракции, содержащей люциферазу, солюбилизировали в 10 мкл 2% DDM и добавляли 80 мкл 10 мМ калий-фосфатного буфера, рН 7.0. Затем 20 мкл раствора люциферазы добавляли к 10 мкл реакционной смеси для гидроксилирования гиспидина и сразу же проводили измерение интенсивности биолюминесценции на планшетном ридере CLARIOstar (BMG Labtech, Германия) в спектральном диапазоне 500 ± 50 нм. В качестве положительного контроля использовали очищенный препарат 3-гидроксигиспидина (люциферин грибов – субстрат люциферазы) в эквивалентной концентрации [18]. В качестве отрицательного контроля вместо раствора nnH3H использовали раствор бычьего сывороточного альбумина с концентрацией 0.1 мг/мл. Kd для FAD определяли по методу Loomans et al. [11]. Все эксперименты по определению активности проводили в 3–5 независимых повторах.
Методика расчета константы диссоциации SUMO-nnH3H и FAD. Специфичность взаимодействия FAD и SUMO-nnH3H определяли путем оценки Kd комплекса. Исследование проводили в 96-луночных планшетах по методу, предложенному Loomans et al. [12]. Равновесие в системе FAD–SUMO-nnH3H в случае нековалентного бимолекулярного взаимодействия можно описать с помощью закона действующих масс:
При условии, что с SUMO-nnH3H связалось количество FAD, равное половине максимального, и подставляя все найденные значения, приходим к выражению для константы аффинности (Kа):
При этом зависимость активности фермента, прямо пропорциональная количеству образовавшегося гиспидина, а значит, и интенсивности биолюминесценции, от концентрации кофактора будет иметь характерную сигмовидную форму. При заданных сериях разведений фермента E, E1, E2, …, En константа реакции не будет зависеть от его концентрации и легко выражается в следующее тождество:
В ходе выполнения данной работы были подобраны оптимальное разведение, которое составило 4, и концентрация FAD – 0–80 мкМ (табл. 2). Далее зависимость связывания анализировали при помощи программы SigmaPlot (http://www.sigmaplot.co.uk/) и определяли концентрации FAD при интенсивности биолюминесценции, равной половине максимальной. Для образца без разведения концентрация FAD составила 12.18 мкМ, а для разведения в 4 раза – 3.63 мкМ. Таким образом, оценочное значение Kd составляет (4 × 3.63 – 12.18)/3 = 0.78 мкМ.
ЗАКЛЮЧЕНИЕ
Показано, что гиспидин-3-гидроксилаза тропического гриба Neonothopanus nambi может эффективно нарабатываться в клетках E. coli в виде гибрида с белком-партнером SUMO в функционально активном состоянии. При оптимизации условий культивирования и коэкспрессии с геном шаперона GroEL/ES удалось достичь уровня биосинтеза ~1 г/л, причем ~30% целевого продукта находились в растворимой форме. Очищенный гибридный белок проявляет свою специфическую активность в отношении гиспидина. Оказалось, что добавление FAD на несколько порядков увеличивает активность фермента, в отличие от его флавиновых предшественников. По нашим оценкам, Kd для FAD составляет ~0.78 мкМ. Более того, мы впервые показали, что молекула FAD не только связывается с молекулой фермента в качестве кофактора, но и выступает субстратом.
Полученные результаты – важный шаг на пути изучения биохимических и структурных особенностей гидроксилаз данного семейства, а также механизмов биолюминесценции.
Список литературы
Kaskova Z.M., Tsarkova A.S., Yampolsky I.V. // Chem. Soc. Rev. 2016. V. 45. P. 6048–6077. https://doi.org/10.1039/C6CS00296J
Oba Y., Stevani C.V., Oliveira A.G., Tsarkova A.S., Chepurnykh T.V., Yampolsky I.V. // Photochem. Photobiol. 2017. V. 93. P. 405–415. https://doi.org/10.1111/php.12704
Kotlobay A.A., Sarkisyan K.S., Mokrushina Y.A., Marcet-Houben M., Serebrovskaya E.O., Markina N.M., Somermeyer L.G., Gorokhovatsky A.Y., Vvedensky A., Purtov K.V., Petushkov V.N., Rodionova N.S., Chepurnyh T.V., Fakhranurova L.I., Guglya E.B., Ziganshin R., Tsarkova A.S., Kaskova Z.M., Shender V., Abakumov M., Abakumova T.O., Povolotskaya I.S., Eroshkin F.M., Zaraisky A.G., Mishin A.S., Dolgov S.V., Mitiouchki-na T.Y., Kopantzev E.P., Waldenmaier H.E., Oliveira A.G., Oba Y., Barsova E., Bogdanova E.A., Gabaldón T., Stevani C.V., Lukyanov S., Smirnov I.V., Gitelson J.I., Kondrashov F.A., Yampolsky I.V. // Proc. Natl. Acad. Sci. USA. 2018. V. 115. P. 12728–12732. https://doi.org/10.1073/pnas.1803615115
Medvedev K.E., Kinch L.N., Schaeffer R.D., Grishin N.V. // PLoS Comput. Biol. 2019. V. 15. P. e1007569. https://doi.org/10.1371/journal.pcbi.1007569
Rosano G.L., Ceccarelli E.A. // Front. Microbiol. 2014. V. 5. P. 172. https://doi.org/10.3389/fmicb.2014.00172
McCoy J., Lavallie E. // Curr. Protoc. Mol. Biol. 2001. Chapter 16. Unit 16.8. https://doi.org/10.1002/0471142727.mb1608s28
Zhang W., Lu J., Zhang S., Liu L., Pang X., Lv J. // Microb. Cell Fact. 2018. V. 17. P. 50. https://doi.org/10.1186/s12934-018-0894-y
Butt T.R., Edavettal S.C., Hall J.P., Mattern M.R. // Protein Expr. Purif. 2005. V. 43. P. 1–9. https://doi.org/10.1016/j.pep.2005.03.016
Kobayashi H., Yoshida T., Inouye M. // Appl. Environ. Microbiol. 2009. V. 75. P. 5356–5362. https://doi.org/10.1128/AEM.00691-09
Lamppa J.W., Tanyos S.A., Griswold K.E. // J. Biotechnol. 2013. V. 164. P. 1–8. https://doi.org/10.1016/j.jbiotec.2012.11.007
Mossessova E., Lima C.D. // Mol. Cell. 2000. V. 5. P. 865–876. https://doi.org/10.1016/s1097-2765(00)80326-3
Loomans E.E., Roelen A.J., Van Damme H.S., Bloemers H.P., Gribnau T.C., Schielen W.J. // J. Immunol. Methods. 1995. V. 184. P. 207–217. https://doi.org/10.1016/0022-1759(95)00089-s
Sambrook J., Fritsch E.F., Maniatis T. // Molecular Cloning. A Laboratory Manual. 4th edition. Cold Spring Harbor Laboratory Press, 2012.
Kurien B.T., Scofield R.H. // Biotechniques. 1995. V. 18. P. 1023–1026.
Laemmli U.K. // Nature. 1970. V. 227. P. 680–685. https://doi.org/10.1038/227680a0
Zeder-Lutz G., Cherouati N., Reinhart C., Pattus F., Wagner R. // Protein Expr. Purif. 2006. V. 50. P. 118–127. https://doi.org/10.1016/j.pep.2006.05.017
Goffin P., Dewerchin M., De Rop P., Blais N., Dehottay P. // Biotechnol. J. 2017. V. 12. https://doi.org/10.1002/biot.201700168
Bubyrev A.I., Tsarkova A.S., Kaskova Z.M. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 183–185. https://doi.org/10.1134/S106816201902002X
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия