

Биоорганическая химия, 2022, T. 48, № 3, стр. 266-278
Антибиотики группы ауреоловой кислоты: перспективы биологически активного класса соединений
А. К. Исагулиева 1, 2, *, А. Н. Тевяшова 1, 3, А. А. Штиль 4
1 Научно-исследовательский институт по изысканию новых антибиотиков имени Г.Ф. Гаузе
119021 Москва, ул. Б. Пироговская, 11, стр. 1, Россия
2 Институт биологии гена РАН
119334 Москва, ул. Вавилова, 34/5, Россия
3 Российский химико-технологический университет имени Д.И. Менделеева
125047 Москва, Миусская пл., 9, Россия
4 Национальный медицинский исследовательский центр онкологии имени Н.Н. Блохина Минздрава России
115478 Москва, Каширское шоссе, 23, Россия
* E-mail: kia2303@ya.ru
Поступила в редакцию 06.05.2021
После доработки 05.06.2021
Принята к публикации 16.06.2021
- EDN: STCCXG
- DOI: 10.31857/S0132342322020129
Аннотация
Антибиотики группы ауреоловой кислоты – митрамицин, хромомицин А3, оливомицин А – ароматические гликозилированные поликетиды, продуцируемые актиномицетами. Молекула ауреоловой кислоты состоит из трициклического агликона с двумя олигосахаридными цепями в качестве заместителей, а также пентильной боковой цепи в положении С3. Для митрамицина и хромомицина А3 детально исследованы механизм биосинтеза в штаммах-продуцентах, роль ферментов, необходимость каждого этапа биосинтеза и их влияние на биологическую активность продукта. Благодаря этому стал возможен рациональный поиск новых, более специфичных и менее токсичных аналогов существующих антибиотиков группы ауреоловой кислоты. В настоящем обзоре обобщены базовые и новейшие знания о биосинтезе ауреоловых антибиотиков, современных подходах к химическим и полусинтетическим модификациям, взаимосвязях “структура–активность”. Проанализированы молекулярные механизмы цитотоксичности ауреоловой кислоты и перспективы развития этого класса соединений как “кандидатов” в противоопухолевые препараты.
СОДЕРЖАНИЕ
ВВЕДЕНИЕ……………...................................266
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ И ПРИМЕНЕНИЕ АНТИБИОТИКОВ ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ............................267
Химиотерапия опухолей………………………….…267
Молекулярные механизмы действия: связывание с GC-богатыми областями ДНК...................269
Влияние на транскрипционные факторы и структуру хроматина...................................269
Нейропротекторные свойства…………………..271
ВЛИЯНИЕ СТРУКТУРЫ АНТИБИОТИКОВ ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ НА БИОЛОГИЧЕСКУЮ АКТИВНОСТЬ.........271
Модификации олигосахаридных цепей………...272
Модификации агликона и боковой пентильной цепи................................................................273
БИОСИНТЕЗ АНТИБИОТИКОВ ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ........................273
ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ АНТИБИОТИКОВ ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ............................................................275
ЗАКЛЮЧЕНИЕ……………………………………..275
СПИСОК ЛИТЕРАТУРЫ………………………276
ВВЕДЕНИЕ
Среди противоопухолевых препаратов – синтетических антиметаболитов, алкилирующих агентов, природных и полусинтетических алкалоидов, антрациклинов и др. – антибиотики занимают важное место [1, 2]. Побочные эффекты противоопухолевых препаратов (нефротоксичность, гепатотоксичность, миелосупрессия, кардиотоксичность [3, 4] как наиболее частые) существенно снижают возможности терапии. Поиск новых противоопухолевых агентов осложняется продолжительностью исследований и не всегда дает значимые результаты: практическое значение приобретают отдельные соединения из тысяч исходных. В связи с этим актуальной становится оптимизация структуры и свойств применяемых сегодня антибиотиков [5], для которых установлены (или активно изучаются) механизмы действия. Производные ауреоловой кислоты (митрамицин, хромомицин А3, оливомицин А и их аналоги) в настоящий момент вновь достаточно активно исследуются в качестве препаратов для терапии онкологических и других заболеваний.
Ауреоловая кислота (АК) – первый антибиотик, выделенный из данной группы соединений и получивший название митрамицин (синонимы: LA-7017 или PA-144) [6]. Параллельные независимые исследования нескольких научных групп, а также наличие в выделяемых культуральных средах промежуточных метаболитов и производных обусловили трудности наименования и идентификации отдельных соединений [7]. Впоследствии были получены другие представители группы: 1) хромомицин А3, выделенный из штаммов Streptomyces griseus и Streptomyces cavourensis, имеющий идентичный митрамицину хромофор – хромомицинон; 2) оливомицин А, получаемый из штамма Actinomyces olivoreticuli; 3) хромоцикломицин, продуцируемый штаммом Streptomyces atroolivaceus и не проявляющий существенной цитотоксичности на линиях опухолей, т.к. не способен проникать в клетки про- и эукариот (его тетрациклический агликон хромоциклин получен как отдельное производное, которое также не проявило активность из-за отсутствия связывания с ДНК) [8].
Различия в структурах ауреоловых антибиотиков определяются следующими параметрами: 1) количеством циклов в агликоне; 2) размером и составом олигосахаридных остатков в положениях С-2 и С-6; 3) длиной и составом алифатической цепи в положении С-3; 4) наличием алкильного заместителя в положении С-7. Все АК, кроме хромоцикломицина, представляют собой трициклические ароматические гликозилированные поликетиды, имеющие две олигосахаридные цепи разной длины и состава в положениях С-2 и С-6 агликона (рис. 1, табл. 1). Хромоцикломицин имеет в качестве агликона тетрациклический хромоциклин. Митрамицин и хромомицин А3 имеют общую структуру агликона и отличаются лишь составом сахаров. Оливомицин А схож с хромомицином А3, за исключением Е-сахара (4-О-изобутирил-оливомикоза вместо 4-О-ацетил-L-хромозы) и алкильного заместителя в положении С-7 (метильная группа у хромомицинона; у агликона оливомицина А, оливина, заместитель отсутствует).
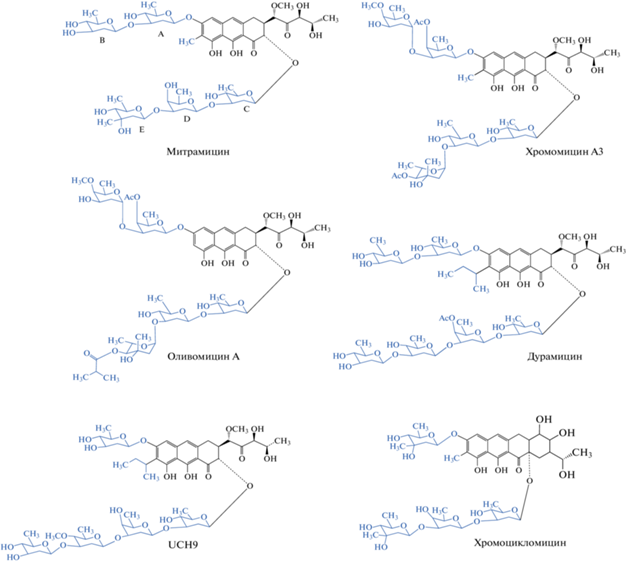
Таблица 1.
Состав олигосахаридных цепей представителей группы ауреоловой кислоты
| Сахар | Антибиотик | |||||
|---|---|---|---|---|---|---|
| митрамицин | хромомицин А3 | оливомицин А | дурамицин | UCH9 | хромоцикло-мицин | |
| A | D-оливоза | 4-О-ацетил- D-олиоза | 4-О-ацетил- D-олиоза | D-оливоза | D-оливоза | D-оливоза |
| B | D-оливоза | Оливомоза | Оливомоза | D-оливоза | D-оливоза | D-оливоза |
| C | D-оливоза | D-оливоза | D-оливоза | D-оливоза | D-олиоза | D-оливоза |
| D | D-оливоза | D-оливоза | D-оливоза | 4-О-ацетил- D-олиоза | 4-О-метил- D-олиоза | D-оливоза |
| E | D-микароза | 4-О-ацетил- L-хромоза В | 4-О-изобутирил-оливомикоза | D-оливоза | D-оливоза | D-микароза |
| F | – | – | – | D-оливоза | – | – |
Для основных представителей класса – хромомицина А3, митрамицина и оливомицина А – и их производных ниже рассмотрены биологические свойства, взаимосвязь структуры и активности, пути биосинтеза, а также предполагаемые механизмы действия.
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ И ПРИМЕНЕНИЕ АНТИБИОТИКОВ ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ
Первоначальное внимание исследователей было обращено на антибактериальный эффект антибиотиков группы АК. Хромомицин А3, оливомицин А и митрамицин показали ингибирующую активность в отношении штаммов Staphylococcus aureus и Bacillus subtilis: минимальная ингибирующая концентрация (МИК) на обоих штаммах составляла не более 0.15 мкг/мл для хромомицина А3, 0.5 мкг/мл для оливомицина А и 0.039 мкг/мл для митрамицина. Однако в отношении грамотрицательных бактерий и грибов соединения были неактивны [9–12]. Кроме этого, была исследована противовирусная активность соединений. Митрамицин в концентрации 50 нМ ингибировал индуцированную TNFα реактивацию вируса иммунодефицита человека (HIV-1) в клетках Jurkat [13]. Действие оливомицина А и его дегликозилированных производных исследовано на вирусах-возбудителях оспы, гриппа, иммунодефицита человека, герпеса и др., но активность проявлялась только в отношении штаммов HIV-1 и HIV-2 [14]. Хромомицин А3 также показал противовирусную активность в отношении HIV-1 [15].
Химиотерапия опухолей
Основное применение антибиотики группы АК нашли в химиотерапии злокачественных новообразований; рациональное использование этих соединений в лечении инфекционных заболеваний ограничивается их высокой общерезорбтивной токсичностью. Митрамицин успешно применяли в лечении рака яичек, острого и хронического миелоидного лейкоза [16]. Кроме этого, до появления бисфосфонатов митрамицин в малых дозах использовали в лечении гиперкальциемии у онкологических больных [17, 18] и при раке Педжета [19]. Хромомицин А3 активно исследовали и применяли в Японии и США как монопрепарат и в комбинированной терапии рака толстой кишки, желудка и других солидных опухолей [20]. Оливомицин А нашел широкое клиническое применение в СССР и использовался в лечении хорионкарциномы [21], опухолей яичка, сарком мягких тканей [12] и рака желудка [20].
В настоящее время клиническое применение митрамицина, хромомицина А3 и оливомицина А прекращено из-за выраженных побочных эффектов, в основном нефро- и гепатотоксичности [22]. Вместе с тем представляется целесообразным продолжить исследования столь активного химического класса соединений. Знания механизмов действия антибиотиков группы АК послужат рациональному дизайну производных, применимых в клинической практике.
Молекулярные механизмы действия: связывание с GC-богатыми областями ДНК
Интерес к АК как химиотерапевтическим агентам объясняется их способностью взаимодействовать с ДНК и нарушать транскрипцию генов. Особенность этих узкобороздочных лигандов – высокая аффинность к GC-богатым участкам ДНК. Такие участки присутствуют преимущественно в регуляторных областях генома (короткие GC-повторы и CpG-островки) [23]. Связываясь с ними, антибиотики способны ингибировать транскрипцию. Механизмы и последствия такого действия в настоящий момент не до конца ясны. Предполагается, что, связываясь с GC-повторами, антибиотики группы АК блокируют сайты посадки транскрипционных факторов, например, Sp1 (specificity protein 1) [24–26].
В ряде работ исследованы предпочтительные сайты связывания антибиотиков группы АК [27–29]. В экспериментах с мечеными короткоцепочечными олигонуклеотидами показано, что молекулы антибиотика способны образовывать комплекс, координированный ионом двухвалентного металла (1 : 1 или 2 : 1); комплекс стабилизируется в малой бороздке ДНК водородными связями с аминогруппой гуанина. Комплекс взаимодействует с сайтом длиной как минимум 4 нуклеотида, причем центральные основания имеют ключевое значение для связывания антибиотика с ДНК и стабильности комплекса (GG ≥ GC > CG) [28].
Такие повторы встречаются в том числе в регуляторных областях генов и могут выполнять роль сайтов связывания транскрипционных факторов (ТФ), например, фактора SP1 с сайтом связывания 5'-CGGGGCCCCG-3'. В геноме человека с помощью иммунопреципитации хроматина и высокопроизводительного скрининга обнаружено >12 тыс. подобных консенсусов [30], более половины из них расположены в 5'- или 3'-нетранслируемых областях, где зачастую находятся регуляторные элементы (промоторы, энхансеры, сайленсеры, инсуляторы). SP1-опосредованная транскрипция отмечена, в частности, для генов, участвующих в ответе на повреждения ДНК, в регуляции ангиогенеза, апоптоза, пролиферации, метастазирования и ремоделирования хроматина [31]. Кроме того, уровень SP1 повышен в клетках рака легкого, желудка, поджелудочной железы и др. [32, 33], что обосновывает возможность использования этого ТФ в качестве лекарственной мишени. Обработка клеток аденокарциномы легкого и яичников антибиотиками группы АК приводила к ингибированию экспрессии онкогенов с-Src, c-Myc и MDR1, имеющих сайты связывания ТФ семейства SP в промоторных областях (рис. 2) [34–37].
Рис. 2.
Предполагаемые механизмы ингибирования транскрипции антибиотиками группы ауреоловой кислоты и возможности их применения.
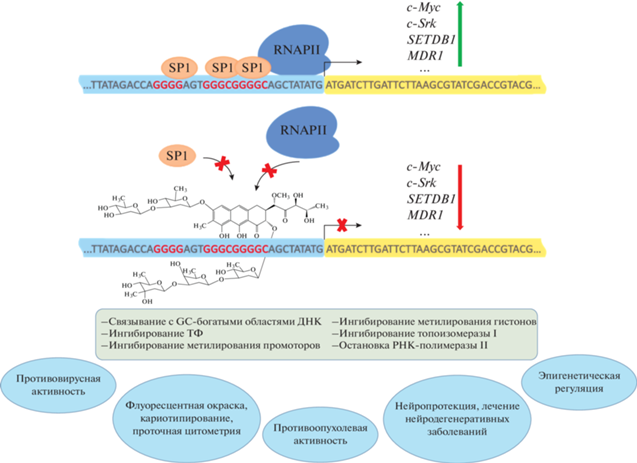
Помимо ингибирования генов-мишеней SP1, митрамицин способен существенно подавлять транскрипцию генов, зависимых от ТФ EWS-FLI1, в клетках саркомы Юинга [38]. Сайт связывания EWS-FLI1 не содержит предпочтительного для митрамицина GC-повтора [39], поэтому механизм его действия остается неясным. Митрамицин продолжает исследоваться в качестве монопрепарата или в комбинированной терапии с ингибиторами CDK9 [40].
Производные АК также ингибируют активность топоизомераз I и II [41, 42], создавая пространственные препятствия для связывания ферментов с ДНК и матричных синтезов. Связывание ауреоловых антибиотиков с ДНК может препятствовать образованию преинициаторного транскрипционного комплекса, связыванию и/или процессированию РНК-полимеразы II [28]. В настоящее время детально исследуется ингибирование функций РНК-полимеразы II при действии оливомицина А и его производного, оливамида, на опухолевые и неопухолевые клетки [43]. Интересно, что оливомицин А вызывает не только ингибирование транскрипции, но и активацию экспрессии ряда генов. Это подтверждает многосторонний характер влияния АК на важнейшие процессы, обусловливающие жизнеспособность клеток.
Свойства антибиотиков группы АК не ограничиваются подавлением экспрессии онкогенов. Они способны усиливать или восстанавливать транскрипцию части генов, регулирующих клеточный цикл и выживание. В нейронах дрозофилы митрамицин ингибировал экспрессию с-Myc и активировал экспрессию р21waf1/cip1 (негативный регулятор клеточного цикла); этими механизмами обусловливался нейропротекторный эффект митрамицина [44] (см. также подраздел “Нейропротекторные свойства”).
Влияние на транскрипционные факторы и структуру хроматина
Предполагалось, что снижение транскрипции SP1-зависимых генов обусловливается тем, что производные АК блокируют сайт связывания ТФ на ДНК. Однако АК снижают уровень и самого SP1 (по крайней мере, в отдельных клеточных линиях) [45, 46]. Примерно на 20% снижался уровень мРНК SP1 в линиях HEp-2 (карцинома гортани) и KB (эпидермальная карцинома) при действии митрамицина (200 и 80 нМ соответственно) в течение 48 ч. Уровень белка SP1 снижался еще более существенно. Обработка клеток митрамицином в комбинации с циклогексимидом (ингибитор синтеза белка) или MG132 (ингибитор 26S протеасомы) показала, что ингибирующее действие митрамицина на SP1 связано с функцией протеасом [45]. Инкубация клеток карциномы яичников с 200 нМ митрамицина или его аналога DIG-MSK снизила уровень мРНК SP1 более чем на 20% уже после 8 часов [47]. На клетках миеломы мыши 5TGM1 подавление SP1 практически не наблюдалось, что не позволяет сделать однозначный вывод о действии митрамицина на экспрессию SP1 [48].
Долгое время антибиотики группы АК рассматривались главным образом как ингибиторы SP1-транскрибируемых генов; в настоящее время также изучается их влияние на эпигенетическую регуляцию транскрипции. Известно, что регуляция укладки хроматина происходит на эпигенетическом уровне: конденсация вызывается метилированием ДНК и модификациями гистонов. Метилирование остатков цитозина привлекает деацетилазу гистонов HDAC в составе хроматин-ремоделирующего комплекса, что способствует образованию транскрипционно неактивного хроматина. Гиперметилирование в CpG-островках промоторов генов-супрессоров опухолей снижает их экспрессию, что может приводить к злокачественной трансформации клетки.
Снижение метилирования при действии митрамицина показано на клетках рака легкого (линии CL1-5 и A549) [49]. Добавление малых концентраций митрамицина (10 нМ) существенно снижало гиперметилирование генов SLIT2 и TIMP3 на 14-й день инкубации. Иммуноблоттинг показал снижение уровня метилтранферазы DNMT1 после 14 дней инкубации клеток с 10 нМ митрамицином. Интересно, что уровень мРНК метилтрансферазы при тех же условиях оставался практически неизменным, хотя ее экспрессия SP1-зависимая [50]. Уровень белка и мРНК метилтрансферазы гистона H3, SETDB1, снижался при инкубации клеток меланомы с митрамицином или его аналогом EC-8042 [51], при этом экспрессия гена SETDB1 SP1-зависимая.
Не вполне ясны механизмы эпигенетической регуляции производными АК, среди вероятных – подавление экспрессии генов, кодирующих ферменты модификации гистонов и ДНК, а также связывание с этими ферментами и их инактивация. Последнее – ингибирование метилтрансферазной активности – рассмотрено для оливомицина А и его производного оливамида, которые способны подавлять ДНК-метилтрансферазу Dnmt3a [52]. Для присоединения донорной метильной группы по положению С-5 цитозина Dnmt3a должна присоединиться к таргетной GC‑последовательности, экспонировать остаток цитозина из минорной бороздки и расположить его в каталитическом центре. После этого происходит образование стабильного ковалентно связанного интермедиата с последующим метилированием цитозинового остатка. Оливомицин А и оливамид препятствуют метилированию на этапе образования стабильного интермедиата, что, видимо, вызвано затруднением доступа каталитического центра Dnmt3a к цитозину в малой бороздке ДНК. Рассматривая вышеназванные свойства оливомицина А, митрамицина и их аналогов, можно предположить, что они действуют как ингибиторы транскрипции метилтрансфераз и/или непосредственно как ингибиторы их ферментативной активности.
Нейропротекторные свойства
Описанные выше свойства производных АК оказываются важными в новых для этого химического класса областях. Указанные антибиотики неожиданно проявили нейропротекторный эффект. Малые дозы митрамицина (150 мкг/кг) способствовали увеличению продолжительности жизни на 30% у модельных мышей с болезнью Хантингтона и улучшению их двигательных функций. При этом происходило существенное снижение уровня метилированного гистона H3 [53]. Такой эффект митрамицина ранее был описан для линий опухолевых клеток (линии рака легкого CL1-5 и A549, линия клеток меланомы А375); оказалось, что, действуя на нейроны, митрамицин защищает их от внешних стрессовых воздействий. Введение хромомицина мышам с моделируемой болезнью Хантингтона также улучшало двигательную активность и увеличивало время жизни, однако помимо этого снижалось количество повреждений ДНК нейронов, а профиль ацетилирования гистонов был таким же, как у мышей дикого типа [54].
Нейропротекторные свойства митрамицина и его аналогов показаны также на моделях заболеваний, в патогенезе которых играет роль нарушение ответов на стресс (неправильная пространственная организация белков – стресс эндоплазматического ретикулума; изменение баланса окисления-восстановления при повреждении ДНК): болезни Альцгеймера, Паркинсона, Хантингтона, дофаминергическая нейротоксичность вследствие употребления метамфетамина, ишемия головного мозга [55, 56]. Эти состояния связаны с увеличением копийности ряда генов, нарушениями эпигенетической регуляции и повышением экспрессии транскрипционных факторов, в том числе SP1.
Важно отметить, что, хотя основной механизм действия ауреоловых антибиотиков – их способность связывать ДНК и ингибировать транскрипцию, тем не менее не всегда этот механизм объясняет наблюдаемые эффекты. В моделях болезни Хантингтона действие митрамицина и хромомицина, по-видимому, не связано с прямым ингибированием экспрессии гена хантингтина (HTT): последняя сохраняется на исходном уровне при добавлении антибиотика [55]. При этом промотор гена HTT содержит три сайта связывания SP1 вблизи точки старта транскрипции. Нейропротекторный эффект может объясняться, по крайней мере частично, снижением экспрессии Myc, Hif1α и Src [44]. Это позволяет нейронам избежать гибели в условиях экзогенного окислительного стресса.
ВЛИЯНИЕ СТРУКТУРЫ АНТИБИОТИКОВ ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ НА БИОЛОГИЧЕСКУЮ АКТИВНОСТЬ
Биологическая активность антибиотиков группы АК определяется, главным образом, их способностью образовывать комплексы с двуцепочечной ДНК в присутствии Mg2+. АК – узкобороздочные лиганды, предпочтительно связывающиеся с GC-богатыми областями генов. Агликоновая часть антибиотика располагается в малой бороздке ДНК, при этом гидроксильная группа в положении С-8 взаимодействует с аминогруппой гуанина, экспонированной внутрь малой бороздки, образуя водородные связи. А- и B-сахара и боковая цепь С-3 агликона образуют межмолекулярные связи с сахарофосфатным остовом ДНК [57]. Двухвалентный ион магния координируется гидроксильной группой С-9 и соседней карбонильной группой С-1 агликона [27]. Образование устойчивого комплекса антибиотик–ДНК повышает локальную температуру плавления, что приводит к стабилизации вторичной структуры ДНК и нарушению матричных синтезов [58].
Различия в структуре главных представителей антибиотиков группы АК во многом определяют основные пути синтеза их аналогов (рис. 3): 1) изменение профиля ацетилирования и метилирования A-, B- и E-сахаров; 2) изменение длины и состава олигосахаридных остатков; 3) модификации агликона по положениям С-5, С-7 и фенольной группе C-8; 4) модификации 2'-кето и 3'-гидроксигрупп или замена боковой цепи агликона на более короткую (производные SA, SK и SDK).
Рис. 3.
Основные направления структурных модификаций антибиотиков группы ауреоловой кислоты (на примере оливомицина А).

Модификации олигосахаридных цепей
4-О-ацетилирование D-олиозы (А-сахар) у хромомицина А3 и оливомицина А, а также D‑хромозы (Е-сахар хромомицина А3) приводит к увеличению стабильности соединений в комплексе с ДНК, вероятно, за счет образования дополнительной водородной связи между ацильной группой и 2-аминогруппой гуанина. A,E-4-О-дидеацетилированная форма хромомицина А3 показала меньшую цитотоксичность на ряде опухолевых линий клеток [59]. Митрамицин не имеет ацетильных заместителей у сахаров, что делает его более “гибким”, т.е. менее требовательным к пространственной структуре GC-богатых участков малой бороздки, позволяя связывать больше GC-сайтов [60]. Отсутствие изобутирильной группы у производных оливомицина А почти не влияло на формирование магний-координируемого комплекса, однако соединение теряло аффинность к ДНК; антипролиферативная активность снижалась [61]. Кроме этого, такая роль изобутирильной группы для проявления антипролиферативной активности оказалась значительно важнее, чем роль ацетильной группы в положении А4.
Более радикальные модификации базовой структуры, такие как изменение состава олигосахаридных цепей, как правило, приводят к существенному снижению аффинности соединения к ДНК. Так, для хромомицина А3 и митрамицина получены аналоги, отличающиеся наличием Е-сахара. Присутствие Е4-ОН-группы митрамицина стабилизирует молекулу в малой бороздке ДНК, обеспечивая прочность связи и увеличивая время диссоциации комплекса. Отсутствие D-микарозы не исключало возможности образования комплекса 4-кетодемикарозилмитрамицин–ДНК, при этом соединение не ингибировало транскрипцию гена cSRK, активируемую SP1 [34]. Аналогичная зависимость скорости диссоциации и, вследствие этого, биологической активности от длины олигосахаридных цепей показана для производных хромомицина А3 [62, 63]. Аналоги оливомицина А с фенилдиазенильными заместителями, присоединенными по 5-положению агликона, и отсутствующей дисахаридной цепью имели низкую цитотоксичность на клеточных линиях лейкозов [14]. Отсутствие токсичности на опухолевых клетках наблюдалось также и для производных агликона оливина. Эти данные указывают на существенную роль олигосахаридных остатков, в частности Е4-О-заместителей, в способности антибиотиков группы АК образовывать стабильные комплексы с ДНК и влиять на экспрессию генов.
Интересно отметить, что, хотя отсутствие дисахаридной цепи оливомицина А приводило к значительному снижению цитотоксичности, его фенилдиазенильные производные были активны в отношении вируса иммунодефицита человека HIV-1 [14]. Цитотоксическая доза для опухолевых клеток была больше эффективной ингибирующей дозы для штамма HIV-1 в 2 раза (индекс селективности, IC50/EC50), а для отдельных производных – в 30 раз. Это делает подобный путь химических модификаций весьма перспективным для поисков антиретровирусных агентов.
Модификации агликона и боковой пентильной цепи
Модификации агликона ауреоловых антибиотиков, как правило, связаны с изменением состава и длины пентильной цепи в С3-положении или с введением функциональных групп по С7-, С8- и С5-положениям.
Например, по С7-положению хромофора возможно введение алкильной группы разной длины. Модификация не так существенна для биосинтеза в клетке продуцента, однако может влиять на биоактивность: так, для UCH9 и дурамицина показано, что 2-метилбутильная группа вытесняет D-сахар из малой бороздки ДНК, приводя к нестабильности структуры и снижению связывания антибиотика с ДНК. Наличие же небольшой метильной группы в том же положении у митрамицина и хромомицина А3 повышает связывание с ДНК и стабильность образующегося комплекса [64], а ее отсутствие снижает аффиность 7-дезметилмитрамицина к ДНК более чем в 150 раз [65].
С5-фенилдиазенильные производные оливомицина А не показали значимого эффекта на опухолевых клетках, однако в их случае происходил одновременный гидролиз дисахаридной цепи, поэтому нельзя однозначно сказать, какой вклад вносят С5-заместители агликона в биологическую активность [14]. Производное оливомицина А с ацетилированной С8-фенильной группой показало схожую с исходным антибиотиком активность на линиях лейкоза [14].
Один из самых успешных подходов к синтезу новых производных АК – модификация пентильной цепи в С3-положении. Подобные производные были впервые получены для митрамицина путем инактивации гена mtmW, кодирующего соответствующую кеторедуктазу, в штамме-продуценте Streptomyces argillaceus. На последних стадиях биосинтеза она отвечает за восстановление 4'‑кетогруппы алкильной боковой цепи после расщепления 4-го кольца агликона. Ее инактивация привела к получению трех новых производных: митрамицина SK (short ketone), митрамицина SA (short acid) и демикарозилмитрамицина SK [66]. Те же производные хромомицин А3 с добавлением еще одного продукта, дикетона (SDK), были получены схожим образов при инактивации гена кеторедуктазы cmmWI [67]. Формы SK и SDK показали схожую или большую активность на ряде опухолевых линий клеток в сравнении с митрамицином или хромомицином А3, а SK-форма митрамицина показала еще и в разы меньшую цитотоксичность и лучший терапевтический индекс. Для оливомицина А полусинтетически была создана SA-форма, однако она не вызывала гибель опухолевых клеток. Другое производное – оливамид, содержащий N,N-диметиламиноэтиламидную группу в боковой цепи, показал схожую с оливомицином А цитотоксичность на серии линий опухолевых клеток. В экспериментах на мышах с трансплантированной меланомой В16 переносимость оливамида превышала таковую оливомицина А [68].
Таким образом, на аффинность АК к ДНК и, как следствие, на биологические свойства, влияют наличие ацетокси- и метоксигрупп в олигосахаридных цепях, длина и состав последних, наличие небольшой алкильной группы в положении С-7, а также тип боковой цепи в положении С-3 агликона.
БИОСИНТЕЗ АНТИБИОТИКОВ ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ
Для митрамицина и хромомицина А3 подробно описан механизм биосинтеза в штаммах-продуцентах Streptomyces argillaceus и Streptomyces griseus. Биосинтез центральной части молекулы на первых стадиях аналогичен получению тетрациклинов и антрациклинов и начинается с конденсации ацетатно-малонатных единиц. Далее у ауреоловых антибиотиков происходит последовательное гликозилирование с образованием моно- и олигосахаридных боковых цепей, а также окислительное расщепление тетрацикла до дигидроантраценового агликона. Дополнительные модификации включают метилирование и/или ацетилирование A-, B-, D- или E-сахаров, а также алкилирование по положению С-7 агликона.
Первый тетрациклический предшественник, 4-деметилпремитрамицинон (4-DMPC), образуется при действии ароматазы (на примере синтеза митрамицина, рис. 4) mtmQ для первых двух колец митрамицина и циклазы mtmY для 3-го кольца из линейного декакетида, после чего под действием циклазы mtmX завершается образование 4-го кольца. При действии метилтрансферазы mtmMI происходит метилирование гидроксильной группы по положению 4 с образованием премитрамицинона (PMC) – это решающий шаг для последующего синтеза [64]. Тетрациклический премитрамицинон – общий предшественник всех антибиотиков этой группы.

Для первого этапа гликозилирования тетрациклического интермедиата необходима D-оливоза, а ее предшественник 4-кето-2,6-дидезокси-D-оливоза – промежуточное звено в синтезе всех остальных мономеров олигосахаридных цепей агликона. Гликозилирование PMC начинается с присоединения первой молекулы D-оливозы, общей для всех антибиотиков группы АК, к гидроксильной группе при положении С-12а агликона. К образованному премитрамицину A1 (РМА1) соответствующие гликозилтрансферазы присоединяют второй сахар D-оливозу (хромомицин А3, оливомицин А) или D-олиозу (митрамицин, UCH9). Синтез трисахаридной цепи завершается присоединением 4-кето-D-микозы (для митрамицина) с последующим восстановлением кеторедуктазой mtmTIII, или L-хромозы (для хромомицина). Е-сахар у оливомицина А представлен 4-О-изобутирил-L-оливомикозой. Трисахаридная цепь может также содержать 4-О-ацетильные формы оливозы и олиозы (дурамицин, UCH9). Гликозилирование по положению С-6 агликона происходит по схожему механизму с последовательным присоединением сахаров. Чаще всего это дисахаридная цепь, содержащая D-оливозу, D-олиозу или их 4-О-ацетильные или метильные производные.
Важный этап синтеза антибиотиков группы АК – расщепление 4-го кольца агликона. В отсутствие этой модификации образуется премитрамицин В, который не проявляет биологической активности на опухолевых клетках [34]. Размыкание цикла происходит под действием монооксигеназы Байера–Виллигера mtmOIV через образование лактонного кольца с его последующим декарбоксилированием и восстановлением кетогруппы при 4'-положении боковой пентильной группы агликона [69, 70]. Интересно, что, хотя действие монооксигеназы, как правило, необходимо для проявления биологической активности, в отдельных случаях оно может не влиять на связывание антибиотика с ДНК. Так, выделенный в результате инактивирующей мутации по гену cmmGII тетрацикличный прехромомицин А3 вызывал гибель опухолевых клеток в субмикромолярных концентрациях [71].
Помимо перечисленных основных преобразований для антибиотиков группы АК существуют дополнительные модификации: алкилирование по положению С-7 агликона, 4-О-метилирование и ацетилирование сахарных остатков. Такие модификации влияют на биологическую активность соединений и используются при поиске новых аналогов. Нужно заметить, что отдельные реакции, помимо придания биологической активности соединению, могут также защищать клетки продуцента от воздействия антибиотика. Например, ацетилирование А- и Е-сахара хромомицина А3 происходит на последних этапах синтеза; мембранная локализация ацетилтрансферазы cmmA, по-видимому, играет существенную роль в устойчивости штамма-продуцента к итоговому продукту биосинтеза [72].
ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ АНТИБИОТИКОВ ГРУППЫ АУРЕОЛОВОЙ КИСЛОТЫ
Хотя антибиотики группы АК в настоящий момент не используются в терапии, они все же нашли практическое применение в медицинских и научных исследованиях. Например, флуоресценция в видимом спектре и аффинность к GC-богатым областям позволяет использовать хромомицин А3 для кариотипирования [73–75]. Комбинация с AT-селективными флуорофорами (DAPI, Хехст 33342) дает возможность визуализировать хромосомы и выявлять их структуру, что важно, в частности, для выявления внутриутробных патологий [76]. Окрашивание хромомицином А3 позволяет определить нарушения конденсации хроматина в сперматозоидах из-за недостаточной протаминизации, что используется в лабораторных исследованиях фертильности [77].
Для применимости производных АК как противоопухолевых агентов один из ключевых вопросов – преодоление общерезорбтивной токсичности. В литературе описаны аналоги митрамицина и хромомицина А3, модифицированные по боковой цепи агликона (SK и SDK) и полученные в результате комбинаторного биосинтеза. Схожее направление (модификация боковой пентильной цепи) было выбрано и для оливомицина А. Поиск основывался на знании механизмов взаимодействия оливомицина А с мишенью – малой бороздкой ДНК. Цель создания нового производного – несколько ослабить связывание с ДНК, сохраняя противоопухолевые свойства. Действительно, у нового производного, оливамида, приемлемое соотношение токсической и эффективной дозы (терапевтическое окно) и улучшенная переносимость in vivo (см. выше), что позволяет рассуждать о его возможном клиническом применении и о векторе дальнейших модификаций структуры. Получение менее токсичных аналогов с сохранением их биологической эффективности служит прекрасным примером того, насколько подобный рациональный подход к мишень-направленному дизайну противоопухолевых препаратов (в том числе на основе АК) оказывается важен в практическом отношении.
ЗАКЛЮЧЕНИЕ
Открытый в 1960-х гг. класс антибиотиков-производных АК проявляет разносторонние свойства – антибактериальные, противоопухолевые и противовирусные. В настоящее время детально изучен биосинтез основных представителей класса – митрамицина и хромомицина А3. Многолетние исследования биосинтеза производных АК обосновали рациональный поиск оптимизированных аналогов митрамицина, хромомицина А3 и оливомицина А, основанный на знании механизмов биологического действия. Наиболее активные производные с модифицированной боковой цепью агликона выделены из продуцентов, полученных в результате инактивирующих мутаций генов кеторедуктазы. У таких производных повышена специфичность к внутриклеточным мишеням – GC-богатым областям в малой бороздке ДНК, снижена общерезорбтивная токсичность. Таким образом, имеются возможности направленной модификации структуры АК для получения производных с улучшенным терапевтическим индексом. Описаны первые оптимизированные соединения для клеток млекопитающих.
Молекулярные механизмы действия антибиотиков группы АК определяются образованием стабильных комплексов с мишенью, следовательно, ведущий механизм – множественные (genome wide) нарушения матричных процессов, главным образом, транскрипции. Это может происходить вследствие эпигенетической регуляции структуры хроматина, подавления экспрессии генов, кодирующих ТФ, а также из-за нарушений конформации ДНК, препятствующих связыванию ТФ и/или функционированию РНК-полимеразы II.
Установление механизмов действия на отдельные гены позволит использовать производные АК для исследований в фундаментальной биологии. Детальное изучение свойств антибиотиков этой группы перспективно для разработки противоопухолевых средств и, возможно, нейропротекторов.
Список литературы
Турсунова Н.В., Чурин Б.В., Клинникова М.Г. // Современные проблемы науки и образования. 2018. Т. 5. С. 1–12. https://doi.org/10.17513/spno.28056
Vardanyan R., Hruby V. // In: Synthesis of Best-Seller Drugs. Academic Press, 2016. P. 495–574. https://doi.org/10.1016/b978-0-12-411492-0.00028-6
Martins-Teixeira M., Carvalho I. // Chem. Med. Chem. 2020. V. 15. P. 933–948. https://doi.org/10.1002/cmdc.202000131
Efferth T., Oesch F. // Semin. Cancer Biol. 2019. V. 26. P. 143–163. https://doi.org/10.1016/j.semcancer.2019.12.010
Тренин А.С. // Антибиотики и химиотерапия. 2015. Т. 60. С. 34–46.
Grundy E., Goldstein W., Rickher J., Hanes M.E., Warren H.B., Sylvester J.C. // Antibiot. Chemother. (Northfield). 1953. V. 3. P. 1215–1217.
Berlin Y., Kiseleva O., Kolosov M., Shemyakin M.M., Soifer V.S., Vasina I., Yartseva I.V., Kuznetsov V.D. // Nature. 1968. V. 218. P. 193–194. https://doi.org/10.1038/218193a0
Кущ А.А., Федосеева Г.Е., Киселева О.А., Зеленин А.В. // Антибиотики. 1972. Т. 17. С. 504–513.
Gause G., Ucholina R., Sveshnikova M. // Antibiotiki. 1962. V. 7. P. 34–38.
Rao K., Cullen W., Sobin B. // Antibiot. Chemother. (Northfield). 1962. V. 12. P. 182–186.
Kaziwara K., Watanabe J., Komeda T., Usui T. // Ann. Rep. Takeda Research Laboratory. 1960. V. 19. P. 68.
Gause G. // In: Antineoplastic and Immunosuppressive Agents / Ed. Sartorelli A.C. Berlin, Heidelberg: Springler-Verlag, 1975. P. 615–622.
Hotter D., Bosso M., Jønsson K.L., Krapp C., Stürzel C.M., Das A., Littwitz-Salomon E., Berkhout B., Russ A., Wittmann S., Gramberg T., Zheng Y., Martins L.J., Planelles V., Jakobsen M.R., Hahn B.H., Dittmer U., Sauter D., Kirchhoff F. // Cell Host Microbe. 2019. V. 25. P. 858–872.E13. https://doi.org/10.1016/j.chom.2019.05.002
Tevyashova A.N., Olsufyeva E.N., Turchin K.F., Balzarini J., Bykov E.E., Dezhenkova L.G., Shtil A.A., Preobrazhenskaya M.N. // Bioorg. Med. Chem. 2009. V. 17. P. 4961–4967. https://doi.org/10.1016/j.bmc.2009.05.076
Bianchi N., Rutigliano C., Passadore M., Tomassetti M., Pippo L., Mischiati C., Feriotto G., Gambari R. // Biochem. J. 1997. V. 326. P. 919–927. https://doi.org/10.1042/bj3260919
Kennedy B., Torkelson J. // Med. Pediatr. Oncol. 1995. V. 24. P. 327–328. https://doi.org/10.1002/mpo.2950240511
Zojer N., Keck A., Pecherstorfer M. // Drug Safety. 1999. V. 21. P. 389–406. https://doi.org/10.2165/00002018-199921050-00004
Perlia C.P., Gubisch N.J., Wolter J., Edelberg D., Dederick M.M., Taylor S.G. // Cancer. 1970. V. 25. P. 389–394. https://doi.org/10.1002/1097-0142(197002)25:2<389: :aid-cncr2820250217>3.0.co;2-x
Condon J.R., Reith S.B., Nassim J.R., Millard F.J., Hilb A., Stainthorpe E.M. // British Med. J. 1971. V. 1. P. 421–423.
Ogawa M. // Appl. Cancer Chemotherapy. 1978. V. 24. P. 149–159. https://doi.org/10.1159/000401511
Новикова И., Савинова В. // Вестник АМН СССР. 1968. Т. 6. С. 53–57.
Sissung T.M., Huang P.A., Hauke R.J., McCrea E.M., Peer C.J., Barbier R.H., Strope J.D., Ley A.M., Zhang M., Hong J.A., Venzon D., Jackson J.P., Brouwer K.R., Grohar P., Glod J., Widemann B.C., Heller T., Schrump D.S., Figg W.D. // Mol. Pharmacol. 2019. V. 96. P. 158–167. https://doi.org/10.1124/mol.118.114827
Antequera F. // CMLS, Cell. Mol. Life Sci. 2003. V. 60. P. 1647–1658. https://doi.org/10.1007/s00018-003-3088-6
Ray R., Snyder R., Thomas S., Koller C.A., Miller D.M. // J. Clin. Invest. 1989. V. 83. P. 2003–2007. https://doi.org/10.1172/jci114110
Blume S., Snyder R., Ray R., Koller C.A., Miller D.M. // J. Clin. Invest. 1991. V. 88. P. 1613–1621. https://doi.org/10.1172/jci115474
Lambert M., Jambon S., Depauw S., David-Cordonnier M. // Molecules. 2018. V. 23. P. 1479. https://doi.org/10.3390/molecules23061479
Hou M. // Nucleic Acids Res. 2004. V. 32. P. 2214–2222. https://doi.org/10.1093/nar/gkh549
Beniaminov A.D., Chashchina G.V., Livshits M.A., Kechko O.I., Mitkevich V.A., Mamaeva O.K., Tevyashova A.N., Shtil A.A., Shchyolkina A.K., Kaluzhny D.N. // Int. J. Mol. Sci. 2020. V. 21. P. 5299. https://doi.org/10.3390/ijms21155299
Carpenter L., Marks N., Fox R. // Eur. J. Biochem. 1993. V. 215. P. 561–566. https://doi.org/10.1111/j.1432-1033.1993.tb18066.x
Cawley S., Bekiranov S., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Wheeler R., Wong B., Drenkow J., Yamanaka M., Patel S., Brubaker S., Tammana H., Helt G., Struhl K., Gingeras T.R. // Cell. 2004. V. 116. P. 499–509. https://doi.org/10.1016/s0092-8674(04)00127-8
Beishline K., Azizkhan-Clifford J. // FEBS J. 2015. V. 282. P. 224–258. https://doi.org/10.1111/febs.13148
Jiang N.Y., Woda B.A., Banner B.F., Whalen G.F., Dresser K.A., Lu D. // Cancer Epidemiology Biomarkers & Prevention. 2008. V. 17. P. 1648–1652. https://doi.org/10.1158/1055-9965.epi-07-2791
Wang L., Wei D., Huang S., Peng Z., Le X., Wu T.T., Yao J., Ajani J., Xie K. // Clin. Cancer Res. 2003. V. 9. P. 6371–6380.
Remsing L., Bahadori H., Carbone G., McGuffie E.M., Catapano C.V., Rohr J. // Biochemistry. 2003. V. 42. P. 8313–8324. https://doi.org/10.1021/bi034091z
Ritchie S., Boyd F., Wong J., Bonham K. // J. Biol. Chem. 2000. V. 275. P. 847–854. https://doi.org/10.1074/jbc.275.2.847
Albertini V. // Nucleic Acids Res. 2006. V. 34. P. 1721–1734. https://doi.org/10.1093/nar/gkl063
Durandin N., Vinogradov A., Shtil A., Kuzmin V. // FEBS J. 2013. V. 280. P. 86–87.
Grohar P.J., Woldemichael G.M., Griffin L.B., Mendoza A., Chen Q.R., Yeung C., Currier D.G., Davis S., Khanna C., Khan J., McMahon J.B., Helman L.J. // JNCI: J. Nat. Cancer Inst. 2011. V. 103. P. 962–978. https://doi.org/10.1093/jnci/djr156
Guillon N., Tirode F., Boeva V., Zynovyev A., Barillot E., Delattre O. // PLoS One. 2009. V. 4. P. 4932. https://doi.org/10.1371/journal.pone.0004932
Flores G., Everett J.H., Boguslawski E.A., Oswald B.M., Madaj Z.B., Beddows I., Dikalov S., Adams M., Klumpp-Thomas C.A., Kitchen-Goosen S.M., Martin S.E., Caplen N.J., Helman L.J., Grohar P.J. // Mol. Cancer Ther. 2020. V. 19. P. 1183–1196. https://doi.org/10.1158/1535-7163.mct-19-0775
Hou M., Lu W., Lin H., Yuann J. // Biochemistry. 2008. V. 47. P. 5493–5502. https://doi.org/10.1021/bi701915f
Tevyashov A.N., Olsufyeva E.N., Balzarini J., Shtil A.A., Dezhenkova L.G., Bukhman V.M., Zbarsky V.B., Preobrazhenskaya M.N. // J. Antibiot. 2009. V. 62. P. 37–41. https://doi.org/10.1038/ja.2008.7
Isagulieva A., Beniaminov V., Tatarskiy V., Soshnikova N.V., Tevyashova A.N., Kaluzhny D.N., Shtil A.A. // FEBS Open Bio. 2019. V. 9. P. 65–431. https://doi.org/10.1002/2211-5463.12675
Sleiman S.F., Langley B.C., Basso M., Berlin J., Xia L., Payappilly J.B., Kharel M.K., Guo H., Marsh J.L., Thompson L.M., Mahishi L., Ahuja P., MacLellan W.R., Geschwind D.H., Coppola G., Rohr J., Ratan R.R. // J. Neurosci. 2011. V. 31. P. 6858–6870. https://doi.org/10.1523/jneurosci.0710-11.2011
Choi E.S., Nam J.S., Jung J.Y., Cho N.P., Cho S.D. // Sci. Rep. 2014. V. 4. P. 1–8. https://doi.org/10.1038/srep07162
Tominaga T., Tsuchiya T., Mochinaga K., Arai J., Yamasaki N., Matsumoto K., Miyazaki T., Nagasaki T., Nanashima A., Tsukamoto K., Nagayasu T. // BMC Cancer. 2016. V. 16. P. 1–9. https://doi.org/10.1186/s12885-016-2392-0
Fernández-Guizán A., López-Soto A., Acebes-Huerta A., Huergo-Zapico L., Villa-Álvarez M., Núñez L.E., Morís F., Gonzalez S. // PLoS One. 2015. V. 10. P. 1–19. https://doi.org/10.1371/journal.pone.0140786
Otjacques E., Binsfeld M., Rocks N., Blacher S., Vanderkerken K., Noel A., Beguin Y., Cataldo D., Caers J. // PLoS One. 2013. V. 8. P. 1–11. https://doi.org/10.1371/journal.pone.0062818
Lin R., Hsu C., Wang Y. // Anticancer Drugs. 2007. V. 18. P. 1157–1164. https://doi.org/10.1097/cad.0b013e3282a215e9
Kishikaw, S., Murata T., Kimura H., Shiota K., Yokoyama K.K. // Eur. J. Biochem. 2002. V. 269. P. 2961–2970. https://doi.org/10.1046/j.1432-1033.2002.02972.x
Federico A., Steinfass T., Larribère L., Novak D., Morís F., Núñez L.E., Umansky V., Utikal J. // Mol. Therapy. Oncolytics. 2020. V. 18. P. 83–99. https://doi.org/10.1016/j.omto.2020.06.001
Cергеев А.Л., Тевяшова А.Н., Воробьев А.П., Громова Е.С. // Биохимия. 2019. Т. 84. С. 229–239. [Sergeev A., Tevyashova A., Vorobyov A., Gromova E. // Biochemistry (Moscow). 2019. V. 84. P. 62–70.] https://doi.org/10.1134/s0006297919010085
Ferrante R. // J. Neurosci. 2004. V. 24. P. 10335–10342. https://doi.org/10.1523/jneurosci.2599-04.2004
Stack E.C., Del Signore S.J., Luthi-Carter R., Soh B.Y., Goldstein D.R., Matson S., Goodrich S., Markey A.L., Cormier K., Hagerty S.W., Smith K., Ryu H., Ferrante R.J. // Hum. Mol. Genet. 2007. V. 16. P. 1164–1175. https://doi.org/10.1093/hmg/ddm064
Osada N., Kosuge Y., Ishige K., Ito Y. // J. Pharmacol. Sci. 2013. V. 122. P. 251–256. https://doi.org/10.1254/jphs.13r02cp
Chatterjee S., Zaman K., Ryu H., Conforto A., Ratan R.R. // Ann. Neurol. 2001. V. 49. P. 345–354. https://doi.org/10.1002/ana.71
Sastry M., Patel D. // Biochemistry. 1993. V. 32. P. 6588–6604. https://doi.org/10.1021/bi00077a012
Andreeva E., Vinogradov A., Tevyashova A., Olsufyeva E.N., Burova T., Grinberg N.V., Grinberg V., Skuridin S., Preobrazhenskaya M., Shtil A., Kuzmin V. // Dokl. Biochem. Biophys. 2010. V. 435. P. 334–338. https://doi.org/10.1134/s1607672910060141
Menéndez N., Nur-E-Alam M., Braña A.F., Rohr J., Salas J.A., Méndez C. // Mol. Microbiol. 2004. V. 53. P. 903–915. https://doi.org/10.1111/j.1365-2958.2004.04166.x
Chakrabarti S. // Indian J. Biochem. Biophys. 2001. V. 38. P. 64–70.
Tevyashova A.N., Durandin N.A., Vinogradov A.M., Zbarsky V.B., Reznikova M.I., Dezhenkova L.G., Bykov E.E., Olsufyeva E.N., Kuzmin V.A., Shtil A.A., Preobrazhenskaya M.N. // J. Antibiot. 2013. V. 66. P. 523–530. https://doi.org/10.1038/ja.2013.39
Behr W., Honikel K., Hartmann G. // Eur. J. Biochem. 1969. V. 9. P. 82–92. https://doi.org/10.1111/j.1432-1033.1969.tb00579.x
Hayasaka T., Inoue Y. // Biochemistry. 1969. V. 8. P. 2342–2347. https://doi.org/10.1021/bi00834a014
Lozano M.J., Remsing L.L., Quirós L.M., Braña A.F., Fernández E., Sánchez C., Méndez C., Rohr J., Salas J.A. // J. Biol. Chem. 2000. V. 275. P. 3065–3074. https://doi.org/10.1074/jbc.275.5.3065
Rodríguez D., Quirós L., Salas J. // J. Biol. Chem. 2003. V. 279. P. 8149–8158. https://doi.org/10.1074/jbc.m312351200
Remsing L.L., González A.M., Nur-e-Alam M., Fernández-Lozano M.J., Braña A.F., Rix U., Oliveira M.A., Méndez C., Salas J.A., Rohr J. // J. Am. Chem. 2003. V. 125. P. 5745–5753. https://doi.org/10.1021/ja034162h
Menéndez N., Nur-e-Alam M., Braña A. F., Rohr J., Salas J. A., Méndez C. // Chem. Biol. 2004. V. 11. P. 21–32. https://doi.org/10.1016/j.chembiol.2003.12.011
Tevyashova A.N., Shtil A.A., Olsufyeva E.N., Luzikov Y.N., Reznikova M.I., Dezhenkova L.G., Isakova E.B., Bukhman V.M., Durandin N.A., Vinogradov A.M., Kuzmin V.A., Preobrazhenskaya M.N. // Bioorg. Med. Chem. 2011. V. 19. P. 7387–7393. https://doi.org/10.1016/j.bmc.2011.10.055
Gibson M., Nur-e-alam M., Lipata F., Oliveira M.A., Rohr J. // J. Am. Chem. Soc. 2005. V. 127. P. 17594–17595. https://doi.org/10.1021/ja055750t
Beam M.P., Bosserman M.A., Noinaj N., Wehenkel M., Rohr J. // Biochemistry. 2009. V. 48. P. 4476–4487. https://doi.org/10.1021/bi8023509
Menéndez N., Nur-e-Alam M., Fischer C., Braña A.F., Salas J.A., Rohr J., Méndez C. // Appl. Environ. Microbiol. 2006. V. 72. P. 167–177. https://doi.org/10.1128/aem.72.1.167-177.2006
García B., González-Sabín J., Menéndez N., Braña A.F., Núñez L.E., Morís F., Salas J.A., Méndez C. // Microb. Biotechnol. 2010. V. 4. P. 226–238. https://doi.org/10.1111/j.1751-7915.2010.00229.x
Ng B.L., Fu B., Graham J., Hall C., Thompson S. // Cytometry. Part A. 2018. V. 95. P. 323–331. https://doi.org/10.1002/cyto.a.23692
Arndt-Jovin D., Jovin T. // Cytometry. 1990. V. 11. P. 80–93. https://doi.org/10.1002/cyto.990110110
Schweizer D. // Chromosoma. 1976. V. 58. P. 307–324. https://doi.org/10.1007/bf00292840
Gray J.W., Trask B., van den Engh G., Silva A., Lozes C., Grell S., Schonberg S., Yu L.C., Golbus M.S. // Am. J. Hum. Genet. 1998. V. 42. P. 49–59.
Iranpour F., Nasr-Esfahani M., Valojerdi M., Taki Al-Taraihi T. // J. Assist. Reprod. Genet. 2000. V. 17. P. 60–66. https://doi.org/10.1023/a:1009406231811
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия