

Биоорганическая химия, 2022, T. 48, № 4, стр. 479-485
Нитроазолопиримидины: влияние на аденозиновый рецептор А1 и внутриглазное давление у крыс
К. В. Саватеев 1, *, В. Л. Русинов 1, С. К. Котовская 1, А. А. Спасов 3, Л. В. Науменко 2, А. С. Таран 3, А. А. Бригадирова 2, Д. С. Яковлев 3, К. Т. Султанова 3, Н. М. Щербакова 2
1 ФГАОУ ВО “Уральский федеральный университет имени первого Президента России Б.Н. Ельцина”
620002 Екатеринбург, ул. Мира, 19, Россия
2 ФГБОУ ВО “Волгоградский государственный медицинский университет” Минздрава России
400131 Волгоград, пл. Павших Борцов, 1, Россия
3 ГБУ “Волгоградский медицинский научный центр”
400131 Волгоград, пл. Павших Борцов, 1, Россия
* E-mail: i-krafttt@yandex.ru
Поступила в редакцию 04.10.2021
После доработки 31.10.2021
Принята к публикации 11.11.2021
- EDN: TORXGG
- DOI: 10.31857/S0132342322040182
Аннотация
На основании роли аденозинового рецептора А1 в модуляции внутриглазного давления, повышение которого – важный фактор развития глаукомы, и структурного анализа среди известных ингибиторов данного рецептора были отобраны и синтезированы шесть соединений ряда 5(7)-алкиламино-6-нитроазолопиримидинов и 8-алкилазоло[5,1-b]пуринов. Показано, что данные гетероциклы проявляют слабое сродство к аденозиновому рецептору А1 на модели аденозинзависимого изменения хронотропного эффекта in vitro с использованием изолированных предсердий белых мышей. С другой стороны, тиадиазоло[3,2-a]пиримидины и триазоло[5,1-b]пурин продемонстрировали гипотензивное действие в экспериментах in vivo на крысах: при использовании соединения-лидера (5-метил-8-(гидроксиэтил)триазоло[5,1-b]пурин, 0.2%-ный раствор) офтальмотонус снижался на 34% через 3 ч, нежелательный резорбтивный эффект не наблюдался. Кроме того, с помощью МТТ-теста на линии клеток гепатоцеллюлярной карциномы человека HepG2 было показано, что гетероциклы, оказывающие влияние на ВГД, демонстрируют 1–2 порядка меньшую цитотоксичность, чем препарат сравнения доксорубицин.
ВВЕДЕНИЕ
В настоящее время глаукома – одна из ведущих причин необратимой слепоты в мире. Количество больных глаукомой даже по самым оптимистичным прогнозам будет непрерывно расти [1], что коррелирует с тенденцией по увеличению числа инвалидов по зрению вследствие глаукомы [2].
Несмотря на определенные успехи в изучении патогенеза и разработке схем лечения данного заболевания, снижение внутриглазного давления (ВГД) остается первоочередной задачей при терапии глаукомы [3].
В местной медикаментозной терапии глаукомы используются препараты с различным механизмом действия: как снижающим продукцию ВГД (β-адреноблокаторы, ингибиторы карбоангидразы II, α-адреномиметики), так и улучшающим ее отток (аналоги простагландинов, м-холиномиметики, α-адреномиметики). В последнее время в литературе обсуждается появление новых классов препаратов, снижающих ВГД путем воздействия на определенные биологические мишени. К ним относятся препараты, снижающие офтальмотонус путем повышения трабекулярного оттока – ингибиторы Rho-киназы [4], соединения, обладающие сродством к III подтипу мелатониновых рецепторов, способные снижать ВГД концентрационно-зависимым образом [5], доноры оксида азота и препараты, воздействующие на глобулярные белки, содержащиеся в клетках трабекулярной сети и шлеммова канала, а также антагонисты рецепторов аденозина.
Было показано, что аденозиновый рецептор А1 (А1 АР) экспрессируется в переднем сегменте человеческого глаза и представляет собой привлекательную мишень для контроля за ВГД [6]. Новые исследования в этой области установили, что данный рецептор активно представлен в дренажной системе глаза, в частности в протоках с водянистой влагой, и играет значительную роль в модуляции ВГД [7, 8].
Так, избыток аденозина – эндогенного агониста А1-рецептора – в передней камере глаза приводит к развитию глазной гипертензии [9]. Соответственно, поиск новых ингибиторов данного подтипа аденозиновых рецепторов – перспективное направление для снижения ВГД.
Цель данной работы – синтез гетероциклов азолоазинового ряда и исследование их влияния на аденозиновый рецептор А1 и внутриглазное давление у крыс для установления возможной взаимосвязи между ингибирующей активностью и гипотензивным действием.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Синтез гетероциклов азолоазинового ряда. Ранее нами было показано, что нитросодержащие азолоазины – это близкие структурные аналоги известных антагонистов аденозинового рецептора А2а [10]. При этом известно, что сайты связывания А2а- и А1-рецепторов близки по структуре, что, с одной стороны, диктует необходимость поиска селективных эффекторов, с другой стороны, можно заключить, что эффекторы А2а-рецептора будут иметь сродство и к сайту связывания аденозинового рецептора А1. Для исследования антагонистического эффекта были синтезированы алкиламин-содержащие нитроазолопиримидины (III–V) по разработанному нами ранее методу последовательного хлордезоксигенирования с помощью смеси хлористого фосфорила и пиридина и нуклеофильного замещения атома галогена на алкиламины (схема 1 ) [11–13]. Подобная стратегия была использована и при синтезе 5-алкиламино-6-нитро-1,3,4-тиадиазоло[3,2-a]пиримидин-8-онов, содержащих фрагменты п-хлорфенилэтиламина (VIII) и тирамина (IX) (схема 2 ).
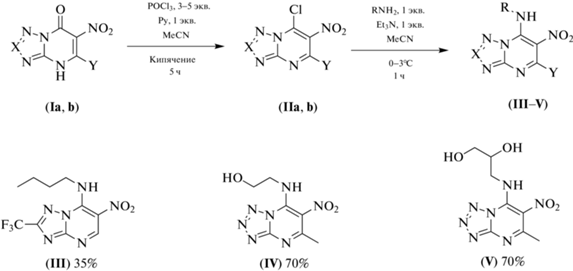
Схема 1 . Двухстадийный синтез 6-нитро-7-алкиламиноазоло[1,5-a]пиримидинов (III–V).
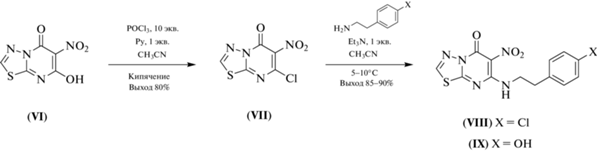
Схема 2 . Синтез 5-алкиламино-6-нитро-1,3,4-тиадиазоло[3,2-a]пиримидин-7-онов (VIII) и (IX).
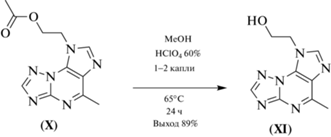
Схема 3 . Снятие ацильной защиты с гидроксильной группы гетероцикла (X) с получением 5-метил-8-(2-гидроксиэтил)-1,2,3-триазоло[5,1-b]пурина (XI).
Кроме того, был получен 5-метил-8-(2-гидроксиэтил)триазоло[5,1-b]пурин (XI), который представляет собой структурный аналог трициклических ингибиторов аденозиновых рецепторов. С этой целью было выполнено снятие ацильной защиты с гидроксигруппы полученного ранее триазоло[5,1-b]пурина (X) с помощью каталитического количества хлорной кислоты при кипячении в метаноле (схема 3 ).
Фармакологические свойства синтезированных гетероциклов. Антагонистическая активность по отношению к аденозиновому рецептору А1 in vitro. Исследование антагонистического действия синтезированных азолоазинов (III–V), (VIII), (IX) и (XI) в концентрации 10 мкМ в отношении аденозинового рецептора А1 проведено на модели аденозинзависимого изменения хронотропного эффекта in vitro с использованием изолированных предсердий белых мышей. Было обнаружено, что наибольшую антагонистическую активность проявляют гетероциклы 6-нитроазолопиримидинового ряда (III–V), в то время как соответствующие тиадиазолопиримидины (VIII), (IX) и триазолопурин (XI) фактически не оказывали статистически значимого действия на А1 АР. Однако все исследуемые соединения уступали по данному показателю препарату сравнения кофеину (табл. 1). Тем не менее полученные результаты позволяют рассматривать триазоло[1,5-a]пиримидиновый скаффолд как перспективную основу для поиска структур с более высоким сродством к данной биологической мишени.
Таблица 1.
А1-антагонистическая активность синтезированных гетероциклов
| Соединение или препарат | Подавление отрицательного хронотропного эффекта аденозина, Δ% |
|---|---|
| (III) | 36.9 ± 5.4 |
| (IV) | 32.5 ± 3.8 |
| (V) | 45.9 ± 3.7 |
| (VIII) | 0.5 ± 0.5* |
| (IX) | 2.5 ± 2.5* |
| (XI) | 2.5 ± 1.1* |
| Кофеин | 60.5 ± 3.7 |
Примечание. Концентрация исследуемых соединений и кофеина составляла 10 мкМ. Данные представлены в виде среднего арифметического значения ± стандартная ошибка среднего арифметического значения. * p < 0.05 по сравнению с показателями, полученными для кофеина (критерий Краскела–Уоллиса с постестом Данна для множественных сравнений).
Офтальмогипотензивные свойства соединений in vitro. Влияние соединений на уровень ВГД изучали на беспородных интактных крысах методом тонометрии. Показано, что все исследуемые соединения обладают офтальмогипотензивными свойствами. Так, при изучении действия гетероцикла (VIII) выявлено повышение офтальмотонуса на 10% в течение 60 мин, после чего происходило незначительное снижение ВГД до исходных значений (рис. 1а) как в тестовом, так и в контрольном глазах, что выступает показателем нежелательного резорбтивного эффекта. Однако к третьему часу ВГД тестового глаза снижалось на 20%, а в контрольном глазу не изменялось.
Рис. 1.
Влияние соединений (VIII) (а), (IX) (б) и (XI) (в) на внутриглазное давление интактных крыс.
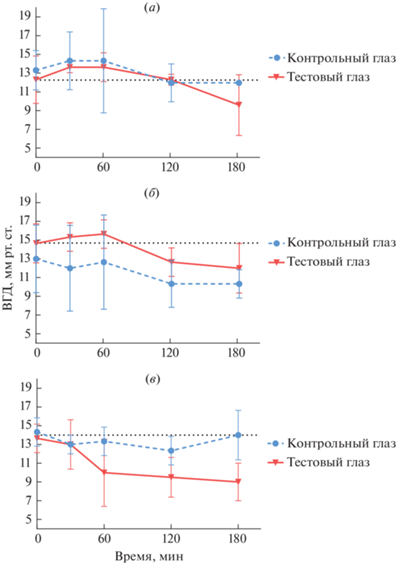
Исследования соединения (IX) продемонстрировали, что в течение 60 мин после его закапывания в тестовый глаз ВГД практически не изменялось. Однако ко второму часу исследования наблюдалось снижение ВГД на 13% и к третьему часу – на 18% (рис. 1б). В контрольном глазу динамика ВГД находилась в пределах нормы, что свидетельствует об отсутствии системного действия изучаемого соединения.
Закапывание в тестовый глаз азоло[5,1-b]пурина (XI) приводило к снижению офтальмотонуса на 26% уже к первому часу исследования, на 30% через 2 ч и достигала максимального эффекта (34%) через 3 ч (рис. 1в). В контрольном глазу ВГД изменялось в диапазоне 1–2 мм рт. ст., что свидетельствует об отсутствии системного действия изучаемого соединения (XI).
При этом оказалось, что наиболее активные в отношении А1 АР гетероциклы (III–V) не приводят к снижению ВГД в исследуемых концентрациях. По всей видимости, влияние соединений (VIII), (IX) и (XI) на ВГД не связано с ингибированием А1 АР, а задействует другой механизм биологического действия.
Цитотоксичность соединений. На заключительном этапе с целью предварительной оценки токсикологических характеристик наиболее активных соединений (VIII), (IX) и (XI) была изучена их цитотоксичность в МТТ-тесте на линии клеток человека HepG2. Для 6-нитротиадиазоло[3,2-a]пиримидинов (VIII) и (IX) цитотоксичность LC50 составила 0.073 и 0.072 мМ соответственно, что в 30–40 раз меньше, чем у препарата сравнения доксорубицина. Наименьшее цитотоксическое действие выявлено для триазоло[5,1-b]пурина (XI), уровень LC50 для которого превышает 1 мМ и находится за пределами максимальных исследованных концентраций (табл. 2), превосходя по безопасности доксорубицин более чем в 500 раз.
Таблица 2.
Цитотоксичность соединений (VIII), (IX) и (XI) в сравнении с доксорубицином на клетках гепатоцеллюлярной карциномы человека HepG2 (МТТ-тест)
| Соединение или препарат | LC50, мМ |
|---|---|
| (VIII) | 0.073 |
| (IX) | 0.072 |
| (XI) | >1 |
| Доксорубицин | 0.002 |
Было показано, что с точки зрения сродства к А1 АР наиболее выгоден 1,2,4-триазоло[1,5-a]пиримидиновый (III) и тетразоло[1,5-a]пиримидиновый скаффолд (IV,V), тогда как 1,3,4-тиадиазоло[3,2-a]пиримидины (VIII), (IX) и 1,2,4-триазоло[5,1-b]пурин (XI) не проявили статистически значимого ингибирующего эффекта в отношении этого рецептора. С другой стороны, полученные 6-нитроазоло[1,5-a]пиримидины уступали в сродстве к А1 АР препарату сравнения кофеину, что свидетельствует о необходимости дальнейшей структурной модификации для создания более мощных ингибиторов.
Обратная закономерность наблюдалась при исследовании офтальмогипотензивных свойств полученных гетероциклов: азоло[1,5-a]пиримидины (III–V) не оказывали влияния на ВГД крыс, в то время как тиадиазоло[3,2-a]пиримидины (VIII), (IX) и триазоло[5,1-b]пурин (XI) продемонстрировали гипотензивное действие. При этом соединение-лидер, 5-метил-8-(гидроксиэтил)триазоло[5,1-b]пурин (XI), снижало офтальмотонус на 34% через 3 ч, не проявляя нежелательного резорбтивного эффекта. Кроме того, было показано, что гетероциклы, оказывающие влияние на ВГД, демонстрируют значительно меньшую цитотоксичность, чем препарат сравнения доксорубицин.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез гетероциклов азолоазинового ряда. Спектры 1Н- и 13С-ЯМР полученных соединений регистрировали на спектрометре Avance II (400 и 100 МГц соответственно; Bruker, Германия) при температуре 25°С, внутренний стандарт ТМС, растворитель DMSO-d6 и CDCl3. Элементный анализ выполняли на анализаторе 2400 CHN (PerkinElmer, США). Контроль за ходом реакций осуществляли при помощи ТСХ на пластинках Silufol UV-254 (Imid LTD, РФ, Краснодар). Температуру плавления измеряли на приборе Stuart SMP3. Гетероциклы (III) [14], (IV) и (V) [12], (VIII) и (IX) [13] и (X) [11] были синтезированы по ранее разработанным методикам.
Синтез 5-метил-8-(2-гидроксиэтил)триазоло-[5,1-b]пурина (XI). К раствору 2.18 г (0.01 моль) 5‑метил-8-(2-ацетоксиэтил)триазоло[5,1-b]пурина в 40 мл MeOH добавляли 0.5 мл 60%-ной HClO4, полученную смесь кипятили в течение 24 ч. Реакционную массу упаривали в вакууме при 35°С, остаток перекристаллизовывали из изо-бутанола.
Белый порошок. Rf (EtOAc) 0.4, Т. пл. 214–216°С. Выход 89%. Найдено, %: С 49.44; Н 4.50; N 38.80. Брутто-формула: C9H10N6O. Вычислено, %: С 49.54; Н 4.62; N 38.51. Спектр 1H-ЯМР (400 МГц, DMSO-d6), δ (м.д.): 2.84 (3Н, c, CH3), 3.87 (2Н, т, J 4.0, OСН2), 4.69 (2Н, т, J 4.0, NСН2), 8.40 (1Н, c, С7Н), 8.84 (1Н, c, C2H). Спектр 13С-ЯМР (100 МГц, CDCl3), δ (м.д.): 20.2 (CH3), 49.0 (NCH2), 59.8 (OCH2), 127.5 (C5a), 134.3 (C8a), 142.6 (C7), 151.2 (C3a), 152.3 (C2), 159.4 (C5).
Фармакологические исследования полученных соединений. Антагонистическая активность соединений по отношению к аденозиновому рецептору A1. Исследование антагонистической активности соединений (III–V), (VIII), (IX) и (XI) по отношению к A1 АР проводили на изолированных предсердиях 20 белых мышей обоих полов возрастом 4 месяца (питомник “Рапполово”, Ленинградская область) на модели аденозинзависимого изменения хронотропного эффекта in vitro в модификации Бригадировой с соавт. [15] с использованием буферного раствора Кребса–Хенселейта (состав, мМ): NaCl – 118.0; KCl – 4.7; KH2PO4 – 1.18; MgSO4 – 1.2; CaCl2 – 2.5; NaHCO3 – 25.0; глюкоза – 5.55; pH 7.4) при постоянной оксигенации (95% O2 – 5% СО2) и термостатировании при 37°С. Исследуемые вещества в концентрации 10 мкМ вносили в ванночку с изолированными предсердиями, после чего добавляли аденозин (10 мкМ; Sigma-Aldrich, США). Активность соединений оценивали по степени подавления индуцированного аденозином снижения хронотропизма изолированных предсердий, работающих в собственном ритме (без стимуляции), в сравнении с контрольным эффектом аденозина (в Δ%). Сокращения предсердий регистрировали с использованием изометрического датчика TSD125C при изометрической нагрузке в 1 г в 4-канальной установке поддержания жизнедеятельности изолированных тканей DA100C (Biopac Systems, Inc., США) и программного обеспечения AcqKnowledge 4.0 (Biopac Systems, Inc., США). Количество сокращений изолированных предсердий измеряли за 30-секундный интервал с последующим расчетом частоты сердечных сокращений (ЧСС). В качестве препарата сравнения использовали неселективный антагонист аденозиновых рецепторов А1/2а – кофеин (10 мкМ; Sigma-Aldrich, США).
Величину подавления хронотропизма предсердий (Δ%) рассчитывали по формуле:
Статистическую обработку результатов проводили с использованием непараметрического критерия Краскела–Уоллиса с постестом Данна для множественных сравнений в программе GraphPad Prism 7.0 (GraphPad Software, США).
Офтальмогипотензивные свойства соединений in vitro. Изучение влияния соединений на уровень ВГД проводили на 48 взрослых беспородных интактных крысах обоих полов возрастом 2 месяца (питомник “Рапполово”, Ленинградская область) методом тонометрии с использованием прибора TonoVet (Финляндия) [16]. Исследование соединений производили по методике Marcus et al. [17]. Производные азолопиримидинов (III–V), (VIII), (IX) и (XI) закапывали в правый (тестовый) глаз лабораторного животного в концентрации 0.2% однократно в объеме 50 мкл, а в левый глаз – деионизированную воду в том же объеме. В качестве препаратов сравнения использовали 0.5%-ные глазные капли тимолол (Тимолол-СОЛОфарм; Гротекс, Россия) и 0.1%-ные глазные капли бримонидин (Сантибрим; Сентисс Фарма Пвт. Лтд., Индия) с доказанным ВГД-снижающим действием, применяемые в клинической практике. Препараты сравнения также закапывали в правый (тестовый глаз) лабораторных животных в объеме 50 мкл, в левый глаз – деионизированную воду в том же количестве. Левый глаз, в свою очередь, служил для оценки возможного системного воздействия исследуемых соединений. Измерение ВГД проводили в пяти временных точках (0, 30, 60, 120, 180 мин), где 0 мин – исходное значение. Наличие ВГД-снижающей активности оценивали по максимальному уменьшению ВГД от исходных значений.
Цитотоксичность соединений. Оценку цитотоксичности наиболее активных соединений (VIII), (IX) и (XI) проводили с помощью МТТ-теста [18] в модификации на клетках линии HepG2 (гепатоцеллюлярной карциномы человека) (ATCC® HB-8065™). Культивирование клеток проводили в полной ростовой среде F-12 (Gibco, США) с добавлением 10% эмбриональной телячьей сыворотки (Gibco, США), 1% пенициллина-стрептомицина (Gibcо, США), 1% незаменимых аминокислот (NEAA) (Sigma-Aldrich, США), 2 мМ пирувата натрия (Sigma-Aldrich, США) в СО2-инкубаторе Galaxy 170R (New Brunswick, Германия) при температуре 37°С в атмосфере 5% СО2. Соединения и наиболее широко используемый цитостатик сравнения доксорубицин (Sigma-Aldrich, США) исследовали в диапазоне концентраций от 0.1–1.0 мМ с инкубацией в течение 48 ч. Жизнеспособность клеток, коррелирующую со способностью митоходриальных дегидрогеназ превращать МТТ-реагент (бромид 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия) в формазан, определяли по данным оптической плотности при 555 нм (референсная длина волны 650 нм) с использованием планшетного ридера CLARIOstar (BMG LABTECH, США). Обработку данных и расчет LC50 (концентрация, подавляющая жизнедеятельность клеток на 50% относительно интактного контроля) проводили с помощью программного обеспечения MARS Data Analysis Software (BMG LABTECH, США) и GraphPad Prism v.7.0 (GraphPad Software, США).
ЗАКЛЮЧЕНИЕ
Таким образом, были синтезированы шесть соединений ряда азолоазинов. На модели аденозинзависимого изменения хронотропного эффекта in vitro с использованием изолированных предсердий белых мышей показано, что с точки зрения сродства к аденозиновому рецептору А1 наиболее выгоден 1,2,4-триазоло[1,5-a]пиримидиновый (III) и тетразоло[1,5-a]пиримидиновый (IV), (V) скаффолд, тогда как 1,3,4-тиадиазоло[3,2-a]пиримидины (VIII), (IX) и 1,2,4-триазоло[5,1-b]пурин (XI) не проявили статистически значимого ингибирующего эффекта в отношении этого рецептора. С другой стороны, полученные 6-нитроазоло[1,5-a]пиримидины уступали препарату сравнения кофеину в сродстве к аденозиновому рецептору А1, что свидетельствует о необходимости дальнейшей структурной модификации для создания более мощных ингибиторов. Обратная закономерность наблюдалась при исследовании офтальмогипотензивных свойств полученных гетероциклов: азоло[1,5-a]пиримидины (III–V) не оказывали влияния на внутриглазное давление крыс, в то время как тиадиазоло[3,2-a]пиримидины (VIII), (IX) и триазоло[5,1-b]пурин (XI) продемонстрировали гипотензивное действие. При этом при использовании соединения-лидера, 5-метил-8-(гидроксиэтил)триазоло[5,1-b]пурина (XI), офтальмотонус снижался на 34% через 3 ч, нежелательный резорбтивный эффект не наблюдался. Кроме того, в МТТ-тесте на линии клеток гепатоцеллюлярной карциномы человека HepG2 было показано, что гетероциклы, оказывающие влияние на ВГД, демонстрируют на 1–2 порядка меньшую цитотоксичность, чем препарат сравнения доксорубицин.
Список литературы
Chou S.F., Luo L.J., Lai J.Y. // Sci Rep. 2017. V. 7. P. 42344. https://doi.org/10.1038/srep42344
Нероев В.В., Киселева О.А., Бессмертный А.М. // Российский офтальмологический журнал. 2013. Т. 6. № 3. С. 4–7.
Национальное руководство по глаукоме: для практикующих врачей (3-е изд., испр. и доп.) / Под ред. Егорова Е.А., Астахова Ю.С., Еричева В.П. Москва: ГЭОТАР-Медиа, 2015. 456 с.
Rao P.V., Pattabiraman P.P., Kopczynski C. // Exp. Eye Res. 2017. V. 158. P. 23–32. https://doi.org/10.1016/j.exer.2016.08.023
Guglielmi P., Carradori S., Campestre C., Poce G. // Exp. Opin. Therap. Pat. 2019. V. 29. P. 769–780. https://doi.org/10.1080/13543776.2019.1653279
Kvanta A., Seregard S., Sejersen S., Kull B., Fredholm B.B. // Exp. Eye Res. 1997. V. 165. P. 595–602. https://doi.org/10.1006/exer.1996.0352
Li A., Leung C.T., Peterson-Yantorno K., Daniel Stamer W., Civan M.M. // Investig. Ophthalmol. Visual Sci. 2011. V. 52. P. 7996–8005. https://doi.org/10.1167/iovs.11-8170
Avila M.Y., Stone R.A., Civan M.M. // Br. J. Pharmacol. 2001. V. 134. P. 241–245. https://doi.org/10.1038/sj.bjp.0704267
Agarwal R., Agarwal P. // Exp. Opin. Therap. Targ. 2014. V. 18. P. 527–539. https://doi.org/10.1517/14728222.2014.888416
Savateev K.V., Ulomsky E.N., Butorin I.I., Charushin V.N., Rusinov V.L., Chupakhin O.N. // Russ. Chem. Rev. 2018. V. 87. P. 636–669. https://doi.org/10.1070/RCR4792
Savateev K.V., Ulomsky E.N., Borisov S.S., Voinkov E.K., Fedotov V.V., Rusinov V.L. // Chem. Heterocycl. Comp. 2014. V. 50. P. 880–887. https://doi.org/10.1007/s10593-014-1542-z
Savateev K.V., Fedotov V.V., Ulomskiy E.N., Rusinov V.L. // Chem. Heterocycl. Comp. 2018. V. 54. P. 197–204. https://doi.org/10.1007/s10593-018-2254-6
Savateev K., Fedotov V., Butorin I., Eltsov O., Slepukhin P., Ulomsky E., Rusinov V., Litvinov R., Babkov D., Khokhlacheva E., Radaev P., Vassilie P., Spasov A. // Eur. J. Med. Chem. 2020. V. 185. P. 111808. https://doi.org/10.1016/j.ejmech.2019.111808
Spasov A.A., Babkov D.A., Sysoeva V.A., Litvinov R.A., Shamshina D.D., Ulomsky E.N., Savateev K.V., Fedotov V.V., Slepukhin P.A., Chupakhin O.N., Charushin V.N., Rusinov V.L. // Archiv der Pharmazie. 2017. V. 350. P. 1700226. https://doi.org/10.1002/ardp.201700226
Бригадирова А.А., Агацарская Я.В., Салихов Д.А., Нагих А.С. // Вестник ВолгГМУ. 2019. Т. 70. С. 55–57. https://doi.org/10.19163/1994-9480-2019-2(70)-55-57
Pease M.E., Hammond J.C., Quigley H.A. // J. Glaucoma. 2006. V. 15. P. 512–519. https://doi.org/10.1097/01.ijg.0000212276.57853.19
Marcus A.J., Iezhitsa I., Agarwal R., Vassiliev P., Spasov A., Zhukovskaya O., Anisimova V., Mohd Ismail N. // Data Brief. 2018. V. 18. P. 523–554. https://doi.org/10.1016/j.dib.2018.03.019
Яковлев Д.С., Султанова К.Т., Золотова Е.А., Гасайниева А.Г., Спасов А.А. // Волгоград. науч.-мед. журн. 2020. № 1. С. 58–61. https://doi.org/10.24412/1995-7225-2020-1-58-61
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия