

Биоорганическая химия, 2022, T. 48, № 6, стр. 694-706
Взаимодействие фторфенильного аналога ретиналя с протеородопсином из Exiguobacterium sibiricum
Н. Е. Беликов 1, Л. Е. Петровская 2, Е. А. Крюкова 2, Д. А. Долгих 2, 3, Е. П. Лукашев 3, А. Ю. Лукин 4, О. В. Демина 1, С. Д. Варфоломеев 1, В. В. Чупин 5, А. А. Ходонов 1, *
1 ФГБУН “Институт биохимической физики им. Н.М. Эмануэля” РАН
119334 Москва, ул. Косыгина, 4, Россия
2 ФГБУН “Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова” РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
3 ФГБОУ ВО “Московский государственный университет им. М.В. Ломоносова”, биологический факультет
119991 Москва, Ленинские горы, 1, стр. 12, Россия
4 ФГБОУ ВО МИРЕА – Российский технологический университет
119571 Москва, просп. Вернадского, 78, Россия
5 ФГАОУ ВО Московский физико-технический институт
141701 Долгопрудный, Институтский пер., 9, Россия
* E-mail: khodonov@gmail.com
Поступила в редакцию 12.04.2022
После доработки 07.05.2022
Принята к публикации 31.05.2022
- EDN: PMZUIU
- DOI: 10.31857/S0132342322060070
Аннотация
Разработан альтернативный вариант метода синтеза аналога природного ретиналя, у которого триметилциклогексеновое кольцо молекулы замещено на п-фторфенильный фрагмент. Показано, что предложенная нами схема синтеза целевого соединения с использованием С5-фосфоната с терминальной нитрильной группой в условиях реакции Хорнера–Эммонса более эффективна и дает более высокий суммарный выход целевого продукта, чем вариант синтеза, описанный нами ранее. Разработана процедура получения аналога микробного протеородопсина ESRh из Exiguobacterium sibiricum с модифицированным хромофором. Установлено, что, как и в случае бактериоопсина из Halobacterium salinarum, замена триметилциклогексенового кольца в природном хромофоре на п-фторфенильный фрагмент не блокирует возможность образования из протеородопсина ESRh искусственного пигмента F-Phe-ESRh, сохраняющего цикл фотохимических реакций. Обнаружены определенные различия в свойствах нативного рекомбинантного ESRh и его аналога F-Phe-ESRh, включающие сдвиг максимума поглощения в коротковолновую область, образование интермедиата М при более низких значениях рН, наличие “долгоживущего М” и общее замедление фотоцикла. Также была продемонстрирована пониженная стабильность полученного аналога протеородопсина F-Phe-ESRh к продолжительному воздействию видимого света.
ВВЕДЕНИЕ
Ретиналь-содержащие белки играют ключевую роль в ряде важнейших биологических процессов: зрении, светозависимом транспорте протонов и других ионов (хлора, натрия), фототаксисе. Ретиналь-содержащие белки подразделяются на несколько семейств, основные из которых – зрительные пигменты палочек и колбочек высших животных и микробные родопсины. Более 1000 представителей этих хромопротеинов были найдены во всех биологических царствах – от высших животных до архей, грибов, водорослей, эубактерий и вирусов [1–3].
Молекулы ретиналь-содержащих белков имеют следующие общие особенности: 1) они представляют собой мембранные белки, состоящие из семи α-спиральных трансмембранных тяжей (7TM), соединенных между собой петлями; 2) в качестве хромофорной группы они содержат определенный изомер альдегида витамина А – ретиналя (all-E- в пигментах микроорганизмов и 11Z- в зрительных пигментах), который соединен с белком через протонированную альдиминную связь с ε-аминогруппой остатка Lys; 3) их общая функция связана с преобразованием энергии светового кванта в различные химические или физиологические ответы [1–6].
В 2022 г. исполнилось 17 лет с начала “эры оптогенетики” (нейробиофотоники), цель которой состоит в использовании генетически кодируемых фоточувствительных белков для регуляции активности клеток человека и животных. Для реализации этого перспективного направления исследований необходимо создание технологий получения фотопереключаемых компонентов для оптогенетики с заданными и/или прогнозируемыми спектрально-кинетическими параметрами на основе новых модифицированных микробных родопсинов и других фоточувствительных белков [5, 6].
Направленные изменения фотохимических свойств ретиналь-содержащих белков могут быть достигнуты путем использования следующих подходов: 1) замещением одного или нескольких аминокислотных остатков в определенных положениях молекулы белка методами сайт-специфичного мутагенеза; 2) замещением остатка природного ретиналя на его различные аналоги; 3) сочетанием вышеперечисленных методов.
Один из возможных вариантов исследования взаимосвязи структура–функция в ретиналь-содержащих белках заключается в замене природного хромофора на его аналоги и всестороннее изучение свойств новых гибридных продуктов [7–14]. Аналоги нативного хромофора уже дали ценную структурную, спектроскопическую и функциональную информацию о структуре основного состояния хромофора в микробных родопсинах до того, как стали доступны 3D-структуры этих молекул с высоким разрешением, и продолжают широко использоваться для определения структуры промежуточных продуктов их фотоциклов. Процедура направленной модификации хромофора позволяет варьировать в широких пределах как положение максимума поглощения основного состояния пигмента, так и другие важные фотохимические параметры.
Таким образом, замена природного ретиналя на его аналоги – один из наиболее перспективных подходов к изучению особенностей участка связывания хромофора у различных семейств микробных родопсинов. Успешное применение данного метода было продемонстрировано для хорошо изученного светозависимого протонного насоса бактериородопсина (BRh) из экстремально галофильного микроорганизма Halobacterium salinarum, на котором в течение последних 40 лет был апробирован широкий арсенал современных методов исследования для определения взаимосвязи структура–функция. Искусственные аналоги BRh были получены с целым рядом модификаций молекулы его хромофора [7, 8].
Объект настоящего исследования – протеородопсин (ESRh) – это новый представитель ретиналь-содержащих белков из психротрофного микроорганизма Exiguobacterium sibiricum, ген которого был выделен из образцов почвы вечной мерзлоты возрастом 3 млн лет [15]. Протеородопсин (ESRh) по своей структуре представляет собой мембранный белок, функционирующий как светозависимый протонный насос. Пространственная 3D-структура ESRh с разрешением 2.3 Å (PDB: 4HYJ) была впервые исследована группой ученых под руководством В. Горделия (Институт структурной биологии, Гренобль, МФТИ). Установлено, что ESRh обладает структурой, типичной для семейства ретиналь-содержащих белков, включая наличие семи α-спиральных сегментов и молекулы ретиналя в all-E-конфигурации, ковалентно связанной с остатком К225 посредством протонированной альдиминной связи [16].
К особенностям строения протеородопсина ESRh относятся: 1) наличие нестандартного донора протонов для основания Шиффа – остатка K96 – и его расположение в гидрофобной полости ближе к поверхности белка; 2) нарушение α-спиральной структуры в средней части спирали F; 3) наличие водородной связи между остатками H57 и D85; 4) наличие ряда специфических особенностей фотоцикла ESRh, включая освобождение протона на поздних стадиях фотоцикла, ускоренное образование М-подобного интермедиата и сдвиг его pK в щелочную область pH; 5) отсутствие спектральных различий в формах ESRh, адаптированных к свету и темноте.
Большое число работ было посвящено изучению механизма функционирования ESRh и его мутантных вариантов [15–22], однако исследования, связанные с заменой хромофорной группы, ранее не проводились и являются пионерскими.
Существование ряда отличий в механизмах фотоцикла и протонного транспорта ESRh от других ретиналь-содержащих белков делает интересным и актуальным сравнительное изучение влияния типа модификации их хромофорных групп на спектральные параметры этих белков и на их функционирование [8, 22].
Для исследования влияния природы хромофорной группы ESRh на его функционирование и структуру в настоящей статье нами представлены: 1) альтернативный метод синтеза фторфенильного аналога ретиналя (II); 2) результаты изучения процесса взаимодействия аналога ретиналя (II) с апобелком ESR-опсином из E. sibiricum; 3) фотохимические характеристики полученного искусственного пигмента.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Синтез фторфенильного аналога ретиналя (II). В данной работе для модификации ESRh нами был выбран фторфенильный аналог ретиналя (II), поскольку ранее он был успешно использован для получения фторфенильного аналога BRh из H. salinarum [23]. Кроме того, присутствие атома фтора в п-фторфенильном фрагменте открывает возможность использования метода ЯМР-спектроскопии на ядрах 19F для изучения структуры этого белка. Проведенное в работе Драчева с соавт. [24] исследование цикла фотореакций фторфенильного аналога BRh показало, что хотя этот искусственный пигмент и сохранил эффективность фотоцикла на уровне ~50% от эффективности цикла природного BRh, но у него была на несколько порядков замедлена обратная реакция свето-темновой адаптации.
В указанных работах [22, 23] all-E- и 13Z-изомеры фторфенильного аналога ретиналя (II) были синтезированы при помощи 4-стадийной процедуры наращивания полиеновой цепи ретиноида с использованием в качестве ключевой стадии реакции олефинирования по Виттигу карбонильного С10-предшественника (IV) с илидом, генерированным из 4-фторбензилтрифенилфосфонийбромида (III), с суммарным выходом 41% (схема 1 ).
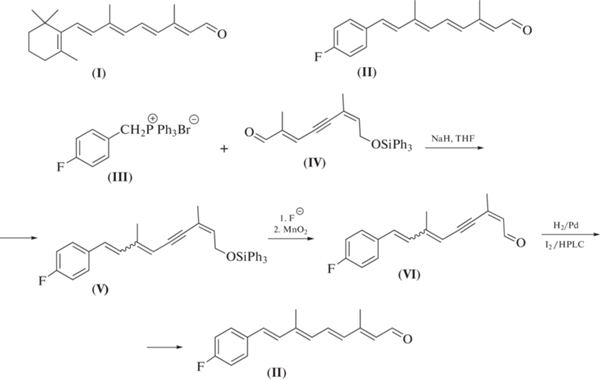
Схема 1 . Синтез целевого ретиноида (II) при помощи реакции олефинирования карбонильных соединений по Виттигу.
В настоящей работе нами был предложен и исследован альтернативный классический вариант наращивания полиеновой цепи ретиноида олефинированием исходного альдегида (XIV) анионом С5-фосфоната (XIIIa, b) в условиях реакции Хорнера–Эммонса (схемы 2, 3 ) [7, 25–29].
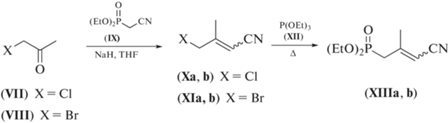
Схема 2 . Синтез С5-фосфоната (XIIIa, b) с терминальной нитрильной группой.
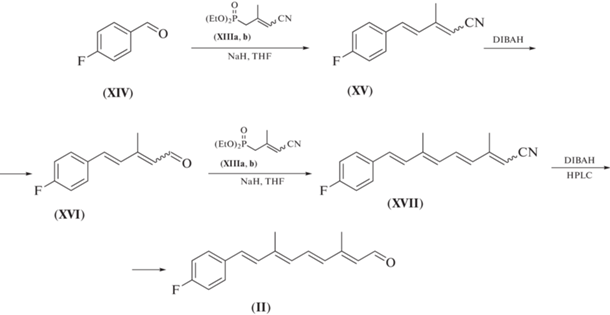
Схема 3 . Синтез целевого ретиноида (II) при помощи реакции олефинирования карбонильных соединений по Хорнеру–Эммонсу.
Сравнение двух синтетических подходов к получению целевого ретиноида (II). Для решения задачи создания системы сопряженных двойных связи в полиеновой цепи молекулы ретиноида (II) ранее [22, 23] и в рамках настоящего исследования в качестве ключевых реакций были использованы два варианта реакции олефинирования карбонильных соединений: 1) реакция олефинирования по Виттигу (схема 1 ); 2) реакция олефинирования по Хорнеру–Эммонсу (схема 3 ).
Ниже представлена критическая оценка преимуществ и недостатков двух синтетических подходов к получению целевого ретиноида (II), изображенных на схемах 1 и 3 .
Схема 1 (ключевая стадия – реакция олефинирования по Виттигу):
1) выход – высокий;
2) стереоселективность вновь образованной двойной связи – низкая, образуется смесь E- и Z-изомеров (1 : 1);
3) трудноотделяемый побочный продукт – трифенилфосфиноксид;
4) необходимость применения препаративной ВЭЖХ – да;
5) проблема потери регио- и стереоселективности процедуры полувосстановления тройной связи потребовала дополнительных исследований;
6) требуется дополнительная стадия – изомеризация 13Z-изомера под действием иода.
Схема 3 (ключевая стадия – реакция олефинирования по Хорнеру–Эммонсу):
1) выход – высокий;
2) стереоселективность вновь образованной двойной связи – высокая (98% E-изомера);
3) необходимость применения препаративной ВЭЖХ – да;
4) необходимость разработки и поиска оптимальной процедуры синтеза С5-фосфоната (XIIIa, b).
Таким образом, для схем синтеза 1 и 3 характерно наличие высокого выхода желаемого продукта, а также использование препаративной ВЭЖХ для получения индивидуальных соединений. Преимуществами схемы 3 являются высокая стереоселективность вновь образованной двойной связи (98%), а также разработка и оптимизация только одной синтетической процедуры, тогда как для схемы 1 существенными недостатками являются низкая стереоселективность вновь образованной двойной связи (E-/Z- 1 : 1), наличие трудноотделимого побочного продукта – трифенилфосфиноксида и разработка с последующей оптимизацией двух синтетических процедур.
Синтез С5-фосфоната (XIIIa, b) с терминальной нитрильной группой. Синтез исходного С2-фосфоната (IX) проводили конденсацией коммерчески доступного хлорацетонитрила с триэтилфосфитом (XII) по Арбузову. Выход соединения (IX) после перегонки составил 87%.
Для осуществления синтеза C5-фосфоната (XIIIa, b) были исследованы два пути, показанные на схеме 2 . В качестве галогенкетона использовали коммерчески доступный хлорацетон (VII) или бромацетон (VIII), синтезированный в одну стадию бромированием ацетона (выход 50%).
Конденсация кетона (VII) с С2‑фосфонатом (IX) в условиях реакции Хорнера–Эммонса дает изомерную смесь галогенонитрила (Xa, b) с общим выходом 52%. Конденсация кетона (VIII) с С2-фосфонатом (IX) в тех же условиях приводит к образованию изомерной смеси галогенопроизводного (XIa, b) с общим выходом 30%. Низкий выход реакции по второму варианту, через бромокетон (VIII), объясняется его низкой стабильностью, поэтому выгоднее использовать хлорацетон (VII). Кроме того, бромосодержащий нитрил (XIa, b) обладает меньшей термостабильностью, чем соответствующий хлорозамещенный нитрил (Xa, b). Полученные галогенонитрилы (Xa, b) и (XIa, b) вводили в дальнейшие превращения без дополнительной очистки. Конденсация соединения (Xa, b) с триэтилфосфитом (XII) по Арбузову дает после перегонки изомерную смесь С5-фосфоната (XIIIa, b) с выходом 81%. Соотношение изомеров (XIIIa, b) (E-/Z- 60 : 40) было получено из данных 1Н-ЯМР-спектра путем сравнения интегральной интенсивности сигналов метильных групп при δ 1.94 и 2.04 м.д. для Z- (XIIIb) и Е- (XIIIa) изомеров соответственно. Однородность изомерной смеси (XIIIa, b) оценивали по данным 31Р-ЯМР-спектроскопии. 31Р-ЯМР-спектр содержит два сигнала, относящиеся к ядрам пентакоординированного фосфора при δ 23.8 и 23.0 м.д. в слабом поле (относительно внешнего стандарта – 85% Н3РО4) для Е- (XIIIa) и Z- (XIIIb) изомеров соответственно.
К сожалению, С5-синтон (XIIIa, b) не удалось получить, исходя из бромонитрила (XIa, b), из-за склонности последнего к полимеризации в условиях проведения реакции Арбузова.
Таким образом, нами было показано, что наиболее эффективный вариант синтеза С5-фосфоната (XIIIa, b) – путь в две стадии из коммерчески доступного хлорацетона (III). Параметры 1Н- и 13С-ЯМР-спектров для E- и Z-изомеров фосфоната (XIIIa, b) приведены в разделе “Эксперим. часть”.
Ключевые стадии синтеза фторфенильного аналога ретиналя (II) представлены на схеме 3 . На первой стадии нами было осуществлено олефинирование по Хорнеру–Эммонсу исходного 4-фторбензальдегида (XIV) анионом С5-фосфонатного синтона с нитрильной терминальной полярной группой (XIIIa, b). В качестве основания при генерировании аниона С5-фосфоната использовали NaH в THF, при этом было показано, что в результате реакции Хорнера–Эммонса вновь образующаяся связь С=С в продукте (XV) имела Е-конфигурацию, что подтверждалось значениями констант спин‑спинового взаимодействия (16.2 Гц). Затем следовала стадия восстановления нитрильной функции DIBAH при температуре от –70 до –80°С.
Повторение указанной последовательности операций – олефинирования альдегида (XVI) по Хорнеру–Эммонсу и последующего восстановления нитрильной функции у соединения (XVII) – приводила к получению целевого ретиноида (II) с общим выходом 47% на исходный альдегид (XIV). Индивидуальный all-E-изомер ретиноида (II) был выделен с чистотой 98–99% при помощи препаративной ВЭЖХ. Структура all-E-изомера соединения (II) была подтверждена набором физико-химических методов анализа (УФ-, 1Н-ЯМР-спектроскопией и масс-спектрометрией). В 1Н-ЯМР-спектре при наличии сигналов протонов, отвечающих п-замещенному ароматическому фрагменту (δ 7.03 и 7.42 м.д.), также были зарегистрированы полный набор дублетов протонов полиеновой цепи (δ 6.05–7.15 м.д.), сигнал протона альдегидной группы (дублет при δ 10.1 м.д. (J 8.2 Гц)) и два сигнала протонов двух метильных групп (при δ 2.10 и 2.35 м.д.), что строго подтверждало структуру целевого альдегида (II).
Масс-спектр содержал молекулярный ион ([М+] 256.1), соответствующий рассчитанному значению.
Таким образом, было показано, что схема синтеза all-E-изомера ретиноида (II) (схема 3 ) с использованием С5-фосфоната (XIIIa, b) с терминальной нитрильной группой более эффективна, чем вариант синтеза, использованный нами ранее [22, 23] (схема 1 ).
Протеородопсин (ESRh) и его аналог F-Phe-ESRh. Для получения препаратов рекомбинантного ESRh была использована сконструированная ранее система экспрессии в клетках Escherichia coli, обеспечивающая встраивание белка во внутреннюю мембрану бактерий [15]. Выделение целевого белка ESRh или его производного F-Phe-ESRh осуществляли после добавления природного хромофора all-E-ретиналя (I) или его фторфенильного аналога (II) к мембранной фракции клеток бактерий в мицеллах неионного детергента н-додецилмальтозида (DDM), обеспечивающего сохранение нативной конформации и функциональных свойств препаратов. Для очистки белка, содержащего С-концевую гексагистидиновую последовательность, был использован метод металл-аффинной хроматографии на никель-содержащей смоле (Ni-Sepharose FastFlow).
Электронные спектры поглощения препаратов (ESRh и его аналога F-Phe-ESRh). Образцы рекомбинантного ESRh и его аналога F-Phe-ESRh были получены двумя альтернативными путями: 1) добавлением к препарату апобелка ESRh, солюбилизированного в буфере А (0.2% DDM, 50 мM NaH2PO4, 200 мM NaCl), pH 8.0, 20°C, 1.5 кратного молярного избытка all-E-ретиналя (I) или его аналога (II) в виде раствора в этаноле (не более 0.01% по объему, чтобы минимизировать побочные процессы: денатурацию и агрегацию) или 2) добавлением 1.5-кратного молярного избытка all-E-ретиналя (I) или его аналога (II) в виде раствора в этаноле в среду культивирования E. coli BL21(DE3)pLysS, с последующим выделением препаратов (ESRh) и его аналога c помощью солюбилизации в буфере А, pH 8.0, и металл-хелатной хроматографии. В дальнейшем при проведении спектральных исследований нами при получении препаратов (ESRh и его аналога) было изменено соотношение белка и производного ретиноида до 1 : 0.8 (рис. 1а, кривые 3 и 4).
Рис. 1.
(а) – Электронные спектры поглощения образцов: 1 – фторфенильного аналога ретиналя (II) в этаноле; 2 – препарата рекомбинантного ESRh в мицеллах DDM (буфер А, рН 8.0); 3 – препарата F-Phe-ESRh, полученного в условиях 0.8 экв. фторфенильного аналога ретиналя (II) в мицеллах DDM (буфер А, рН 8.0); 4 – препарата F-Phe-ESRh, полученного в условиях 1.5 экв. избытка фторфенильного аналога ретиналя (II) в мицеллах DDM (буфер А, рН 8.0); (б) – электронные спектры поглощения образца препарата F-Phe-ESRh, полученного в условиях 0.8 экв. фторфенильного аналога ретиналя (II) в мицеллах DDM (буфер А) при различных значениях рН (рН 5.0–9.0).
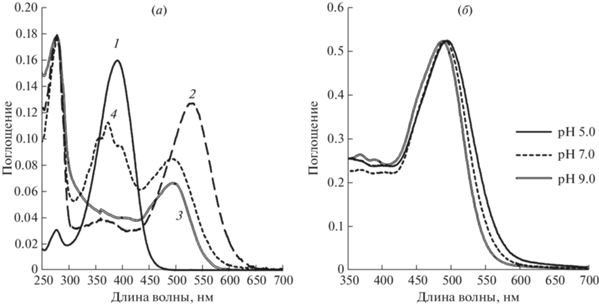
Спектры поглощения полученных препаратов ESRh и его аналога F-Phe-ESRh были исследованы при помощи стационарной и импульсной спектроскопии в диапазоне 250–700 нм. Анализ процесса образования искусственных пигментов на основе ESRh и F-Phe-ESRh показал, что различия в кинетике встраивания all-E-изомера ретиналя и его фторфенильного аналога в ESRh-опсин, солюбилизированный в мицеллах DDM, практически отсутствуют (τ ≤ 2 мин). В то же время при встраивании all-E-изомера ретиналя и его фторфенильного аналога в препарат апомембран, содержащих бактериоопсин H. salinarum, кинетика этих процессов существенно различается (τ ~ 30 мин и ~6 ч соответственно) [8, 22].
Эти различия, вероятно, можно объяснить структурными отличиями ближайшего окружения участка связывания хромофора ESRh и BRh [3]. На небольшом расстоянии от основания Шиффа в молекуле ESRh находится остаток H57, определяющий свойства первичного акцептора [3, 17] и отсутствующий в молекуле BRh. Кроме того, боковая цепь остатка R82, в отличие от BRh, ориентирована противоположным образом и не оказывает существенного влияния на свойства белка. Для установления точных причин наблюдаемых эффектов необходимо проведение дальнейших исследований, в том числе с мутантными формами ESRh.
При рН 7.0 спектр поглощения полученного препарата F-Phe-ESRh демонстрирует максимум при 496 нм, что на 36 нм короче, чем у рекомбинантного ESRh (532 нм) [15–20]. При изменении до рН 9.0 он сдвигается в коротковолновую область до 490 нм (рис. 1б). Сравнение спектров поглощения F-Phe-ESRh (λmax = 496 нм) со спектрами F-Phe-BRh ($\lambda _{{\max }}^{{{\text{DA}}}}$ = 524 нм и $\lambda _{{\max }}^{{{\text{LA}}}}$ = 510 нм) показало, что для белковой матрицы ESRh максимум спектра поглощения сдвинут в коротковолновую область на 28 нм [8, 24]. Аналогичные сдвиги максимума поглощения в коротковолновую область были обнаружены у большинства ароматических производных BRh и ряда BRh с модифицированным триметилциклогексеновым кольцом хромофора [8, 30].
“Опсиновый сдвиг” рассчитывали по формуле:
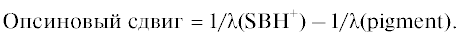
“Опсиновый сдвиг” для образцов F-Phe-ESRh и F-Phe-BRh составил 1963 и 3040 см–1 соответственно [8].
λmax модельного хлоргидрата альдимина аналога ретиналя (II) с н-бутиламином (SBH+) в метаноле равна 452 нм.
У препаратов F-Phe-ESRh и ESRh также наблюдается отсутствие различий в спектрах образцов, адаптированных к свету и темноте ‒ явление свето-темновой адаптации, которое характерно для природного BRh из H. salinarum и большинства его аналогов [8, 30].
Срок хранения препаратов F-Phe-ESRh при 4°С был не менее 6 месяцев без заметного изменения своих характеристик.
Особенности фотоцикла аналога протеородопсина F-Phe-ESRh. Фотоцикл аналога F-Phe-ESRh исследовали методом импульсной лазерной спектроскопии (флэш-фотолиза). Были получены кинетические кривые изменений поглощения F-Phe-ESRh на четырех характерных длинах волн в суспензии мицелл DDM при pH 7.0 и 9.0 (рис. 2a, 2б).
Рис. 2.
Особенности фотоцикла рекомбинантного F-Phe-ESRh. Представлена кинетика светоиндуцированных изменений поглощения белка в мицеллах DDM при рН 7.0 (a) и 9.0 (б) на характерных длинах волн. Врезка на панели (а) ‒ изменения поглощения при 390 нм, вызванные образованием М-интермедиата.
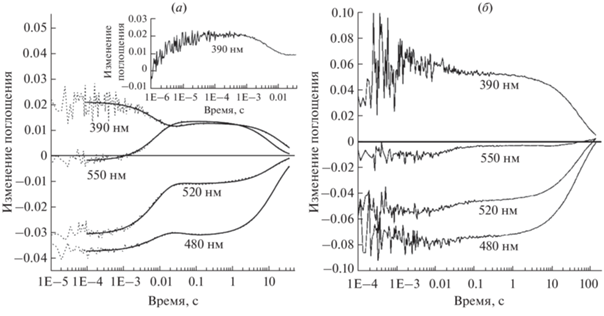
В ответ на вспышку света при pH 7.0 детектируется быстрое (τ = 50–70 мкс) образование “короткоживущей” формы М-интермедиата, соответствующей депротонированному основанию Шиффа, с максимумом поглощения 390 нм (рис. 2а). Это отличает фотоцикл F-Phe-ESRh от дикого типа ESRh, содержащего all-E-ретиналь, в котором при рН 7.0 в мицеллах DDM образование М-интермедиата практически не детектируется [15–21]. Таким образом, можно утверждать, что pKa образования М-интермедиата в производном F-Phe-ESRh сдвинут в кислую область рН по сравнению с ESRh дикого типа. Кроме того, наблюдается формирование более “долгоживущей” М-формы (τ = 34 ± 7 мс) во второй фракции пигмента (~40%).
В результате распада “короткоживущей” М-формы интермедиата c τ = 7.7 мс происходит репротонирование основания Шиффа, часть пигмента образует состояние, аналогичное N/О-интермедиату фотоцикла BRh, с максимумом поглощения 550 нм. Другая часть, предположительно, не образует длинноволновую форму, а принимает участие в обратимой реакции репротонирования основания Шиффа со стороны акцептора протонов D85. Как следствие, присутствие существенной доли “долгоживущей” формы М-интермедиата наблюдается в F-Phe-ESRh вплоть до конца фотоцикла, общая продолжительность которого значительно замедлена и составляет ~27 с. При pH 9.0 наблюдается еще более значительное замедление фотоцикла (до ~100 с), без заметного накопления N/О-интермедиата (рис. 2б).
Стабильность и доступность альдиминной связи у F-Phe-ESRh к действию различных условий и реагентов. Для определения строения продуктов взаимодействия серии арилполиеновых альдегидов с апомембранами, содержащими бактериоопсин из H. salinarum, А.М. Шкробом с соавт. был разработан целый набор химических тестов в комбинации со спектроскопическими методами в ставшей классикой работе [31].
Для оценки доступности протонированной альдиминной связи к действию различных стимулов и систем реагентов был проведен следующий ряд экспериментов: 1) засветка образцов F-Phe-ESRh видимым cветом галогеновой лампы ThorLabs OSL1-EC, светофильтр ЖС-12 (λ ≥ ≥ 400 нм) в течение 2–10 мин; 2) воздействие на образец F-Phe-ESRh раствора 50 мкМ NaBH4, рН 9.0, в темноте и на свету; 3) реакция замещения образца F-Phe-ESRh раствором all-E-ретиналя в этаноле (5 мМ) в темноте и на свету; 4) воздействие на образец F-Phe-ESRh раствора NH2OH (50 мМ) в темноте и на свету.
В результате проведенных исследований было обнаружено, что при продолжительном освещении образца F-Phe-ESRh видимым cветом галогеновой лампы его протонированная альдиминная связь подвергалась частичному гидролизу с появлением продукта распада, спектрально идентичного образцу фторфенилретиналя (II) в водном буфере А. При выдерживании “засвеченного видимым cветом образца F-Phe-ESRh” в темноте более 2–3 ч наблюдалась частичная реконструкция пигмента с увеличением оптической плотности полосы поглощения F-Phe-ESRh (496 нм). Спектр поглощения продукта распада демонстрировал тонкую структуру: основная полоса 374 нм, и два плеча 355 нм (пл.) и 398 нм (пл.) (рис. 3а, кривые 2, 3). Дополнительным доказательством строения продукта распада была его трансформация в соответствующий спирт действием раствора 50 мкМ NaBH4, рН 9.0, в темноте. Спектр поглощения восстановленного продукта распада в буфере А был весьма близок к спектру реперного образца фторфенилретинола (основная полоса 343 нм, два плеча 328 и 361 нм), который был получен обработкой NaBH4 фторфенилретиналя (II) в водном этаноле. Было проведено контрольное восстановление образца F-Phe-ESRh NaBH4 в темноте, которое показало, что протонированная альдиминная связь в образце F-Phe-ESRh достаточно стабильна к действию раствора NaBH4 в темноте (τ1/2 ≈ 10 мин). После освещения “образца после NaBH4” в течение 2 мин полоса пигмента почти полностью исчезала.
Рис. 3.
Спектры поглощения F-Phe-ESRh. (а): 1 – Исходный образец, полученный в условиях 1.5 экв. избытка фторфенильного аналога ретиналя (II); 2 – он же после 2 мин освещения видимым светом с длиной волны >450 нм (фильтр ЖС-12); 3 – он же после 12 мин освещения видимым светом с длиной волны >450 нм (фильтр ЖС-12); 4 – засвеченный в течение 15 мин образец после добавления раствора NaBH4 (50 мкМ NaBH4, рН 9.0); (б): 1 – исходный образец, полученный в условиях 1.5 экв. избытка фторфенильного аналога ретиналя (II); 2 – “темновой” исходный образец через 15 мин после добавления раствора NaBH4 (50 мкМ NaBH4, рН 9.0); 3 – образец через 15 мин после добавления раствора NaBH4, после 2 мин освещения видимым светом с длиной волны >450 нм (фильтр ЖС-12).
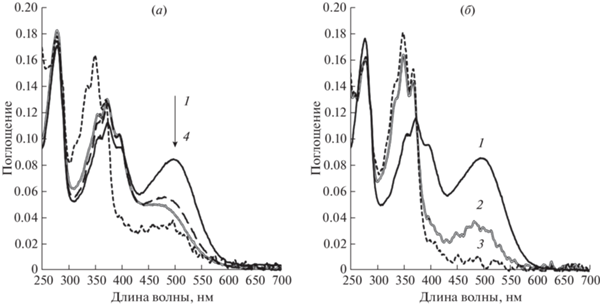
В то же время препарат рекомбинантного ESRh в мицеллах DDM (буфер А, рН 8.0) оказался устойчивым к воздействию освещения видимым светом [15–21].
Реакция замещения аналога хромофора F-Phe-ESRh раствором all-E-ретиналя в темноте протекала очень медленно (τ1/2 > 18 ч). Этот факт говорит о том, что в белке отсутствуют свободные сайты посадки хромофора на ε-аминогруппу остатка лизина К225 или другие возможные остатки лизина, способные к формированию протонированной альдиминной связи.
Также образец F-Phe-ESRh был достаточно стабилен в темноте в реакции замещения гидроксиламином (τ1/2 > 3 ч).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Тонкослойную хроматографию осуществляли на пластинках Kieselgel 60 F254 (Merck, Германия) в системе растворителей: (А) гексан/эфир (1 : 1). Пятна веществ детектировали выдерживанием пластинки в парах иода. Препаративную флэш-хроматографию проводили на силикагеле Kieselgel 60 (Merck, Германия). Растворители очищали и высушивали: диэтиловый эфир и тетрагидрофуран – перегонкой над алюмогидридом лития; метанол абсолютизировали с использованием магниевой стружки.
Препаративную ВЭЖХ-хроматографию проводили на ВЭЖХ-хроматографе SmartLine 1000 (Knauer, Германия) в изократическом режиме, колонка Knauer Eurospher 100-10 Si, 20 × 250 мм, элюент гексан/диэтиловый эфир 7 : 1 по объему, УФ-детектор К-2500 с детекцией при 370 нм, скорость потока 5 мл/мин.
В работе использовали хлороацетонитрил (Fluka, Швейцария), триэтилфосфит (Fluka, Швейцария), хлороацетон, 4-фторбензальдегид (Merck, Германия), а также реагенты и растворители марок “х.ч.” и “ч.д.а.” отечественного производства.
Все операции с реагентами, чувствительными к влаге и кислороду, проводили в тщательно высушенной аппаратуре в атмосфере сухого аргона. Упаривание растворов осуществляли на роторном испарителе при температуре не выше 35°С и давлении 12 мм рт. ст.
Все спектральные исследования проводили при 20°С. ЯМР-спектры растворов в дейтерохлороформе регистрировали на спектрометре Avance III 500 (Bruker, Германия) со следующими рабочими частотами: 1Н-ЯМР-спектры – 500 МГц, 13С-ЯМР-спектры – 126 МГц, 31Р-ЯМР-спектры – 203 МГц. Химические сдвиги приведены в миллионных долях (м.д.) относительно внутреннего стандарта: тетраметилсилана (δ 0.00) или дейтерохлороформа (δ (1Н-ЯМР) – 7.25 м.д. и (13С-ЯМР) – 77.2 м.д.), для 31Р-ЯМР-спектров – относительно внешнего стандарта (85%-ного раствора ортофосфорной кислоты в D2O). Величины констант спин-спинового взаимодействия измерены в герцах (Гц).
При описании ЯМР-спектров полиенового альдегида (II) принята нумерация атомов, соответствующая нумерации атомов полиеновой цепи природного ретиналя (I).
При описании спектров приняты следующие сокращения: с – синглет, д – дублет, т – триплет, кв – квартет, м – мультиплет.
Масс-спектры получали на спектрометре Finnigan 4021 (США) при прямом вводе образца и ионизации электронным ударом (ЭИ 70 эВ).
Измерения спектральных и фотохимических характеристик растворов соединений и пигментов проводили в кварцевых кюветах толщиной 10 мм на спектрофотометре UV-2140PC (Shimadzu, Япония) и на специальном стенде, созданном на основе комплекта модульного оптоволоконного спектрофотометрического оборудования (Ocean Optics, США). Для облучения растворов образцов видимым светом (λ ≥ 400 нм) использовали галогеновую лампу OSL1-EC (ThorLabs, США, 25 Вт) в комбинации со светофильтром ЖС-12.
Фотореакции образцов ESRh и F-Phe-ESRh исследовали методом флеш-фотолиза на импульсном однолучевом дифференциальном спектрофотометре с двойной монохроматизацией измеряющего света [17, 19, 20]. В качестве источника светового возбуждения использовали Nd-YAG-лазер LS 2131M (LOTIS TII, Беларусь, 532 нм, 8 нс, 5 мДж). С целью улучшения соотношения сигнал/шум проводили накопление и усреднение 100 одиночных сигналов с помощью аналого-цифрового преобразователя Octopus CS 8327 (GaGe Applied Technologies, США).
Для получения полной кинетической картины фотоцикла F-Phe-ESRh проводили измерения при четырех длинах волн, характерных для превращений различных интермедиатов: 390, 480, 520 и 550 нм. Набор кинетических кривых в логарифмической шкале времени анализировали с помощью программы Mathematica (Wolfram Research, США) методом глобального фитирования с подбором 4–5 характерных экспоненциальных составляющих.
Смесь E- и Z-изомеров 3-метил-4-хлоро-2-бутенонитрила (Xа, b). В четырехгорлый реактор объемом 250 мл, снабженный капельной воронкой, помещали в атмосфере аргона 3.8 г (0.13 моль) 80%-ной суспензии NaH в минеральном масле и промывали абсолютным гексаном (2 раза по 5 мл). Затем добавляли при перемешивании в токе аргона 30 мл свежеперегнанного THF. Реакционную смесь охлаждали до 0°С и при интенсивном перемешивании постепенно добавляли по каплям 15 мл (9.9 ммоль) C2-фосфоната (IX), перемешивали при той же температуре в течение 30 мин до полного растворения NaH. К полученному раствору при перемешивании добавляли по каплям 8.6 мл (0.11 моль) хлорацетона (VII) и выдерживали реакционную смесь при 20°С в течение 1.5 ч. По окончании реакции добавляли 30 мл H2O, 20 мл диэтилового эфира и доводили 0.1 н. раствором HСl до рН 6.0. Экстрагировали Et2O (3 раза по 100 мл). Эфирные фракции объединяли, промывали водой до рН 7.0 и сушили над Na2SO4. Сушитель отфильтровывали, растворитель удаляли, а оставшуюся реакционную массу перегоняли в вакууме (0.1 мм рт. ст.). Получили 5.94 г (52%) галогенонитрила (Xа, b) в виде желто-коричневой маслянистой жидкости, смесь изомеров (E-/Z- 75 : 25), т. кип. 45–60°С (0.1 мм рт. ст.). 1Н-ЯМР-спектр (δ, м.д.): E-изомер (Xa): 2.12 (3Н, д, J 0.5, 3-СН3), 4.05 (2Н, с, –СН2Cl), 5.49 (1Н, дд, J 3.0/1.5, 2-СН); Z-изомер (Xb): 2.03 (3Н, д, J 1.5, 3-СН3), 4.24 (2Н, с, –СН2Cl), 5.27 (1Н, дд, J 3.0/1.5, 2-СН).
Смесь E- и Z-изомеров диэтил(2-метил-3-циано-2-пропенил)фосфоната (XIIIa, b). В трехгорлый реактор объемом 250 мл, снабженный высокоэффективным обратным холодильником и насадкой для перегонки с термометром и холодильником Либиха, помещали в атмосфере аргона 11.55 г (0.1 моль) изомерной смеси хлоронитрила (Xа, b) и 16.84 г (0.1 моль) свежеперегнанного (EtO)3P (XII). Далее при интенсивном перемешивании реакционную смесь постепенно нагревали до 150°С, следя за тем, чтобы не было бурного вспенивания. Окончание реакции контролировали по окончанию выделения этилхлорида. Остаток перегоняли в вакууме (0.1 мм рт. ст.). Получили 17.58 г (81%) целевого С5-фосфоната (XIIIa, b) в виде изомерной смеси (E-/Z- 60 : 40), т. кип. 78–98°С (0.1 мм рт. ст.). 1Н-ЯМР-спектр (δ, м.д.): E-изомер (XIIIa): 1.14 (6Н, т, J 7.0, (–ОСН2СН3)2), 2.04 (3Н, дд, J 3.4/1.3, –СН3), 2.55 (2Н, д, J 23.5, –СН2), 3.95 (4Н, кв, J 7.0, (–ОСН2СН3)2), 5.12 (1Н, м, =СН); Z-изомер (XIIIb): 1.15 (6Н, т, J 7.0, (–ОСН2СН3)2), 1.94 (3Н, дд, J 3.8/1.7, –СН3), 2.81 (2Н, д, J 24.0, –СН2), 3.96 (4Н, кв, J 7.0, (–ОСН2СН3)2), 5.12 (1Н, м, =СН). 13С-ЯМР-спектр (δ, м.д.): E-изомер (XIIIa): 16.2 (c, (–ОСН2СН3)2), 22.0 (c, –ССН3), 36.8 (д, J 81.4, –СН2), 62.3 (c, (–ОСН2СН3)2), 116.1 (c, –ССН3), 155.6 (д, J 11.1, –CN); Z-изомер (XIIIb): 16.1 (c, (–ОСН2СН3)2), 23.9 (c, –ССН3), 34.1 (д, J 81.4, –СН2), 62.2 (c, (–ОСН2СН3)2), 115.9 (c, –ССН3), 155.2 (д, J 11.1, –CN). 31Р-ЯМР-спектр (δ, м.д.): E-изомер: 23.84; Z-изомер: 23.01.
Стандартные методики олефинирования по Хорнеру–Эммонсу карбонильных предшественников (XIV) и (XVI) с С5-фосфонатом (XIIIa, b) и последующего восстановления нитрильной группы DIBAH в промежуточных нитрилах (XV) и (XVII). В трехгорлый реактор объемом 100 мл помещали в атмосфере аргона 0.06 г 80%-ной суспензии NaH в минеральном масле и промывали абсолютным гексаном (3 раза по 3 мл). Затем при интенсивном перемешивании и 0°С прибавляли 10 мл абсолютного THF и 0.30 мл (1.56 ммоль) С5-фосфоната (XIIIa, b). Смесь перемешивали в течение 1 ч до полного растворения NaH и постепенно, при помощи шприца, прибавляли раствор 1.3 ммоль альдегида ((XIV) или (XVI)) в 10 мл абсолютного THF. По окончании реакции прибавляли по каплям 5 мл Н2О, 10 мл Et2O и нейтрализовали 0.1 н. раствором HCl до pH 6.0. Органический слой отделяли, остаток экстрагировали Et2O (3 раза по 50 мл). Эфирные экстракты объединяли с органическим слоем, промывали водой до рН 7.0 и сушили над безводным Na2SO4. Растворитель удаляли, остаток хроматографировали на колонке с 10 г силикагеля, элюируя гексаном и увеличивая полярность элюента добавлением Et2O от 0 до 15%. Фракции, содержащие нитрил ((XV) или (XVII)), объединяли, растворитель удаляли, остаток сушили в вакууме 1 ч при 0.1 мм рт. ст., растворяли в 10 мл абсолютного толуола и помещали в трехгорлый реактор объемом 100 мл в атмосфере аргона. Реакционную смесь охлаждали до температуры в диапазоне от –70 до –80°С, затем постепенно, при помощи шприца прибавляли 1.5 экв. 20%-ного раствора DIBAH в толуоле и оставляли до тех пор, пока температура реакции не достигала 20°С. Далее реакционную массу обрабатывали влажным силикагелем, перемешивали 30 мин, фильтровали через слой целита (1 см), промывая слой сорбента Et2O (50 мл). Фильтрат упаривали досуха, остаток хроматографировали на колонке с 10 г силикагеля, элюируя вещество гексаном и увеличивая полярность элюента добавлением Et2O от 0 до 10%. Фракции, содержащие смесь изомеров промежуточного или целевого альдегида ((XVI) или (II)), объединяли, растворитель удаляли, остаток сушили в вакууме 1 ч при давлении 0.1 мм рт. ст.
all-E-Изомер целевого альдегида (II) выделяли препаративной ВЭЖХ на хроматографе SmartLine 1000 (Knauer, Германия) в изократическом режиме, колонка Knauer Eurospher 100-10 Si, 20 × × 250 мм, элюент гексан/диэтиловый эфир 7 : 1 по объему, УФ-детектор К-2500 с детекцией при 370 нм, скорость потока 5 мл/мин.
Выход all-E-изомера целевого альдегида (II): (47% в расчете на исходный альдегид (XIV)). Rf 0.44 (A). 1Н-ЯМР-спектр (CDCl3, δ, м.д.): 2.10 (3H, с, 9-СН3), 2.35 (3H, д, J 1.5, 13-СН3), 6.05 (1H, д, J 8.2, 14-Н), 6.36 (1H, д, J 11.5, 10-Н), 6.43 (1H, д, J 15.5, 12-Н), 6.68 (1H, д, J 16.2, 8-Н), 6.82 (1H, д, J 16.2, 7-Н), 7.03 (2H, дд, J2(4)H,F 8.5, J 8.5, 2,4-Н), 7.15 (1H, дд, J 15.5, J 11.5, 11-Н), 7.42 (2H, J1(5)H,F 5.5, J 8.5, 1,5-Н), 10.12 (1H, д, J 8.2, 15-Н). УФ-спектр (метанол, λmax, нм, [ε, M–1 cм–1]): 387.5 [47 700]. Масс-спектр (m/z) ([М+] 256.1).
Получение альдимина аналога ретиналя (II) с н‑бутиламином. К раствору 3 мг альдегида (II) в 0.1 мл абсолютного метанола добавляли 0.1 мл н-бутиламина и 10 мг молекулярных сит 3 Å. Реакционную смесь выдерживали 24 ч при 0°С в темноте и в атмосфере аргона. Сита отделяли, растворитель и избыток н-бутиламина удаляли при 20°С и давлении 0.1 мм рт. ст., остаток растворяли в 0.2 мл метанола и хранили при –10°С.
Спектральные характеристики: альдимин аналога ретиналя (II) с н-бутиламином (метанол, λmax, нм, [ε, M–1 cм–1]: 369 [43 500]; хлоргидрат альдимина аналога ретиналя (II) с н-бутиламином (метанол, λmax, нм, [ε, M–1 cм–1]: 452 [56 000].
Получение препаратов рекомбинантного протеородопсина ESRh и его аналога F-Phe-ESRh. Для получения рекомбинантного ESRh с природным или модифицированным хромофором в препаративном количестве использовали штамм E. coli BL21(DE3)pLysS (Novagen Merck, Германия), трансформированный плазмидой pET-ESRh [15]. В среду LB с ампициллином (100 мкг/мл) объемом 200 мл засевали ночную культуру штамма до OD560 = 0.15. Культуру инкубировали в при 37°C в качалке Innova (New Brunswick Scientific, США) при 250 об/мин до OD560 = 0.8, после чего добавляли раствор изопропил-β-D-1-тиогалактопиранозида до 0.2 мМ и одно из производных ретиналя до 6–7 мкМ, продолжали культивирование при 30°C в течение 24 ч. Для солюбилизации в препарат мембранной фракции добавляли раствор 10%-ного н-додецил-β-D-мальтопиранозида (DDM) до 1%, коктейль ингибитора протеаз (Sigma, США) до 0.3%. Суспензию инкубировали при комнатной температуре на качалке в течение 3 ч, после чего центрифугировали в течение 15 мин при 30 000 g. Для выделения белка после солюбилизации проводили металл-аффинную хроматографию на Ni-Sepharose FastFlow (GE Healthcare, США), как описано в работе Petrovskaya et al. [15]. Рекомбинантный ESRh был получен с высокой степенью чистоты (не менее 90%) и выходом 10–15 мг/л.
ЗАКЛЮЧЕНИЕ
Предложен альтернативный вариант наращивания полиеновой цепи целевого ретиноида с двумя ключевыми стадиями: 1) реакция олефинирования по Хорнеру–Эммонсу исходного 4-фторбензальдегида анионом С5-фосфоната; 2) последующее восстановление нитрильной функции в промежуточных нитрилах DIBAH до формильной группы при температуре от –70 до –80°С. Показано, что схема синтеза all-E-изомера ретиноида (II) с использованием С5-фосфоната с терминальной нитрильной группой более эффективна и дает более высокий суммарный выход целевого ретиноида (II), чем вариант синтеза, описанный нами ранее [22, 23].
Установлено, что вариант модификации триметилциклогексенового кольца природного хромофора рекомбинантного протеородопсина ESRh путем замены его на п-фторфенильный фрагмент приводит, как и в случае бактериоопсина из H. salinarum, к образованию искусственного пигмента F-Phe-ESRh, сохраняющего цикл фотохимических реакций. Обнаружены определенные различия в свойствах нативного рекомбинантного ESRh и его аналога F-Phe-ESRh, включающие сдвиг максимума поглощения в коротковолновую область, образование интермедиата М при более низких значениях рН, наличие “долгоживущего М” и общее замедление фотоцикла. Также продемонстрирована пониженная стабильность полученного аналога F-Phe-ESRh к продолжительному воздействию видимого света.
Полученные результаты подтверждают ранее описанные требования к структуре хромофора микробных родопсинов, включая наличие терминальной формильной группы, определенную длину полиеновой цепи и ее конфигурацию, а также отсутствие жестких пространственных ограничений района триметилциклогексенового кольца и наличие определенных препятствий в районе двойной связи С13=С14 [8–13, 23, 29, 30].
Список литературы
Kandori H. // Biophys. Rev. 2020. V. 12. P. 355–361. https://doi.org/10.1007/s12551-020-00645-0
Ernst O.P., Lodowski D.T., Elstner M., Hegemann P., Brown L.S., Kandori H. // Chem. Rev. 2014. V. 114. P. 126−163. https://doi.org/10.1021/cr4003769
Gushchin I., Gordeliy V. // In Subcellular Biochemistry / Eds. Harris J.R., Boekema E.J. 2018. V. 87. Ch. 2. P. 19–56. https://doi.org/10.1007/978-981-10-7757-9_2
Oesterhelt D., Stoeckenius W. // Nature (New Biologist). 1971. V. 233. P. 149–151. https://doi.org/10.1038/newbio233149a0
Deisseroth K. // Nat. Neurosci. 2015. V. 18. P. 1213–1225. https://doi.org/10.1038/nn.4091
Говорунова Е.Г., Коппель Л.А. // Биохимия. 2016. Т. 1. С. 1172–1186. [Govorunova E.G., Koppel L.A. // Biochemistry (Moscow). 2016. V. 81. P. 928–940.] https://doi.org/10.1134/S0006297916090029
Dawson M.I., Okamura W.H. // Chemistry and Biology of Synthetic Retinoids. Boca Raton: CRC Press Inc., 1990. https://doi.org/10.1201/9781351070638
Khodonov A.A., Belikov N.E., Demina O.V. // Properties of Artificial Bacteriorhodopsin Analogs. Version 2, 2020. From 1975 to 2019. Moscow: IBCP/MIPT, Russia. http://biochemphysics.ru/assets/upload/documents/ docs/BRDT_v2.pdf
Мицнер Б.И., Ходонов А.А. // В кн. Светочувствительные биологические комплексы и оптическая регистрация информации / Ред. Иваницкий Г.Р. Пущино: АН СССР, 1985. С. 38–49.
Мицнер Б.И., Ходонов А.А., Звонкова Е.Н., Евстигнеева Р.П. // Биоорг. химия. 1986. Т. 12. С. 5–53.
Mitsner B.I., Khodonov A.A., Zvonkova E.N., Karnauchova E.N. // In Retinal Proteins / Ed. Ovchinnikov Yu.A. Utrecht: VNU Press, 1989. P. 561–569.
Ходонов А.А., Еремин С.В., Локшин Дж.Л., Швец В.И., Демина О.В., Хитрина Л.В., Каулен А.Д. // Биоорг. химия. 1996. Т. 22. С. 745–776.
Barachevsky V.A., Khodonov A.A., Belikov N.E., Laptev A.V., Lukin A.Yu., Demina O.V., Luyksaar S.I., Krayushkin M.M. // Dyes and Pigments. 2012. V. 92. P. 831–837. https://doi.org/10.1016/j.dyepig.2011.05.009
Ходонов А.А., Лаптев А.В., Лукин А.Ю., Беликов Н.Е., Фомин М.А., Демина О.В., Складнев Д.А., Тюрин С.А., Швец В.И. // Вестник МИТХТ. 2011. Т. 6. С. 15–36.
Petrovskaya L.E., Lukashev E.P., Chupin V.V., Sychev S.V., Lyukmanova E.N., Kryukova E.A., Ziganshin R.H., Spirina E.V., Rivkina E.M., Khatypov R.A. // FEBS Lett. 2010. V. 584. P. 4193–4196. https://doi.org/10.1016/j.febslet.2010.09.005
Gushchin I., Chervakov P., Kuzmichev P., Popov A.N., Round E., Borshchevskiy V., Ishchenko A., Petrovskaya L., Chupin V., Dolgikh D.A, Arseniev A.S., Kirpichnikov M., Gordeliy V. // Proc. Natl. Acad. Sci. USA. 2013. V. 110. P. 12631–12636. https://doi.org/10.1073/pnas.1221629110
Balashov S.P., Petrovskaya L.E., Lukashev E.P., Imasheva E.S., Dioumaev A.K., Wang J.M., Sychev S.V., Dolgikh D.A., Rubin A.B., Kirpichnikov M.P., Lanyi J.K. // Biochemistry. 2012. V. 51. P. 5748–5762. https://doi.org/10.1021/bi300409m
Dioumaev A.K., Petrovskaya L.E., Wang J.M., Bala-shov S.P., Dolgikh D.A., Kirpichnikov M.P., Lanyi J.K. // J. Phys. Chem. B. 2013. V. 117. P. 7235–7253. https://doi.org/10.1021/jp402430w
Петровская Л.Е., Балашов С.П., Лукашев Е.П., Имашева Е.С., Гущин И.Ю., Дюмаев А.К., Рубин А.Б., Долгих Д.А., Горделий В.И., Лани Я.К., Кирпичников М.П. // Биохимия. 2015. Т. 80. С. 814–828. [Petrovskaya L.E., Balashov S.P., Lukashev E.P., Imasheva E.S., Gushchin I., Dioumaev A.K., Rubin A.B., Dolgikh D.A., Gordeliy V.I., Lanyi J.K., Kirpichnikov M.P. // Biochemistry (Moscow). 2015. V. 80. P. 688–700.] https://doi.org/10.1134/S000629791506005X
Siletsky S.A., Mamedov M.D., Lukashev E.P., Balashov S.P., Dolgikh D.A., Rubin A.B., Kirpichnikov M.P., Petrovskaya L.E. // Biochim. Biophys. Acta. 2016. V. 1857. P. 1741–1750. https://doi.org/10.1016/j.bbabio.2016.08.004
Smitienko O.A., Feldman T.B., Petrovskaya L.E., Nekrasova O.V., Yakovleva M.A., Shelaev I.V., Gostev F.E., Cherepanov D.A., Kolchugina I.B., Dolgikh D.A., Nadtochenko V.A., Kirpichnikov M.P., Ostrovsky M.A. // J. Phys. Chem. B. 2021. V. 125. P. 995–1008. https://doi.org/10.1021/acs.jpcb.0c07763
Belikov N.E., Melnikova I.A., Demina O.V., Petrovskaya L.E., Kryukova E.A., Dolgikh D.A., Kuzmichev P.K., Chupin V.V., Lukin A.Yu., Shumsky A.N., Chizhov I., Levin P.P., Kirpichnikov M.P., Varfolomeev S.D., Khodonov A.A. // Mendeleev Commun. 2018. V. 28. P. 406–408. https://doi.org/10.1016/j.mencom.2018.07.022
Мицнер Б.И., Ходонов А.А., Звонкова Е.Н., Евстигнеева Р.П. // Биоорг. химия. 1987. Т. 13. С. 238–251.
Драчев Л.А., Зорина В.В., Мицнер Б.И., Хитрина Л.В., Ходонов А.А., Чекулаева Л.Н. // Биохимия. 1987. Т. 52. С. 1559–1569.
Walker B.J. // In Organophosphorus Reagents in Organic Synthesis / Ed. Cadogan J.I.G. London: Academic Press, 1979. Chapter 3. P. 155–206.
Courtin J.M.L., Verhagen L., Biesheuvel P.L., Lugtenburg J., van der Bend R.L., van Dam K. // Recl. Trav. Chim. Pays-Bas. 1987. V. 106. P. 112–119.
Ernst L., Hopf H., Krause N. // J. Org. Chem. 1987. V. 52. P. 398–405.
Groesbeek M., de Vries E.F.J., Berden J.A., Lugtenburg J. // Recl. Trav. Chim. Pays-Bas. 1993. V. 112. P. 303–308.
Миронова Е.В., Лукин А.Ю., Шевяков С.В., Алексеева С.Г., Швец В.И., Демина О.В., Ходонов А.А., Хитрина Л.В. // Биохимия. 2001. Т. 66. С. 1638–1648. [Mironova E.V., Lukin A.Y., Shevaykov S.V., Alexeeva S.G., Shvets V.I., Demina O.V., Khodonov A.A., Khitrina L.V. // Biochemistry (Moscow). 2001. V. 66. P. 1323–1333.] https://doi.org/10.1023/A:1013147722255
Ходонов А.А., Беликов Н.Е., Лукин А.Ю., Петровская Л.Е., Чупин В.В., Демина О.В. // Актуальные вопросы биологической физики и химии. 2020. Т. 5. С. 91–100.
Шкроб А.М., Родионов А.В., Овчинников Ю.А. // Биоорг. химия. 1981. Т. 7. С. 1169–1194.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия