

Биоорганическая химия, 2022, T. 48, № 6, стр. 707-713
Cинтез 2-метилиден-спермидина и его N1-ацетильного производного
М. А. Хомутов 1, *, А. Р. Хомутов 1
1 Институт молекулярной биологии им. В.А. Энгельгардта РАН
119991 Москва, ул. Вавилова, 32, Россия
* E-mail: makhomutov@mail.ru
Поступила в редакцию 12.05.2022
После доработки 07.06.2022
Принята к публикации 17.06.2022
- EDN: LCIOOV
- DOI: 10.31857/S0132342322060148
Аннотация
Предложены простые и удобные методы синтеза неизвестных ранее 2-метилиденовых производных спермидина (1,8-диамино-2-метилиден-4-азаоктан, 2-Met-Spd) и N1-Ас-спермидина (N1-Ас-1,8-диамино-2-метилиден-4-азаоктан, N1-Ас-2-Met-Spd), исходя из коммерчески доступного 2-хлорметил-3-хлорпропена-1. Целевые соединения были получены в семь стадий с высоким суммарным выходом. Обсуждаются перспективы их использования для ингибирования FAD-зависимой N1‑ацетилполиаминоксидазы.
ВВЕДЕНИЕ
Биогенные полиамины спермин (Spm) и спермидин (Spd), представляющие собой низкомолекулярные поликатионы, присутствуют в клетках эукариот в микро- и миллимолярных концентрациях, что определяет множественность и жизненную важность их клеточных функций [1, 2]. Нарушение гомеостаза полиаминов не только связано с возникновением злокачественных трансформаций (опухолевые клетки имеют повышенное содержание полиаминов, а соединения, снижающие их уровень, обладают противоопухолевой активностью [3, 4] и используются в период ремиссии [5]), но и ассоциировано с развитием некоторых типов панкреатита, синдрома Шнайдер–Робинсона, болезней Альцгеймера и Паркинсона, атеросклероза, сердечно-сосудистых заболеваний, инсульта, воспалительных процессов и заболеваний, связанных со снижением иммунного ответа [6–13].
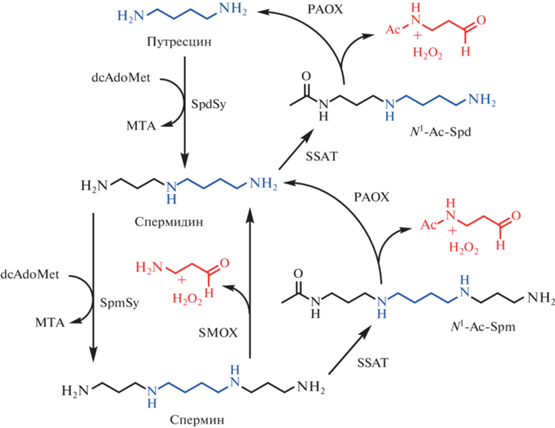
Соответственно, уровень полиаминов в клетке необходимо поддерживать на заданном уровне, что достигается согласованной работой ферментов синтеза и деградации полиаминов, а также системой их транспорта в клетки. Биосинтез, активность и деградация скорость-определяющих ферментов синтеза (декарбоксилазы орнитина и S-аденозил-L-метионина – dcAdoMet, рис. 1) и катаболизма (спермидин/спермин-N1-ацетилтрансфераза – SSAT и сперминоксидаза – SMOX, рис. 1) полиаминов тонко регулируются в ответ на изменения внутриклеточной концентрации полиаминов. Считается, что FAD-зависимая N1‑ацетилполиаминоксидаза (РАОХ, рис. 1) конститутивно экспрессируется в большинстве клеток, и ее активность зависит от активности SSAT, которая синтезирует ацетилированный субстрат. Однако в клетках опухоли молочной железы экспрессия PAOX вариативна, что предполагает существование регуляторных путей, не характерных для других типов клеток [14].
Рис. 1.
Катаболизм и взаимопревращения полиаминов. PАОX – N1-ацетилполиаминоксидаза, SSAT – спермидин/спермин-N1-ацетилтрансфераза, SMOX – сперминоксидаза, SpdSy – спермидинсинтаза, SpmSy – сперминсинтаза.
N1,N4-бис(2,3-Бутадиенил)-1,4-диаминобутан (MDL-72527, рис. 2) необратимо ингибирует изолированную РАОХ (Ki = 0.09 мкМ, τ1/2 = 2.2 мин [15]) и проявляет высокую активность в экспериментах в культуре клеток и in vivo. По данным рентгеноструктурного анализа, движущей силой необратимого торможения РАОХ под действием MDL-72527 оказывается присоединение FAD к алленовой системе ингибитора с образованием ковалентного аддукта [16]. Вместе с тем MDL-72527 достаточно активен и в отношении близкородственной FAD-зависимой SMOX (Ki = 63 мкМ [17]), что в ряде случаев приводит к одновременному ингибированию PAOX и SMOX в культуре клеток и in vivo. Это затрудняет оценку вклада каждого из ферментов в интегральный биологический эффект или развитие полиамин-ассоциированного заболевания. Ряд других ингибиторов РАОХ, созданных на основе α,ω-диаминоалканов и их производных, обладал худшей активностью по сравнению с MDL-72527. При этом фермент-активируемые ингибиторы (suicide inhibitors) PAOX, созданные на основе скелета Spd, до настоящего времени не известны.
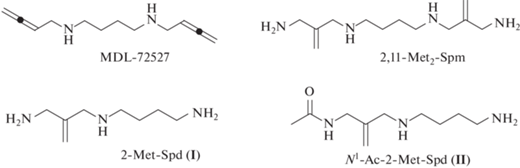
Недавно мы сообщали об использовании для ингибирования SMOX 2,11-бис(метилиден)-1,12-диамино-4,9-диазадодекана (2,11-Met2-Spm, рис. 2), активность которого в отношении SMOX была близка к активности MDL-72527 [18]. В настоящей работе мы используем этот алгоритм для получения нового ингибитора FAD-зависимой РАОХ и описываем простой и удобный способ синтеза не известных ранее 1,8-диамино-2-(метилиден)-4-азаоктана (2-Met-Spd (I), рис. 2) и N1-Ас-1,8-диамино-2-(метилиден)-4-азаоктана (N1-Ас-2-Met-Spd (II), рис. 2), которые представляют интерес для изучения особенностей РАОХ-реакции, а также обладают определенным потенциалом для ингибирования фермента в культуре клеток.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
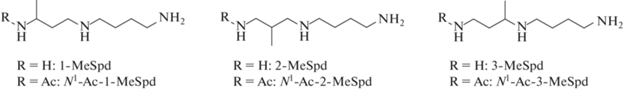
Ранее мы исследовали взаимодействие N1-Ac-производных С-метилированных аналогов Spd (рис. 3) с РАОХ и показали, что продуктивность взаимодействия этих соединений с ферментом можно регулировать, перемещая метильную группу по скелету Spd. Так, N1-Ac-3-MeSpd не расщепляется РАОХ, по-видимому, из-за стерических эффектов метильной группы, а N1-Ac-2-MeSpd и N1-Ac-1-MeSpd оказались субстратами фермента [19]. Таким образом, метильный заместитель во втором положении Spd, как минимум, не препятствует его окислительному расщеплению под действием РАОX.
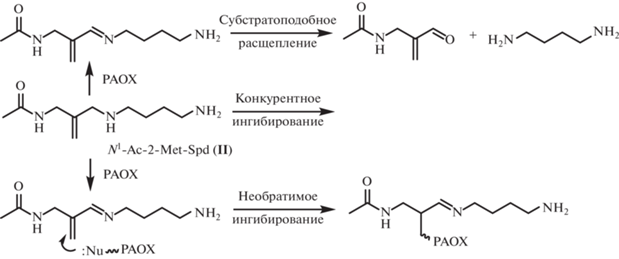
Мы предположили, что N1-Ac-2-Met-Spd (II) должен связываться в активном центре РАОХ и, возможно, претерпевать субстратоподобные превращения, которые приведут к промежуточному образованию основания Шиффа, сопряженного с двойной связью ингибитора (рис. 4). По данным рентгеноструктурного анализа фермент-кофермент-субстратного комплекса, остаток Tyr430 находится на расстоянии 3.4 Å от расщепляемой связи [16], и нельзя исключить возможность присоединения фенольного гидроксила Tyr430 по активированной кратной связи. Наконец, если окислительное расщепление N1-Ac-2-Met-Spd (II) все же приведет к образованию путресцина, то вторым продуктом реакции будет 2-(ацетиламинометилен)акриловый альдегид, который также способен присоединять нуклеофильную группу боковой цепи одной из аминокислот, формирующих активный центр РАОХ (рис. 4).
Синтезированный в настоящей работе 2-Met-Spd (I) (рис. 2) представляет самостоятельный интерес. Исследование взаимодействия С-метилированных аналогов Spd (рис. 3) с SSAT и активности соединений в культуре клеток показало, что субстратные свойства таких аналогов Spd можно регулировать, перемещая метильную группу по скелету Spd [20]. Так, 1-MeSpd и 3-MeSpd не были субстратами SSAT, тогда как 2-MeSpd достаточно эффективно превращается в N1-Ac-2-MeSpd. В то же время все три аналога проникают в клетки DU145, используя систему транспорта полиаминов, и поддерживают рост клеток с истощенным пулом Spd [20]. Соответственно, следует ожидать, что и 2-Met-Spd (I) будет проникать в клетки, и, если он окажется субстратом SSAT, то этот аналог Spd будет представлять собой проингибитор PAOX.
Одной из ключевых стадией синтеза аналогов и производных полиаминов служит создание связей C−N в “скелете” полиаминов, и для этих целей используются разнообразные методы [21]. В настоящей работе в качестве исходного соединения для получения целевых 2-Met-Spd (I) и N1-Ac-2-Met-Spd (II) использовали коммерчески доступный N-(трет-бутилоксикарбонил)-1,4-диаминобутан (III), который превращали в нозильное производное (IV) и затем алкилировали 1-фталимидо-2-метилиден-3-хлорпропаном в DMF при 55°C в течение 8 ч в присутствии K2CO3 (схема 1 ). Затем Ns-защитную группу удаляли “one-pot” действием PhSH/K2CO3 в DMF, и Вос-Pht-триамин (V) выделяли колоночной хроматографией на силикагеле (алкилирование избытка 1,4-диаминобутана 1-фталимидо-2-метилиден-3-хлорпропаном в THF при комнатной температуре проходило неоднозначно и приводило к набору трудноразделяемых продуктов). Дальнейшие превращения включали введение Вос-защитной группы по вторичной аминогруппе соединения (V), что привело к получению тризащищенного триамина (VI), и удаление фталильной защитной группы гидразинолизом, что позволило получить ди-Вос-триамин (VII), который выделяли колоночной хроматографией на силикагеле. После удаления Вос-защитных групп действием HCl/EtOH был получен целевой тригидрохлорид 2-Met-Spd (I) с суммарным выходом 41%, в расчете на Вос-диамин (III). Для получения N1-Ac-2-Met-Spd (II) (схема 1 ) свободную аминогруппу ди-Вос-триамина (VII) ацетилировали AcCl и затем удаляли Вос-защитные группы действием HCl/EtOH, что привело к получению целевого дигидрохлорида N1-Ac-2-Met-Spd (II) с суммарным выходом 45%, считая на Вос-диамин (III).
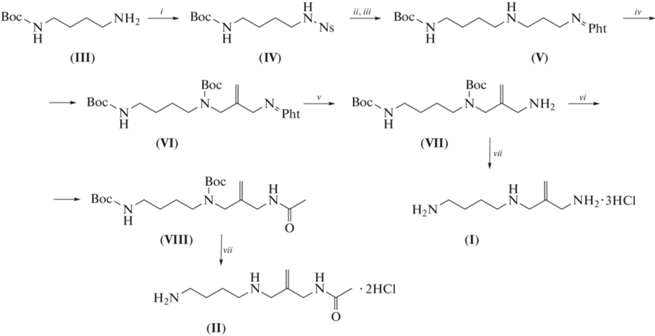
Схема 1 . Синтез 2-Met-Spd (I) и N1-Ac-2-Met-Spd (II). i – NsCl/CH2Cl2/Et3N; ii – Pht=NCH2C(CH2)CH2Cl/DMF/K2CO3/50°C; iii – PhSH/DMF/K2CO3; iv – Boc2O/диоксан; v – H2NNH2. H2O/EtOH/Δ; vi – AcCl/Et3N/CH2Cl2; vii – HCl/EtOH.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
При выполнении работы использовали следующие реактивы: N-(трет-бутилоксикарбонил)-1,4-диаминобутан (Boc-Put), тиофенол (PhSH), хлорангидрид 2-нитробензолсульфокислоты (NsCl), триэтиламин (Et3N), Вос2О и безводный K2CO3 – все реактивы фирмы Aldrich (США). Синтез 1-фталимидо-2-метилиден-3-хлорпропана был осуществлен исходя из 2-хлорметил-3-хлорпропена-1 и фталимида калия согласно методу, описанному ранее [22].
Колоночную хроматографию выполняли на силикагеле Kieselgel (40–63 мкм; Merck, Германия), системы для элюции указаны в тексте. ТСХ проводили на пластинках Kieselgel 60 F254 Plates (Merck, Германия) в следующих системах: EtOAc–гексан, 1 : 2 (А); СH2Cl2–МеOH–NH4OH (25%), 100 : 5 : 0.5 (Б); диоксан–NH4OH (25%), 100 : 1 (В); СH2Cl2–MeOH, 95 : 5 (Г); диоксан–NH4OH (25%), 7 : 3 (Д). Соединения на хроматограммах визуализировали по УФ-поглощению, Boc-производные – при помощи бромфенолового синего, а соединения со свободной аминогруппой – с использованием цветной реакции с нингидрином.
Спектры 1Н- и 13С-ЯМР регистрировали на спектрометре AM-300 (Bruker, Германия) в CDCl3 и D2O, внутренние стандарты – Me4Si (CDCl3) и натриевая соль 3-триметилсилилпропан-сульфокислоты (D2O). Химические сдвиги приведены в миллионных долях, КССВ – в герцах. Температуру плавления определяли в открытом капилляре на приборе Mel-Temp 1202D (Electrotermals, Великобритания).
N1-(2-Нитрофенилсульфонил)-N4-(трет-бутилоксикарбонил)-1,4-диаминобутан (IV). К охлажденному до 4°C раствору 4.85 г (25.8 ммоль) N1-(трет-бутилоксикарбонил)-1,4-диаминобутана (III) в смеси 4.5 мл (33.2 ммоль) Et3N и 50 мл абс. CH2Cl2 добавляли при перемешивании в течение 60 мин раствор 5.21 г (23.5 ммоль) NsCl в 30 мл абс. CH2Cl2, перемешивали в течение 3 ч при 4°C и еще 4 ч при 20°C. Осадок отфильтровывали, фильтрат промывали последовательно 1 М NaHCO3 (4 × 25 мл), H2O (15 мл), 10%-ной лимонной кислотой (5 × 25 мл), H2O (15 мл), 5 М NaCl (2 × 25 мл) и высушивали над MgSO4. Растворитель отгоняли в вакууме. Получили 8.24 г (94%) соединения (IV) в виде густого масла, Rf 0.15 (А).
1H-ЯМР (CDCl3) δ: 8.17–8.09 (1H, м, Ns), 7.89–7.82 (1H, м, Ns), 7.79–7.69 (2H, м, Ns), 5.36 (1H, уш.с, NHNs), 4.53 (1H, уш.с, BocNH), 3.16–3.02 (2H, м, CH2NHNs), 1.61–1.46 (2H, м, CH2NHBoc + + (CH2)2CH2NH), 1.42 (9H, с, C(CH3)3).
13C-ЯМР (CDCl3) δ: 156.1, 148.3, 133.9, 133.7, 132.9, 131.2, 125.5, 79.4, 43.6, 40.0, 28.5 (3C), 27.3, 27.0.
N1-(Фталоил)-N8-(трет-бутилоксикарбонил)-1,8-диамино-2-(метилиден)-4-азаоктан (V). Смесь 7.84 г (21.0 ммоль) соединения (IV), 5.94 г (25.2 ммоль) 1-фталимидо-2-метилиден-3-хлорпропана и 11.6 г (84.1 ммоль) безводного K2CO3 в 50 мл абс. DMF перемешивали в течение 8 ч при 55°C, затем добавляли 4.2 мл (37.5 ммоль) PhSH и 3.15 г (22.5 ммоль) безводного K2CO3, перемешивали еще 3 ч при 20°C. Осадок отфильтровывали, фильтрат упаривали досуха при 0.5 мм Hg, остаток растворяли в абс. CH2Cl2, последовательно промывали H2O (25 мл) и 5 М NaCl (2 × 25 мл), высушивали над MgSO4. Растворитель отгоняли в вакууме, остаток очищали колоночной хроматографией на SiO2 (160 г), элюируя CH2Cl2–MeOH–NH4OH (25%), 100 : 3 : 0.3. Фракции, содержащие соединение (V), объединяли, упаривали в вакууме досуха, высушивали в вакууме над P2O5/KOH. Получили 6.97 г (86%) соединения (V), Rf 0.33 (Б).
1H-ЯМР (CDCl3) δ: 7.88–7.81 (2H м, Pht), 7.75–7.67 (2H м, Pht), 5.09–5.04 (1Н, м, C=СН2), 5.01–4.96 (1Н, м, C=СН2), 4.76 (1H, уш. c, =CCH2NH), 4.30 (2Н, с, СН2N=Pht), 3.25 (2H, c, =CCH2NH), 3.16–3.04 (2H, м, NHCH2CH2), 2.61–2.53 (2H, м, CH2NHBoc), 1.54–1.45 (5Н, м, NH + + CH2CH2CH2NHBoc), 1.42 (9H, c, С(СH3)3).
13C-ЯМР (CDCl3) δ: 168.3, 156.1, 142.0, 134.1, 132.2 123.5, 113.4, 79.1, 53.1, 48.8, 40.9, 40.6, 28.6, 27.9, 27.4.
N4,N8-ди-(трет-Бутилоксикарбонил)-1,8-диамино-2-(метилиден)-4-азаоктан (VII). К раствору 6 г (15.5 ммоль) соединения (V) в 50 мл диоксана добавляли 3.7 г (17 ммоль) Вос2О и перемешивали в течение 4 ч при 20°С. Реакционную смесь упаривали в вакууме досуха, остаток растворяли в 5 мл CH2Cl2 и очищали колоночной хроматографией на SiO2 (60 г), элюируя CH2Cl2, а затем CH2Cl2–MeOH, 100 : 2. Фракции, содержащие соединение (VI), объединяли, упаривали в вакууме досуха, к остатку добавляли раствор 0.6 мл (12 ммоль) N2H4 . H2O в 35 мл EtOH и кипятили при перемешивании 5 ч. Осадок отфильтровывали, фильтрат упаривали в вакууме досуха, остаток очищали колоночной хроматографией на SiO2 (130 г), элюируя смесью диоксан–NH4OH (25%), 100 : 0.5. Фракции, содержащие соединение (VII), объединяли, упаривали в вакууме досуха. После высушивания в вакууме над P2O5/KOH получили 2.1 г (63%, в расчете на соединение (V)) соединения (VII) в виде густого масла, Rf 0.46 (В).
1H-ЯМР (CDCl3) δ: 5.04–4.99 (1Н, м, C=СН2), 4.89–4.83 (1Н, м, C=СН2), 4.56 (1Н, уш.с., NHBoc), 3.86 (2Н, c, BocNCH2С=), 3.25–3.03 (6H, м, BocNНCH2СН2 + NH2CH2С= + + BocNCH2СН2), 1.59–1.35 (24Н, м, NH2 + + N(C(О)О(CH3)3)CH2CH2CH2CH2NHС(О)ОC(CH3)3).
13C-ЯМР (CDCl3) δ: 156.1, 147.3, 110.9, 110.3, 79.8, 79.2, 50.1, 49.5, 46.0, 44.9, 40.4, 28.6, 27.6, 25.5.
N1-Ацетил-N4,N8-ди-(трет-бутилоксикарбонил)-1,8-диамино-2-(метилиден)-4-азаоктан (VIII). К раствору 1 г (2.8 ммоль) соединения (VII) и 0.73 мл (5.25 ммоль) Et3N в 6 мл абс. СH2Cl2 при 4°С добавляли при перемешивании в течение 20 мин раствор 0.25 мл (3.5 ммоль) AcCl в 3 мл абс. СH2Cl2. Реакционную смесь перемешивали в течение 3 ч при комнатной температуре, добавляли 1 мл абс. МеОН, перемешивали при комнатной температуре еще 20 мин, разбавляли вдвое СH2Cl2 и затем последовательно промывали 1 М NaHCO3 (3 × 5 мл), H2O (5 мл), 10%-ной лимонной кислотой (3 × 10 мл), H2O (5 мл), 5 М NaCl (2 × 10 мл), высушивали над MgSO4 и упаривали в вакууме досуха. Получили 1.1 г (98%) соединения (VIII) в виде густого масла, Rf 0.33 (Г).
1H-ЯМР (CDCl3) δ: 6.56 (1H, уш.с., NHAc), 5.12–5.02 (1Н, м, C=СН2), 4.98–4.91 (1Н, м, C=СН2), 4.61 (1Н, уш.с., NHBoc), 3.87–3.74 (4Н, м, AcNHCH2С= + BocNCH2С=), 3.20–3.04 (4H, м, BocNНCH2СН2 + BocNCH2СН2), 1.99 (3H, c, CH3C(O)NH), 1.57–1.36 (22Н, м, N(C(О)О(CH3)3)CH2CH2CH2CH2NHC(О)О(CH3)3).
13C-ЯМР (CDCl3) δ: 170.1, 156.2, 142.1, 114.5, 112.0, 80.1, 79.3, 49.7, 46.3, 42.00, 40.3, 28.5, 27.6, 25.4, 23.4.
Тригидрохлорид 1,8-диамино-2-(метилиден)-4-азаоктана (VIII). К раствору 0.49 г (1.36 ммоль) соединения (VII) в 3 мл абс. EtОН добавляли 2 мл 10 М HCl/EtOH, через 4 ч при 20°С реакционную смесь упаривали в вакууме досуха, остаток соупаривали с абс. EtOH (3 × 10 мл). Остаток растирали со смесью EtOH/Et2O (1 : 3), осадок отделяли центрифугированием. После высушивания в вакууме над P2O5/KOH получили 287 мг (79%) соединения (VIII), Rf 0.28 (Д). Аналитический образец перекристаллизовывали из MeOH/EtOH, т. пл. 178–179°С.
1H-ЯМР (D2O) δ: 5.61–5.57 (1Н, м, C=СН2), 5.56–5.52 (1Н, м, C=СН2), 3.77 (2Н, с, =CСН2NH2), 3.72 (2Н, с, =ССН2NH), 3.20–3.10 (2H, м, CH2CH2NН2), 3.09–2.99 (2H, м, NНCH2CH2), 1.88–1.69 (4H, м, CH2CH2CH2NН2).
13C-ЯМР (D2O) δ: 132.7, 120.4, 49.4, 47.2, 41.4, 38.9, 24.0, 22.7.
HRESIMS: m/z вычислено для C8H19N3 [M + H]+: 158.1657. Найдено: 158.1661.
Дигидрохлорид N1-ацетил-1,8-диамино-2-(метилиден)-4-азаоктана (II). К раствору 1.1 г (2.8 ммоль) соединения (VIII) в 7 мл абс. EtОН добавляли 3 мл 10 М HCl/EtOH, через 4 ч при 20°С реакционную смесь упаривали в вакууме досуха, остаток соупаривали с абс. EtOH (3 × 10 мл). Остаток растирали со смесью EtOH/Et2O (1 : 3), осадок отделяли центрифугированием. После высушивания в вакууме над P2O5/KOH получили 0.66 г (88%) соединения (II), Rf 0.54 (Д). Аналитический образец перекристаллизовывали из MeOH/EtOH, т. пл. 154–155°С.
1H-ЯМР (D2O) δ: 5.35–5.30 (1Н, м, C=СН2), 5.29–5.24 (1Н, м, C=СН2), 3.82 (2Н, с, =CСН2NH2), 3.63 (2Н, с, =ССН2NH), 3.14–2.96 (4H, м, CH2CH2NН2 + NНCH2CH2), 2.01 (3Н, с, СН3С(О)NH), 1.84–1.65 (4H, м, NНCH2CH2CH2CH2NН2).
13C-ЯМР (D2O) δ: 174.7, 136.2, 118.0, 49.3, 46.8, 42.0, 38.9, 24.0, 22.7, 21.8.
HRESIMS: m/z вычислено для C10H21N3O [M + H]+: 200.1763. Найдено: 200.1762.
ЗАКЛЮЧЕНИЕ
Предложен и реализован удобный семистадийный способ синтеза неизвестных ранее 2-метилиденовых производных спермидина 2-Met-Spd и N1-Ac-2-Met-Spd, позволяющий получать данные целевые вещества с высокими суммарными выходами. Полученные соединения могут служить полезными инструментами в исследовании метаболизма полиаминов, а именно представляют интерес для изучения особенностей РАОХ-реакции, а также обладают определенным потенциалом для ингибирования фермента в культуре клеток.
Список литературы
Miller-Fleming L., Olin-Sandoval V., Campbell K., Ralser M. // J. Mol. Biol. 2015. V. 427. P. 3389–3406. https://doi.org/10.1016/j.jmb.2015.06.020
Pegg A.E. // J. Biol. Chem. 2016. V. 291. P. 14904–14912. https://doi.org/10.1074/jbc.R116.731661
Casero R.A., Murray Stewart T., Pegg A.E. // Nature Rev. Cancer. 2018. V. 18. P. 681–695. https://doi.org/10.1038/s41568-018-0050-3
Holbert C.E., Cullen M.T., Casero R.A., Jr., Murray Stewart T. // Nature Rev. Cancer. 2022. https://doi.org/10.1038/s41568-022-00473-2
Gerner E.W., Bruckheimer E., Cohen A. // J. Biol. Chem. 2018. V. 293. P. 18770–18778. https://doi.org/10.1074/jbc.TM118.003343
Alhonen L., Parkkinen J.J., Keinanen T., Sinervirta R., Herzig K.H., Janne J. // Proc. Natl. Acad. Sci. USA. 2000. V. 97. P. 8290–8295. https://doi.org/10.1073/pnas.140122097
Murray-Stewart T., Dunworth M., Foley J.R., Schwartz C.E., Casero R.A. // Med. Sci. (Basel). 2018. V. 6. P. E112. https://doi.org/10.3390/medsci6040112
Inoue K., Tsutsui H., Akatsu H., Hashizume Y., Matsukawa N., Yamamoto T., Toyo’oka T. // Sci. Rep. 2013. V. 3. P. 2364. https://doi.org/10.1038/srep02364
Lewandowski N.M., Ju S., Verbitsky M., Ross B., Geddie M.L., Rockenstein E., Adame A., Muhammad A., Vonsattel J.P., Ringe D., Cote L., Lindquist S., Masliah E., Petsko G.A., Marder K., Clark L.N., Small S.A. // Proc. Natl. Acad. Sci. USA. 2010. V. 107. P. 16970–16975. https://doi.org/10.1073/pnas.1011751107
Guerra G.P., Rubin M.A., Mello C.F. // Pharmacol. Res. 2016. V. 112. P. 99–118. https://doi.org/10.1016/j.phrs.2016.03.023
Eisenberg T., Abdellatif M., Schoeder S., Primessnig U., Stekovic S., Pendl T., Harger A., Schipke J., Zimmermann A., Schmidt A., Tong M., Ruckenstuhl Ch., Dammbrueck Ch., Gross A.S., Herbst V., Magnes Ch., Trausinger G., Narath S., Meinitzer A., Hu Z., Kirsch A., Eller K., Carmona-Gutierrez D., Büttner S., Pietrocola F., Knittelfelder O., Schrepfer E., Rockenfeller P., Simonini C., Rahn A., Horsch M., Moreth K., Beckers J., Fuchs H., Gailus-Durner V., Neff F., Janik D., Rathkolb B., Rozman J., Hrabe de Angelis M., Moustafa T., Haemmerle G., Mayr M., Willeit P., von Frieling-Salewsky M., Pieske B., Scorrano L., Pieber T., Pechlaner R., Willeit J., Sigrist S.J., Linke W.A., Mühlfeld Ch., Sadoshima J., Dengjel J., Kiechl S., Kroemer G., Sedej S., Madeo F. // Nature Med. 2016. V. 22. P. 1428–1438. https://doi.org/10.1038/nm.4222
Igarashi K., Kashiwagi K. // Mol. Nutr. Food Res. 2011. V. 55. P. 1332–1341. https://doi.org/10.1002/mnfr.201100068
Ramani D., De Bandt J.P., Cynober L. // Clin. Nut. 2014. V. 33. P. 14–22. https://doi.org/10.1016/j.clnu.2013.09.019
Wallace H.M., Duthie J., Evans D.M., Lamond S., Nicoll K.M., Heys S.D. // Clin. Cancer Res. 2000. V. 6. P. 3657–3661.
Bey P., Bolkenius F.N., Seiler N., Casara P. // J. Med. Chem. 1985. V. 28. P. 1–2. https://doi.org/10.1021/jm00379a001
Sjorgen T., Wassvik C.M., Snijder A., Aagaard A., Kumanomidou T., Barlind L., Kaminksi T.P., Kasima T.P., Yokota T., Fjellstrom O. // Biochemistry. 2017. V. 56. P. 458–467. https://doi.org/org/10.1021/acs.biochem.6b01140
Bianchi M., Polticelli F., Ascenzi P., Botta M., Federico R., Mariottini P., Cona A. // FEBS J. 2006. V. 273. P. 1115–1123. https://doi.org/10.1111/j.1742-4658.2006.05137.x
Dunston T.T., Khomutov M.A., Gabelli S.B., Stewart T.M., Foley J.R., Kochetkov S.N., Khomutov A.R., Casero R.A., Jr. // Acta Naturae. 2020. V. 12. P. 140–144. https://doi.org/10.32607/actanaturae.10992
Khomutov M.A., Hyvönen M.T, Simonian A.R., Weisell J., Vepsäläinen J., Alhonen L., Kochetkov S.N., Keinänen T.A., Khomutov A.R. // Mendeleev Commun. 2018. V. 28. P. 479–481. https://doi.org/10.1016/j.mencom.2018.09.008
Hyvönen M.T., KeinänenT.A., Khomutov M., Simonian A., Weisell J., Kochetkov S.N., Vepsäläinen J., Alhonen L., Khomutov A.R. // J. Med. Chem. 2011. V. 54. P. 4611–4618. https://doi.org/10.1021/jm200293r
Хомутов M.A., Михура И.В., Кочетков С.Н., Хомутов A.Р. // Биоорг. химия. 2019. Т. 45. С. 588–614. [Khomutov M.A., Mikhura I.V., Kochetkov S.N., Khomutov A.R. // Russ. J. Bioorg. Chem. 2019. V. 45. P. 463–487.] https://doi.org/10.1134/S013234231906023X
Григоренко Н.А., Хомутов M.A., Симонян А.Р., Кочетков С.Н., Хомутов A.Р. // Биоорг. химия. 2016. Т. 42. С. 469–474. [Grigorenko N.A., Khomutov M.A., Simonian A.R., Kochetkov S.N., Khomutov A.R. // Russ. J. Bioorg. Chem. 2016. V. 42. P. 423–427.] https://doi.org/10.7868/S0132342316030088
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия