

Биоорганическая химия, 2022, T. 48, № 6, стр. 732-744
Расчеты свободной энергии взаимодействия широкоспецифичной пиримидиннуклеозидфосфорилазы с потенциальными ингибиторами
П. А. Эйстрих-Геллер 1, *, С. В. Рубинский 1, В. Р. Самыгина 1, 2, А. А. Лашков 1
1 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
119333 Москва, Ленинский проспект, 59, Россия
2 Национальный исследовательский центр “Курчатовский институт”
123182 Москва, ул. Академика Курчатова, 1, Россия
* E-mail: eistrikh.geller@crys.ras.ru
Поступила в редакцию 26.01.2022
После доработки 09.03.2022
Принята к публикации 17.06.2022
- EDN: BNFAJE
- DOI: 10.31857/S0132342322060100
Аннотация
Цель работы – поиск специфичного ингибитора пиримидиннуклеозидфосфорилазы. Использовали методы молекулярного моделирования комплексов белок–лиганд, такие как молекулярный докинг и молекулярная динамика, позволяющие найти энергию связывания белка с лигандом (ΔGbind). В качестве возможных ингибиторов взяты следующие соединения: 2',3'-дидегидро-3'-дезокситимидин (d4T), 1-(2-дезокси-2-фтор-β-D-арабинофуранозил)-5-иодурацил (фиауридин, FIAU), 1-(2-дезокси-2-фтор-β-D-арабинофуранозил)-5-урацил (FAU) и 2-пиримидин-2-ил-1H-имидазол-4-карбоновая кислота (PIA). Предварительную оценку энергии связывания проводили методом линейной интерполяции свободной энергии (LIE), уточнение расчетов – методом возмущения свободной энергии (FEP) в пакете программ GROMACS. Полученные в ходе расчетов данные указывают на то, что соединения PIA и d4T связываются с активным центром белка BsPyNP из Bacillus subtilis с наибольшей аффинностью среди других предполагаемых ингибиторов. PIA к тому же хуже связывается с тимидинфосфорилазой человека, что минимизирует возможные побочные эффекты применения этого соединения в терапевтических целях.
ВВЕДЕНИЕ
Нуклеозидфосфорилазы (в частности, тимидинспецифичная нуклеозидфосфорилаза КФ 2.4.2.4.) – белки-ферменты, играющие важную роль в синтезе нуклеозидов и азотистых оснований [1]. Нуклеозидфосфорилазы катализируют фосфоролитическое расщепление пуриновых и пиримидиновых нуклеозидов. Некоторые белки этого класса обнаружены во многих раковых клетках, что позволяет использовать их в качестве активаторов противоопухолевых препаратов [2, 3]. У некоторых классов прокариот присутствует широкоспецифичная пиримидиннуклеозидфосфорилаза (PyNP), способствующая расщеплению как тимидина, так и уридина примерно с равной каталитической активностью. При этом тимидинфосфорилаза и широкоспецифичная пиримидиннуклеозидфосфорилаза – единственные представители NP-II-семейства нуклеозидфосфорилаз. Показано, что инфицирование клеток некоторых тканей бактериями Mycoplasma hyorhinis препятствует нормальной фармокинетике фторпиримидинов. Это объясняется активностью фермента PyNP в этих бактериях в отношении 5-фтор-2'-дезоксиуридина, 5-трифтортимидина и 5-фтор-5'-дезоксиуридина [1, 2]. Препятствование ферментативой функции PyNP из M. hyorhinis [4] при фторпиримидиновой терапии обусловливает необходимость разработки конкурентных ингибиторов, специфичных к BsPyNP (пиримидиннуклеозидфосфорилаза из Bacillus subtilis) и неактивных в отношении hTP (тимидиннуклеозидфосфорилаза человека) [3].
Цель данного исследования – поиск предполагаемых ингибиторов тимидинфосфорилазы и широкоспецифичной пиримидиннуклеозидфосфорилазы и их сравнение по энергии связывания с белком-мишенью.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Молекулярный докинг и молекулярная динамика. В качестве стартовой трехмерной модели белка взята определенная нами ранее методом рентгеноструктурного анализа структура пиримидиннуклеозидфосфорилазы из Bacillus subtilis (BsPyNP) [5, 6], гомологичной пиримидиннуклеозидфосфорилазе из M. hyorhinis. Для поиска соединений, аффинных активному центру BsPyNP, среди существующих биологически активных соединений, удовлетворяющих правилам “пяти” Липински [7], были отобраны четыре соединения. Первые два взяты из работы [8]: 2',3'-дидегидро-3'-дезокситимидин (d4T) и 1-(2-дезокси-2-фтор-β-D-арабинофуранозил)-5-иодурацил (фиауридин, FIAU) (рис. 1). Поскольку в молекуле FIAU заместителем в пятом положении пиримидинового цикла является атом иода, который может создать дополнительные стерические ограничения при связывании вероятного ингибитора с активным центром фермента, было решено взять в качестве третьего лиганда то же соединение, но без заместителя – атома иода – 1-(2-дезокси-2-фтор-β-D-арабинофуранозил)-5-урацил (FAU). Моделирование связывания двух схожих молекул, но с разными по размеру заместителями по пиримидиновому кольцу, позволяет выяснить степень влияния атома иода на связывание лиганда с белком.
Рис. 1.
Структурные формулы предполагаемых лигандов (ингибиторов) пиримидиннуклеозидфосфорилазы из Bacillus subtilis (BsPyNP): d4T (а), FIAU (б), FAU (в), PIA (г) при pH < 6.5, PIA (д) при pH > 6.5.
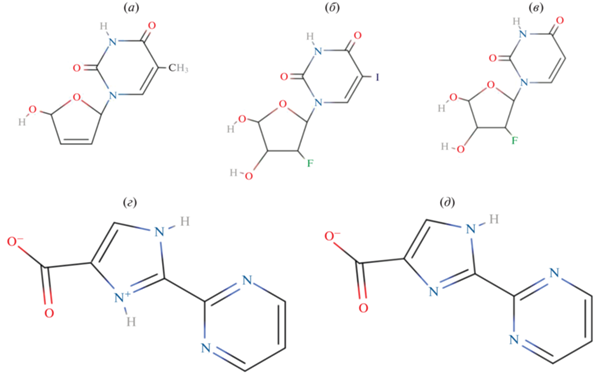
В работе [9] описано соединение 2-пиримидин-2-ил-1H-имидазол-4-карбоновая кислота (PIA) (рис. 1) – предполагаемый специфичный ингибитор BsPyNP, для которого в этой работе был проведен первый этап молекулярного докинга. Однако это соединение при разных значениях pH раствора присутствует в разных формах с разным общим электрическим зарядом: в растворах с нейтральной реакцией среды – в виде в целом не заряженного цвиттер-иона, а при кислой – в виде аниона. Различие общего заряда и состояния протонирования лиганда может кардинальным образом повлиять на его связывание с ферментом.
Результатом первого этапа стал набор конформаций для каждого лиганда. Разница в решениях докинга, полученных с помощью разных программ (Autodock и PLANTS), объясняется разницей в методике определения подходящих конформаций. Близкие друг к другу конформации объединены в кластеры по сходству расположения атомов и углов между связями атомов в конформере. Для d4T найден 1 кластер конформаций, для FIAU – 3 кластера (2 (Autodock) + 1 (PLANTS)), для FAU – два кластера (1 + 1) (рис. 2). Для каждой из конформаций был проведен анализ энергии взаимодействия и устойчивости комплекса (рис. 3) в зависимости от времени молекулярно-динамической симуляции частиц системы и получены, кроме стартовой, конформации с минимальной энергией взаимодействия по методу линейной интерполяции свободной энергии (LIE). Проведено сравнение конформаций лигандов с конформациями похожих лигандов в структурах экспериментально определенных комплексов тимидинфосфорилаз. По трехмерной структуре тимидин- и пиримидинфосфорилазы схожи, однако для последней не изучены структуры комплексов белок–лиганд.
Рис. 2.
Центровая конформация для каждого кластера отобранных конформаций лигандов для d4T (а), FIAU (б) и FAU (в).
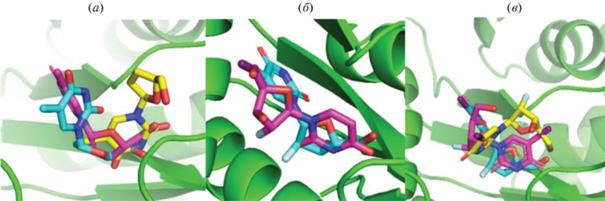
Рис. 3.
Графики зависимости RMSD-координат неводородных атомов лигандов от времени МД-симуляции для d4T (а), FIAU (б) и FAU (в) для разных стартовых конформаций лигандов. Выравнивание структур выполнено по атомам главной цепи фермента BsPyNP.
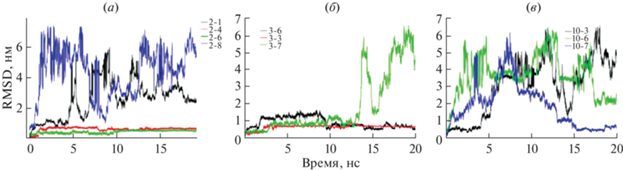
Таким образом, в работе рассмотрены две конформации: start – начальный вариант после молекулярного докинга, min – варианты с наименьшей энергией связывания белок–лиганд, вычисленной по методу LIE. Для лигандов d4T, FAU и FIAU была подобрана одна конформация со стартовым и одна – с минимальным значением энергии связывания по методу LIE (табл. 1).
Таблица 1.
Результаты расчета относительной аффинности связывания (кДж/моль) лигандов BsPyNP методом LIE
| Составляющая свободной энергии Гиббса | FAU (AD) | d4T (AD) | d4T (PL) | FIAU (AD) | PIA (AD) (pH < 6.5) | PIA (PL) (pH < 6.5) | PIA (AD) (pH > 7.0) | PIA (PL) (pH > 7.0) |
|---|---|---|---|---|---|---|---|---|
| 〈ΔGbind〉 | –3.2 | –5.8 | 0.1 | 7.2 | 13.2 | –15.3 | –46.5 | –56.9 |
| σ(ΔGbind) | 15.2 | 18.4 | 11.1 | 9.6 | 15.5 | 16.7 | 22.4 | 21.9 |
| ΔGbind_start | 18.9 | 22.1 | –4.8 | 10.9 | –2.6 | –10.0 | –50.5 | –55.9 |
| ΔGbind_min1 | –95.7 | –55.7 | –50.5 | –38.4 | –79.1 | –87.0 | –136.1 | –140.0 |
| ΔGbind_min2 | – | – | – | – | –73.6 | –80.6 | – | –138.1 |
Для конформаций PIA, полученных в программах Autodock и PLANTS при pH < 6.5, были найдены два минимума энергии с близкими значениями энергии связывания (табл. 1), но сильно отличающимися положениями атомов, поэтому дальнейшие расчеты проводили для двух конформаций: min1 и min2. Аналогичная ситуация характерна для конформаций PIA, полученных с помощью программы PLANTS при pH 7.0. Для них также найдены два минимума со схожими значениями энергий (табл. 1), и для дальнейшей обработки отобраны три конформации (стартовая и две минимальные).
Молекулярная динамика и расчеты методом возмущения свободной энергии (FEP). Полученные величины ΔGbind представлены в табл. 2–5. Для всех конформаций лигандов с ΔGbind > 0 вероятность связывания низкая. Это связано с тем, что при большом значении энергии Гиббса реакция связывания лиганда с активным центром белка не спонтанная. Константа ингибирования в этом случает имеет слишком высокие значения, такие, что концентрация лиганда не является достижимой.
Таблица 2.
Свободная энергия (и ее составляющие) связывания BsPyNP с производными пиримидиновых нуклеозидов (кДж/моль)
| Составляющая свободной энергии Гиббса | d4Tmin | d4Tstart | d4Tmin_PL | FIAU | FAU |
|---|---|---|---|---|---|
| ΔG solv_coul | –85.8 ± 0.5 | –79.5 ± 0.5 | –84.1 ± 0.5 | –86.3 ± 0.5 | |
| ΔG solv_vdw | –0.6 ± 0.3 | –0.2 ± 0.4 | –7.1 ± 0.4 | –4.8 ± 0.4 | |
| ΔG solv(wdv + coul) | –86.4 ± 0.6 | –79.7 ± 0.6 | –91.2 ± 0.6 | –91.1 ± 0.6 | |
| ΔG solv_rest | –33.7 | –35.7 | –38.0 | –34.3 | –33.7 |
| ΔG prot_restr | –9.6 ± 0.2 | –18.4 ± 0.2 | –26.7 ± 0.4 | –13.9 ± 0.2 | –24.0 ± 0.5 |
| ΔG prot_coul | –86.1 ± 0.7 | –72.0 ± 0.4 | –103.5 ± 1.3 | –77.0 ± 0.6 | –66.2 ± 0.5 |
| ΔG prot_vdw | –34.7 ± 0.6 | –10.8 ± 0.9 | –12.7 ± 0.8 | –18.6 ± 0.8 | –20.8 ± 0.8 |
| ΔG prot | –130.3 ± 1.0 | –101.2 ± 1.0 | –143.0 ± 1.6 | –109.5 ± 1.0 | –111.1 ± 1.0 |
| ΔGbind | –10.2 ± 1.2 | 20.9 ± 1.2 | –25.3 ± 1.7 | 16.0 ± 1.2 | 13.7 ± 1.7 |
Таблица 3.
Свободная энергия (и ее составляющие) связывания BsPyNP с различными конформациями цвиттер-ионной формы PIA (кДж/моль) при pH < 6.5
| Составляющая свободной энергии Гиббса | ADstart | ADmin1 | ADmin2 | PLstart | PLmin1 | PLmin2 |
|---|---|---|---|---|---|---|
| ΔG solv_coul | –248.2 ± 0.4 | |||||
| ΔG solv_vdw | 2.3 ± 0.2 | |||||
| ΔG solv(vdw + coul) | –245.9 ± 0.4 | |||||
| ΔG solv_rest | –30.0 | –31.2 | –30.1 | –30.1 | –30.4 | –30.1 |
| ΔG prot_restr | –15.7 ± 0.3 | –8.1 ± 0.1 | –3.7 ± 0.0 | –52.8 ± 1.4 | –8.8 ± 0.2 | –10.7 ± 0.2 |
| ΔG prot_coul | –265.7 ± 1.4 | –269.5 ± 0.8 | –283.9 ± 1.0 | –246.6 ± 1.4 | –259.5 ± 1.5 | –280.4 ± 0.9 |
| ΔG prot_vdw | 18.3 ± 0.9 | –1.4 ± 0.7 | 2.8 ± 0.6 | –0.1 ± 0.6 | –6.9 ± 0.5 | 8.8 ± 0.6 |
| ΔG prot | –263.1 ± 1.7 | –279.0 ± 1.1 | –284.8 ± 1.1 | –299.3 ± 2.0 | –275.2 ± 1.6 | –282.2 ± 1.1 |
| ΔGbind | 12.8 ± 1.7 | –1.9 ± 1.2 | –8.8 ± 1.2 | –23.3 | 1.1 ± 1.6 | –6.2 ± 1.2 |
Таблица 4.
Свободная энергия (и ее составляющие) связывания BsPyNP с различными конформациями анионной формы PIA при pH > 6.5 (кДж/моль)
| Составляющая свободной энергии Гиббса | ADstart | ADmin | PLstart | PLmin1 | PLANTSmin2 |
|---|---|---|---|---|---|
| ΔG solv_coul | –522.1 ± 0.6 | ||||
| ΔG solv_vdw | 2.7 ± 0.2 | ||||
| ΔG solv(vdw+coul) | –519.3 ± 0.7 | ||||
| ΔΔGDSF_solv | –0.3 | ||||
| ΔΔGNET + ΔΔGUSV | 0.7 | ||||
| ΔΔGRIP_solv | ~0.0 | ||||
| ΔG solv_rest | –31.7 | –30.2 | –29.6 | –32.2 | –31.7 |
| ΔG prot_restr | –39.0 ± 0.3 | –4.1 ± 0.0 | –28.2 ± 0.4 | –3.4 ± 0.0 | –7.1 ± 0.0 |
| ΔG prot_coul | –554.5 ± 2.4 | –569.2 ± 2.7 | –553.7 ± 2.6 | –613.3 ± 2.6 | –597.1 ± 2.5 |
| ΔG prot_vdw | 16.3 ± 0.6 | 13.3 ± 0.8 | 14.2 ± 0.7 | 4.2 ± 0.7 | 36.3 ± 0.8 |
| ΔG prot(vdw+coul+restr) | –577.2 ± 2.5 | –560.0 ± 2.9 | –567.6 ± 2.7 | –612.5 ± 2.7 | –567.9 ± 2.6 |
| ΔΔGDSF | –4.2 | –4.4 | –4.4 | –4.4 | –4.6 |
| ΔΔGNET + ΔΔGUSV | 0.3 | ||||
| ΔΔGRIP | 0.6 | 0.3 | 0.2 | 0.0 | 0.0 |
| ΔGbind | –29.9 ± 2.6 | –14.7 ± 3.0 | –23.0 ± 2.7 | –65.5 ± 2.7 | –21.6 ± 2.7 |
Таблица 5.
Свободная энергия (и ее составляющие) связывания мутантной (K108M) BsPyNP с различными конформациями цвиттер-ионной (при pH < 6.5) и анионной (при pH > 6.5) формы PIA (кДж/моль)
| pH | рН < 6.5 | рН > 6.5 | ||
|---|---|---|---|---|
| Конформация | start | min | start | min |
| ΔG solv_rest | –31.8 | –33.1 | –31.3 | –31.1 |
| ΔG prot_restr | –32.4 ± 0.6 | –7.2 ± 0.1 | –11.3 ± 0.1 | –14.0 ± 0.2 |
| ΔG prot_coul | –251.0 ± 1.0 | –277.8 ± 0.9 | –537.4 ± 2.4 | –554.4 ± 2.4 |
| ΔG prot_vdw | –6.3 ± 0.7 | 8.5 ± 0.6 | 6.3 ± 0.8 | –2.7 ± 0.8 |
| ΔG prot(vdw + coul + restr) | –293.5 ± 1.3 | –276.5 ± 1.1 | –542.5 ± 2.5 | –571.1 ± 2.5 |
| ΔΔGDSF | – | – | –4.4 | –4.4 |
| ΔΔGNET+ΔΔGUSV | – | 0.3 | ||
| ΔΔGRIP | – | – | 0.8 | 0.1 |
| ΔGbind | –15.8 ± 1.4 | 2.5 ± 1.2 | 4.4 ± 2.6 | –25.1 ± 2.6 |
Примечание: полужирным выделена общая энергия связывания. Составляющие энергии сольватации лиганда показаны в табл. 3 и 4.
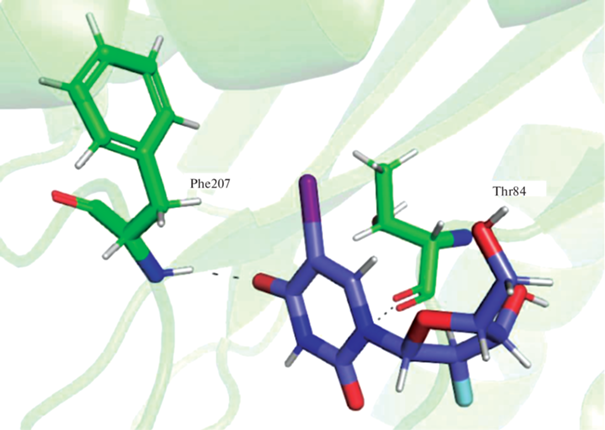
Молекулярное моделирование комплекса белок–лиганд FIAU. Присоединение лиганда осуществляется с помощью трех водородных связей между атомами О1 и О2 лиганда и атомом водорода остатка Gly85, между атомом О2 лиганда и атомом N остатка Phe207 (рис. 4). Величина ∆Gbind = = 16.0 кДж/моль (табл. 2) говорит об очень низкой аффинности связывания данного лиганда с активным центром белка.
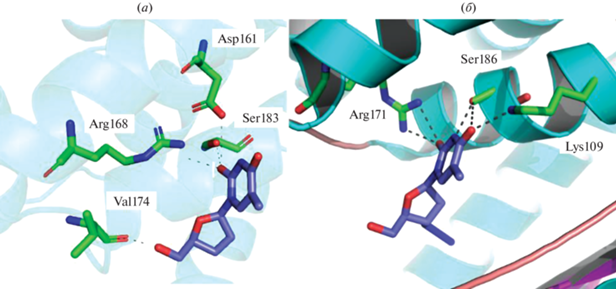
Молекулярное моделирование для комплекса белок–лиганд d4T. Для d4T в конформации с минимальной энергией взаимодействия ∆Gbind = = –10.2 кДж/моль (Ki = 1.4 мкМ) при Т = 300 К (табл. 2). Отрицательное значение свободной энергии связывания говорит о возможности связывания лиганда d4T с активным центром BsPyNP, т.е. d4T – потенциальный ингибитор. В связи с этим проверили специфичность соединения, сравнив комплекс белок–лиганд для BsPyNP и для hTP, соответствующим катализатором ресинтеза азотистых оснований в теле человека. Для этой цели было проведено выравнивание аминокислотной последовательности BsPyNP и hTP (рис. 5). Выявлены аминокислотные остатки активного центра hTP, отличающиеся от соответствующих остатков BsPyNP. Далее был проведен анализ связывания d4T с активными центрами двух ферментов, d4T образует водородные связи с атомами аминокислотных остатков BsPyNP: атомы кислорода d4T связываются с атомами водорода Ser183 (2.2 Å), Val174 (2.1 Å), Arg168 (2.1, 1.9 и 1.7 Å) и Asp161 (1.8 Å) (рис. 6а). В то же время d4T образует связи в активном центре hTP с аминокислотными остатками Arg171 (3 Å), Ser186 (2.7 и 3.5 Å) и Lys109 (2.9 Å) (рис. 6б). Один из остатков – Arg168 (рис. 6а) и этот же остаток под номером Arg171 (рис. 6б) – встречается в обоих случаях.
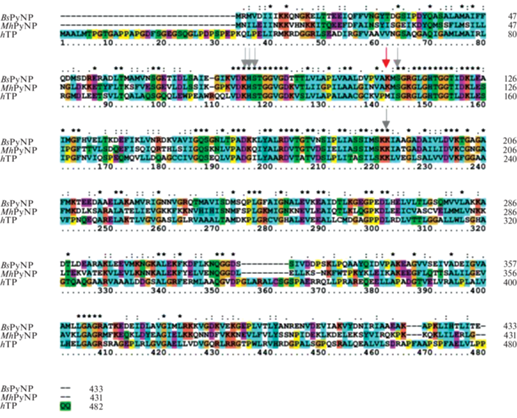
Рис. 5.
Выравнивание аминокислотных последовательностей пиримидиннуклеозидфосфорилазы из Bacillus subtilis (BsPyNP), пиримидиннуклеозидфосфорилазы из Mycoplasma hyorhinis (MhPyNP) и тимидиннуклеозидфосфорилазы человека (hTP). Цвета обозначают степень совпадения аминокислотных остатков, точки обозначают локации полных совпадений, стрелками обозначены аминокислотные остатки, образующие активный центр.
Молекулярное моделирование комплекса белок–лиганд PIA. Молекула PIA была исследована в двух вариантах протонирования: в форме карбоксианиона (нейтральные значения pH) и цвиттер-иона (кислая среда).
При pН < 6.5 ΔGbind = –6.2 кДж/моль для конформации min2, что лучше с точки зрения энергии связывания с белком, чем стартовая конформация, полученная в результате молекулярного докинга, и конформация min1 (табл. 3). Однако это значение намного превышает значение ΔGbind для конформации анионной формы PIA. Для нее проведено моделирование не только с нативной BsPyNP, но и с BsPyNP с мутациями в активном центре, соответствующими активному центру hTP, для сравнения активности PIA в бактериальной клетке и клетках человека. Это сделано не только для определения различий в аффинности связывания лиганда, но и для выявления конкретных остатков активного центра BsPyNP, обусловливающих различие. При pН > 6.5 ΔGbind = –65.5 кДж/моль для конформации min1, что говорит о высокой аффинности связывания PIA с активным центром в этой конформации (табл. 4).
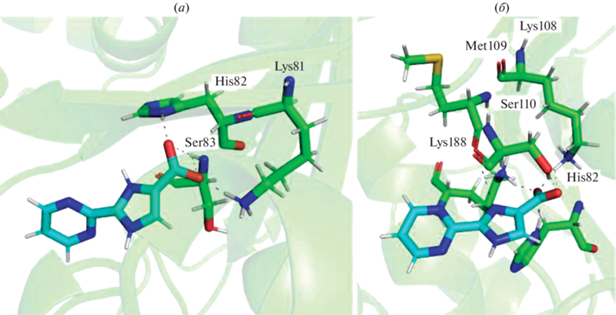
Водородные связи образуются между молекулой предполагаемого ингибитора – PIA – и остатками Lys108, His82, Lys188 и Ser110 белка BsPyNP (рис. 7). Различные найденные конформации PIA отличаются в основном положением карбоксильной группы, которая связывается водородными связями с Lys108 и His82. Эти аминокислотные остатки было решено заменить на соответствующие остатки hTP и провести расчет свободной энергии методом возмущения свободной энергии (FEP).
Созданы структурные модели двух мутантов: с заменой остатка Lys108 на Trp108 и с заменой остатка His82 на Gly82. В результате для получившихся моделей ∆Gbind варьируется от –25.1 до 4.4 кДж/моль (табл. 5), что существенно меньше по модулю энергии для лиганда в активном центре нативной BsPyNP при pH > 6.5.
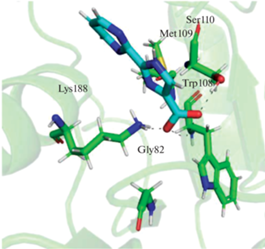
После замены аминокислотных остатков в активном центре белка остатки, образующие водородные связи с лигандом, изменились (рис. 8). Водородные связи образуются между карбоксильной группой лиганда и атомами кислорода для Thr108 (2.9 Å), Lys188 (3.4 и 3.0 Å) и Ser110 (2.6 Å). Также образуется водородная связь между карбоксильной группой и атомом азота в случае Lys188 (3.1 Å). В целом наблюдаются большие расстояния между атомами при связывании PIA с мутантными формами PyNP, что подтверждается меньшей по модулю ∆Gbind.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Молекулярный докинг. В качестве исходной структуры для молекулярного докинга была использована структура белка BsPyNP, полученная в ходе рентгеноструктурного анализа [5]. Для получения наиболее полного набора конфигураций использовали программы молекулярного докинга Autodock [10] и PLANTS [11], работающие с разными алгоритмами поиска глобального минимума: Аutodock использует алгоритм Монте–Карло [12], а PLANTS – алгоритм оптимизации подражания муравьиной колонии [13]. В обеих программах для параметризации атомов использовали полуэмпирическое силовое поле AMBER. В качестве активной области докинга был выбран куб с ребром 22 Å, в который были включены все атомы аминокислотных остатков активного центра белка BsPyNP. Шаг сетки докинга составлял 0.375 Å. Аминокислотные остатки считались неподвижными. В ходе докинга было отобрано по 10 конформаций для каждого лиганда с самыми высокими значениями оценочных функций, используемых по умолчанию в каждой из программ. Конформации объединены в кластеры по параметру среднеквадратичного отклонения координат атомов лиганда (рис. 2).
Молекулярное моделирование методом классической молекулярной динамики. Для исследования устойчивости связывания лигандов с PyNP проводили молекулярно-динамическое (МД) моделирование этого комплекса в пакете программ GROMACS (версия 2018.6) [13, 14]. Компоновку и параметризацию MД-систем комплексов белок–лиганд проводили в web-сервисе CHARMM-GUI [13–15] с использованием набора полноатомных силовых полей CHARMM [16, 17] версии C36m [15]. Модельные ячейки заполняли молекулами воды, которые описывались трехцентровой моделью TIP3P, с добавлением нейтрализующих ионов Na+ и Cl– с концентрацией 0.1 M.
Для интегрирования с шагом 2 фс уравнений движения Ньютона использовали алгоритм “leapfrog”. Длина траектории молекулярной динамики составляла 20 нс. Расчет дальних электростатических взаимодействий проводили методом частица–сетка (PME) [18, 19]. Для описания ван-дер-ваальсовых взаимодействий использовали функцию сглаживания “force–switch” в интервале 10–12 Å. Давление в системе контролировали баростатом Паринелло–Рахмана [19] на уровне 1 бар. Температуру МД-системы поддерживали постоянной с помощью термостата V-rescale [20] на уровне 300 К. Процедуре молекулярной динамики предшествовал этап минимизации энергии системы с использованием выбранного силового поля в виртуальной ячейке методом градиентного спуска и уравновешивание системы путем моделирования с ограничениями, накладываемыми на положения неводородных атомов белка и лиганда в NVT- и NPT-ансамблях в течение 200 пс (использовали баростат Берендсена).
Определение относительной аффинности связывания лигандов методом линейной интерполяции свободной энергии (LIE). Приблизительную оценку аффинности связывания лигандов методом LIE [21] осуществляли в программе gmx lie программного пакета GROMACS (версия 2018.6) [13, 14]. Метод использовали как для оценки энергии связывания лиганда в конформациях, полученных на этапе молекулярного докинга, так и для поиска дополнительных конформаций лиганда с помощью молекулярно-динамической симуляции. Для этого предварительно рассчитывались средние энергии силового поля ван-дер-ваальсовых и электростатических взаимодействий лигандов с водой в течение 20 нс.
Для расчета потенциальной энергии кулоновского взаимодействия лиганда с окружением траектории МД-симуляции белок–лиганд перерабатывали с использованием метода обобщенного поля реакции (Generalized RF) при отсечке 12 Å. Эмпирические константы α и β, определяющие вклад ван-дер-ваальсовых и электростатических взаимодействий в оценку энергии связывания лигандов, выбраны в соответствии со статьей [22]. Коэффициент сдвига γ был принят равным нулю в связи с невозможностью его подбора на данном этапе исследования. Коэффициент γ – дополнительный, он зависит от особенностей модели и гидрофобности активного центра и требуется для согласования данных LIE с экспериментальными данными.
Расчет свободной энергии связывания лиганда с ферментом BsPyNP методом возмущения свободной энергии (FEP). Расчет свободной энергии связывания лигандов с белком BsPyNP проводили, используя термодинамический цикл, описанный Aldeghi et al. [23]. Первый этап заключался в сборе отсчетов производной гамильтониана системы по параметру связи Кирквуда (λ-параметр, [24]) (для метода термодинамического интегрирования (TI) и разницы в потенциальной энергии системы между текущим состоянием и остальными (для методов на основе FEP)).
Для систем белок–лиганд и белок–вода проводили МД-симуляцию в NPT-ансамбле в течение 2.4 нс, используя алгоритм интегрирования “leapfrog” уравнений стохастической динамики для каждого набора λ-параметров. С целью удержания лиганда в области связывания при уменьшении силы взаимодействия с белком вводили дополнительные межмолекулярные ограничения – ориентационные рестрейны [23], используя программу PyFEPRestr [25]. Для системы белок–лиганд параметр связи λ менялись последовательно для λrestr (0, 0.010, 0.025, 0.050, 0.075, 0.100, 0.200, 0.350, 0.500, 0.750, 1.000) и λcoul (0, 0.25, 0.50, 0.75, 1.00); λVdW (0, 0.05, 0.10, 0.20, 0.30, 0.40, 0.50, 0.60, 0.65, 0.70, 0.75, 0.80, 0.85, 0.90, 0.95, 1.00). В случае системы лиганд–вода параметры связи менялись лишь для кулоновских и ван-дер-ваальсовых взаимодействий лиганда с водой. Необходимую для вычисления свободной энергии связывания (ΔGbind) поправку на энергию введенных ограничений для системы лиганд–вода (ΔGsolv_rest) считали аналитически по формуле, приведенной в работе [26]. Ван-дер-ваальсовые взаимодействия рассчитывали, используя потенциал “мягкого ядра” (soft core) Леннарда–Джонса. Для учета как дальних кулоновских, так и ван-дер-ваальсовых взаимодействий использовали метод частица–сетка (PME) [27].
До проведения МД-симуляций в дополнение к общему уравновешиванию системы при каждом значении параметра связи λ проводили процедуры оптимизации геометрии и уравновешивания системы в NVT- и NPT-ансамблях в течение 100 пс.
Обработку данных МД-симуляций при разных параметрах связи Кирквуда осуществляли в написанной в среде Jupyter Notebook программе, с использованием библиотек alchemlib и pymbar. Первый шаг – извлечение разницы редуцированных потенциалов (ΔUij) – выполняли с использованием библиотеки alchemlyb. Затем проводили отбор нескорелированных отсчетов ΔUijи отсечение неуравновешенной части траекторий с использованием функции detectEquilibration библиотеки pymbar [28]. В качестве входного параметра для расчета статистической неэффективности использовали значения ΔUijмежду МД-симуляциями с соседними λ. На третьем шаге статистически независимые отсчеты ΔUijобрабатывали с использованием метода множественного отношения вероятности принятия шага Беннетта (MBAR) [29]. Корректность расчетов контролировали по матрице перекрытия между λ-состояниями, а также сравнением полученных значений ΔG со значениями, полученными другими методами: EXP (метод экспоненциального усреднения) [28], BAR (метод отношения вероятности принятия шага Беннета) [29] и TI (метод термодинамического интегрирования).
Поскольку суммарный заряд атомов PIA при pH > 6.5 равен –1e, необходимо было в этом случае вычислить поправки на суммирование по Эвальду и периодические граничные условия, их рассчитывали с использованием аналитической схемы [30]. Заряд белка был полностью нейтрализован противоионами, эффективный QP = 0. 1/4πεo полагался равным 138.93545585 кДж нм e−2 моль−1, εs для TIP3P воды –82, ρ300K = 997 кг/м3, γS = 0.0764 e нм2. Остаточные интегральные потенциалы (RIP) молекулы белка и лиганда рассчитывали с использованием программы APBS [24].
Свободную энергию связывания белок–лиганд (ΔGbind) рассчитывали по формуле:
В случае заряженного лиганда как к ΔG prot, так и ΔG solv добавляются соответствующие поправки: ΔΔGDSF – часть поправки, связанной с использованием явной (дискретной) модели растворителя, учитывающая конечные размеры ячейки моделирования; ΔΔGNET – поправка на влияние периодических граничных условий на кулоновское взаимодействие бесконечного количества периодических копий точечного заряда, ΔΔGUSV – поправка на влияние эффекта неполной сольвации; ΔΔGRIP – поправка на влияние остаточного интегрированного потенциала. Часть поправки, связанной с использованием явной (дискретной) модели растворителя, для случая системы бесконечного объема ячейки моделирования (DSI) не включена в выходные данные, т.к. ее значение (–74.11 кДж/моль) одинаково для всех рассматриваемых в статье систем белок–лиганд и лиганд–растворитель и обнуляется при расчете ΔGbind [24, 30, 31].
ЗАКЛЮЧЕНИЕ
В ходе работы был проведен поиск специфичного ингибитора пиримидиннуклеозидфосфорилазы. В качестве возможных лигандов были изучены 2',3'-дидегидро-3'-дезокситимидин (d4T), 1-(2-дезокси-2-фтор-β-D-арабинофуранозил)-5-иодурацил (фиауридин, FIAU), 1-(2-дезокси-2-фтор-β-D-арабинофуранозил)-5-урацил (FAU) и 2-пиримидин-2-ил-1H-имидазол-4-карбоновая кислота (PIA) с помощью методов молекулярного моделирования комплексов белок–лиганд, таких как молекулярный докинг и молекулярная динамика, позволяющие найти энергию связывания белка с лигандом (ΔGbind) с использованием методов LIE и FEP. По данным моделирования, а именно по величине свободной энергии связывания белок–лиганд, можно сделать вывод о том, что лиганды d4T и PIA – потенциальные ингибиторы бактериальных широкоспецифичных пиримидиннуклеозидфосфорилаз, т.к. их связывание с активным центром энергетически выгодно.
В настоящее время соединение d4T выпускается под названием Ставудин (ОАО Фармсинтез, Россия) и применяется для лечения ВИЧ [32, 33]. Возможный ингибитор PIA не проходил клинические исследования. Клинические исследования для лиганда FAU не проводились, а для лиганда FIAU в ходе эксперимента была обнаружена высокая токсичность [34].
Полученные в ходе расчетов результаты указывают на то, что оба соединения – PIA и d4T – связываются с активным центром BsPyNP с наибольшей аффинностью среди других предполагаемых субстратов. Причем при моделировании связывания PIA в средах с различным pH энергия принимает более высокие значения по модулю, чем при нейтральном pH. Это, вероятно, объясняется образованием шести водородных связей с 3 а.о. белка при pH > 6.5, в отличие от пяти водородных связей при pH < 6.5 в случае PIA. Дополнительная водородная связь образуется с Lys108. Особенную роль здесь играет значение pH, т.к. в бактериальных клетках значения pH приближены к 7, т.е. химические реакции проходят в нейтральной среде. Следствием специфического связывания белка BsPyNP с выбранными соединениями может стать снижение скорости процессов ресинтеза азотистых оснований, что, в свою очередь, может привести к гибели бактериальных клеток.
В ходе расчетов свободной энергии связывания для комплекса белок–лиганд с замененными аминокислотными остатками в активном центре, имитирующими активный центр тимидинфосфорилазы человека, было обнаружено: PIA хуже связывается с hTP чем с BsPyNP, это минимизирует возможные побочные эффекты применения этого соединения в терапевтических целях.
Список литературы
Friedkin M., Roberts D. // J. Biol. Chem. 1954. V. 207. P. 245–256.
Liekens S., Bronckaers A., Balzarini J. // Lancet Oncol. 2009. V. 10. P. 628–635. https://doi.org/10.1016/s1470-2045(09)70037-3
Bronckaers A., Balzarini J., Liekens S. // Biochem. Pharmacol. 2008. V. 76. P. 188–197. https://doi.org/10.1016/j.bcp.2008.04.019
Vande Voorde J., Gago F., Vrancken K., Liekens S., Balzarini J. // Biochem. J. 2012. V. 445. P. 113–123. https://doi.org/10.1042/bj20112225
Balaev V.V., Prokofev I.I., Gabdoulkhakov A.G., Betzel C., Lashkov A.A. // Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2018. V. 74. P. 193–197. https://doi.org/10.1107/s2053230x18002935
Van Rompay A.R., Johansson M., Karlsson A. // Pharmacol. Ther. 2003. V. 100. P. 119–139. https://doi.org/10.1016/j.pharmthera.2003.07.001
Lipinski C.A. Lombardo F., Dominy B.W., Feeney P.J. // Adv. Drug Deliv. Rev. 2001. V. 46. P. 3–26. https://doi.org/10.1016/s0169-409x(00)00129-0
Balaev V.V., Lashkov A.A., Prokofev I.I., Gabdulkhakov A.G., Seregina T.A., Mironov A.S., Betzel C., Mikhailov A.M. // Crystallogr. Rep. 2016. V. 61. P. 830–841. https://doi.org/10.1134/S1063774516050023
Korb O., Stützle T., Exner T.E. // Swarm Intell. 2007. V. 1. P. 115–134. https://doi.org/10.1007/s11721-007-0006-9
Goodsell D.S., Olson A.J. // Proteins. 1990. V. 8. P. 195–202. https://doi.org/10.1002/prot.340080302
Korb O., Stützle T., Exner T.E. // J. Chem. Inf. Model. 2009. V. 49. P. 84–96. https://doi.org/10.1021/ci800298z
Metropolis N., Ulam S. // J. Am. Stat. Assoc. 1949. V. 44. P. 335–341. https://doi.org/10.1080/01621459.1949.10483310
Dorigo M., Maniezzo V., Colorni A. // IEEE Trans. Syst. Man Cybern. B Cybern. 1996. V. 26. P. 29–41. https://doi.org/10.1109/3477.484436
Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J.C. // J. Comput. Chem. 2005. V. 26. P. 1701–1718. https://doi.org/10.1002/jcc.20291
Jo S., Kim T., Iyer V.G., Im W. // J. Comput. Chem. 2008. V. 29. P. 1859–1865. https://doi.org/10.1002/jcc.20945
MacKerell A.D., Bashford D., Bellott M., Dunbrack R.L., Evanseck J.D., Field M.J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F.T., Mattos C., Michnick S., Ngo T., Nguyen D.T., Prodhom B., Reiher W.E., Roux B., Schlenkrich M., Smith J.C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. // J. Phys. Chem. B. 1998. V. 102. P. 3586–3616. https://doi.org/10.1021/jp973084f
Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B.L., Grubmüller H., MacKerell A.D., Jr. // Nat. Methods. 2017. V. 14. P. 71–73. https://doi.org/10.1038/nmeth.4067
Petersen H.G. // J. Chem. Physics. 1995. V. 103. P. 3668–3679. https://doi.org/10.1063/1.470043
Parrinello M., Rahman A. // J. Appl. Physics. 1981. V. 52. P. 7182–7190. https://doi.org/10.1063/1.328693
Bussi G., Parrinello M. // Computer Physics Comm. 2008. V. 179. P. 26–29. https://doi.org/10.1016/j.cpc.2008.01.006
Hansson T., Marelius J., Åqvist J. // J. Comput. Aided Mol. Des. 1998. V. 12. P. 27–35. https://doi.org/10.1023/a:1007930623000
Aldeghi M., Heifetz A., Bodkin M.J., Knapp S., Biggin P.C. // Chem. Sci. 2016. V. 7. P. 207–218. https://doi.org/10.1039/c5sc02678d
Lashkov A.A., Tolmachev I.V., Eistrikh-Heller P.A., Rubinsky S.V. // Crystallogr. Rep. 2021. V. 66. P. 861–865. https://doi.org/10.1134/S1063774521050126
Rocklin G.J., Mobley D.L., Dill K.A., Hünenberger P.H. // J. Chem. Phys. 2013. V. 139. P. 184103. https://doi.org/10.1063/1.4826261
Boresch S., Tettinger F., Leitgeb M., Karplus M. // J. Phys. Chem. B. 2003. V. 107. P. 9535–9551. https://doi.org/10.1021/jp0217839
Wennberg C.L., Murtola T., Páll S., Abraham M.J., Hess B., Lindahl E. // J. Chem. Theory Comput. 2015. V. 11. P. 5737–5746. https://doi.org/10.1021/acs.jctc.5b00726
Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. // SoftwareX. 2015. V. 1. P. 19–25. https://doi.org/10.1016/j.softx.2015.06.001
Shirts M.R., Chodera J.D. // J. Chem. Phys. 2008. V. 129. P. 124105. https://doi.org/10.1063/1.2978177
Zwanzig R.W. // J. Chem. Phys. 1954. V. 22. P. 1420–1426.
Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L.E., Brookes D.H., Wilson L., Chen J., Liles K., Chun M., Li P., Gohara D.W., Dolinsky T., Konecny R., Koes D.R., Nielsen J.E., Head-Gordon T., Geng W., Krasny R., Wei G., Holst M.J., McCammon J.A., Baker N.A. // Protein Sci. 2018. V. 27. P. 112–128. https://doi.org/10.1002/pro.3280
Balaev V.V., Prokofev I.I., Gabdoulkhakov A.G., Betzel C., Lashkov A.A. // X-Ray Structure of the Complex Pyrimidine-Nucleoside Phosphorylase from Bacillus subtilis at 1.88 A. Worldwide Protein Data Bank, 2018.
Kandil S., Pannecouque C., Chapman F.M., Westwell A.D., McGuigan C. // Bioorg. Med. Chem. Lett. 2019. V. 29. P. 126721. https://doi.org/10.1016/j.bmcl.2019.126721
Institute of Medicine, Committee to Review the Fialuridine (FIAU/FIAC) Clinical Trials. Review of the Fialuridine (FIAU) Clinical Trials. National Academies Press, 1995. 280 p.
Kirkwood J.G. // J. Chem. Phys. Am. Inst. Phys. 1935. V. 3. № 5. P. 300–313.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия